

Abstract
Protein interactions are essential components of signal transduction in cells. With the progress in genome-wide yeast two hybrid screens and proteomics analyses, many protein interaction networks have been generated. These analyses have identified hundreds and thousands of interactions in cells and organisms, creating a challenge for further validation under physiological conditions. The bimolecular fluorescence complementation (BiFC) assay is such an assay that meets this need. The BiFC assay is based on the principle of protein fragment complementation, in which two non-fluorescent fragments derived from a fluorescent protein are fused to a pair of interacting partners. When the two partners interact, the two non-fluorescent fragments are brought into proximity and an intact fluorescent protein is reconstituted. Hence, the reconstituted fluorescent signals reflect the interaction of two proteins under study. Over the past six years, the BiFC assay has been used for visualization of protein interactions in living cells and organisms, including our application of the BiFC assay to the transparent nematode Caenorhabditis elegans. We have demonstrated that BiFC analysis in C. elegans provides a direct means to identify and validate protein interactions in living worms and allows visualization of temporal and spatial interactions. Here we provide a guideline for the implementation of BiFC analysis in living worms and discuss the factors that are critical for BiFC analysis.
Keywords: BiFC, Venus, C. elegans, Fos, Jun, AP-1, protein interaction
Introduction
Protein interactions are key mechanisms regulating many biological processes in cells, and demonstration of interacting proteins has become an important component of biological research. Although many methods are available for study of protein interactions, fluorescent protein-based methods, such as fluorescence resonance energy transfer (FRET) and bimolecular fluorescence complementation (BiFC), allow direct visualization of protein interactions in living cells (1-6). While the FRET assay allows study of dynamic interactions, its low signal-to-noise ratio and relatively expensive setup limit its use in many laboratories. By contrast, BiFC signals can be readily visualized using any fluorescence microscope and have a high signal-to-noise ratio.
The BiFC assay is based on the principle of protein fragment complementation (7, 8). When two non-fluorescent fragments derived from a fluorescent protein are fused to a pair of interacting proteins, the interaction between two proteins brings the two fragments into close proximity and reconstitutes an intact fluorescent protein (5, 9-12). Hence, the reconstituted fluorescence represents the interaction of two proteins. The BiFC signals can be easily visualized using any fluorescence microscope and the assay can be performed without any additional instrumentation. Over the past six years, BiFC assays have become widely used for studies of protein interactions in variety of model systems, such as Drosophila melanogaster (13) and Xenopus (14) and for diverse families of proteins (6, 7, 15, 16).
The nematode Caenorhabditis elegans has been proven to be an excellent model organism for biological and medical research. The sequencing of its entire genome has facilitated genome-wide identification of interacting proteins (17-21). While these studies offer tremendous opportunities for development of testable hypotheses, validation of these interactions under their normal cellular environment remains a challenge. In addition, assays that allow study of protein interactions in living worms remain extremely limited. Thus, development of methods for visualization of protein interactions in living C. elegans will facilitate these studies. For this purpose, we have engineered two worm BiFC cloning vectors for BiFC analysis in living worms (22). Using C. elegans proteins homologous to mammalian Fos and Jun as a model, we have demonstrated that the Venus-based BiFC assay can be used for direct visualization of protein interactions using an inducible heat shock promoter. In addition, our BiFC cloning vectors have also been used for visualization of temporal and spatial interactions of UNC-1 and UNC-9 at the intracellular junctions in living worms when the native promoter of the myo-3 gene is used (23). We recently reported the detailed protocol for applying BiFC analysis to C. elegans (22). To facilitate the BiFC analysis of protein interactions in living C. elegans, here we discuss how to select experimental strategies based on the ultimate research goals and provide general guidelines to help users perform BiFC analysis in living C. elegans. We also provide special considerations of factors that may affect the experimental outcomes.
General design of BiFC analysis in worms
1. Worm BiFC cloning vectors
Determination of protein interactions provides insight into the molecular mechanisms of signal transduction and genetic pathways in C. elegans. Although genetic analysis allows linear arrangement of two genes in a genetic pathway, whether the products of two genes directly interact with each other requires proof using additional experimental approaches. Because of the lack of available imaging methods, biochemical methods such as co-immunoprecipitation or in vitro binding assays are often used to determine if two proteins interact. Often times, two proteins are expressed in different model organisms such as mammalian cells or yeast for further validation of their interaction using the methods available to these organisms. However, results from these analyses must be cautiously interpreted. First, certain C. elegans proteins cannot be well expressed in other organisms. Second, interactions detected using other methods in other organisms or in vitro do not necessarily prove their interactions in worms. Hence, it is desirable to have an assay that allows study of protein interactions in worms. To meet this need, we have recently applied our Venus-based BiFC assay to C. elegans (22). We chose non-fluorescent fragments derived from Venus to develop the worm BiFC assay because chromophore maturation of Venus is less sensitive to low pH and high temperatures (24) and because these fragments produced the highest BiFC signal in mammalian cells when compared with other GFP variants (11). Since BiFC complexes are essentially irreversible (5), we chose the plasmid pPD49.83 (25) that contains the heat shock promoter hsp-16.41(25) to construct two worm BiFC cloning vectors, pCE-BiFCVN173 and pCE-BiFC-VC155. We also incorporated sequences encoding a Myc or HA tag upstream of the multiple cloning sites to allow for detection of BiFC fusion proteins. Sequences encoding a flexible linker were inserted between the multiple cloning sites and the sequences encoding the non-fluorescent fragments VN173 or VC155. Detailed information for these cloning vectors is presented in the worm BiFC protocol (22).
2. Selection of experimental strategies
There are three reasons to visualize protein interactions in C. elegans. First, there is strong genetic evidence that two genes interact with each other and the interaction of the encoded proteins would provide an explanation to the genetic data. In this case, verification of the interaction between two proteins becomes an important part of the research. Second, the interaction of two proteins is already demonstrated using biochemical methods or other methods in other model organisms, and further verification of the interaction in living worms is desirable. Third, visualization of temporal and spatial interactions of two proteins is the goal. Hence, protein interaction studies in living worms can be generally classified into two major experimental goals: validation of protein interactions and visualization of temporal/spatial interactions. The worm BiFC cloning vectors discussed above can be used for both purposes, but it is important to determine your research goal first before you select your experimental strategies. Figure 1 illustrates a general strategy of worm BiFC analysis depending on research purposes. If the experimental goal is to simply determine a potential interaction between two proteins, subclone the cDNAs encoding the two proteins using available restriction enzyme sites in the multiple cloning sites into pCE-BiFC-VN173 and pCE-BiFC-VC155, and then establish transgenic lines by co-injecting the two plasmids along with a co-injection marker, such as pRF4 [rol-6(su1006)] (26), into N2 worms. This allows a rapid and direct validation of two interacting proteins in living worms after heat shock. The use of the heat shock-inducible expression system not only enables the development of transgenic lines but also allows the control of expression levels by varying the heat shock conditions. It is also possible to use biolistic transformation method to establish integrated transgenic lines (26, 27). Although this is not necessary for a simple validation of interacting proteins, the integrated lines have less variation and are useful for quantitative analysis of protein interactions. In addition, the expression levels of BiFC fusion proteins are lower as biolistic transformation usually results in transformants carrying a single or low copy number of transgenes (27).
Figure 1. Strategies for BiFC analysis of protein interactions in C. elegans.
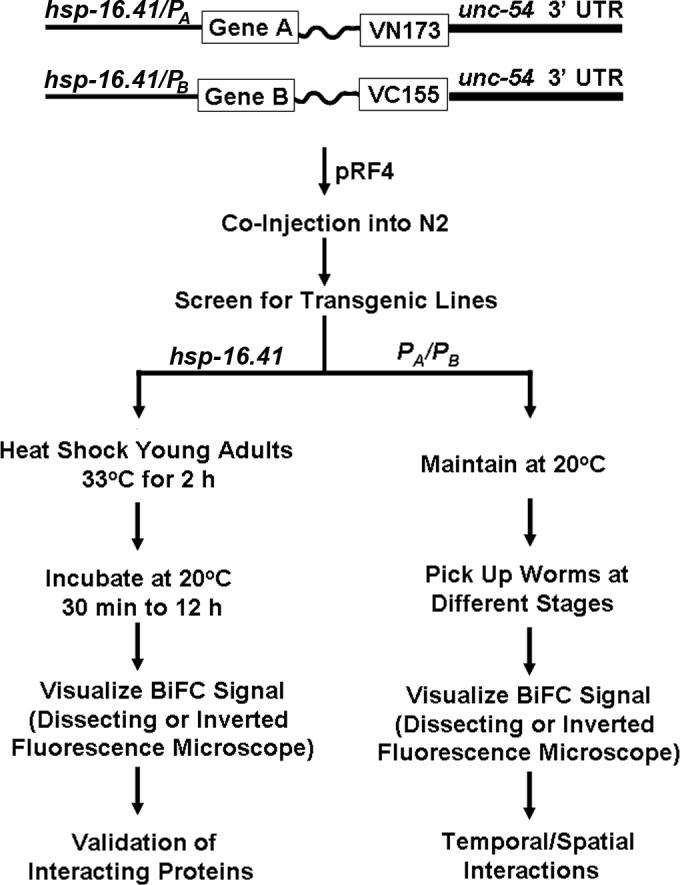
Shown is a flow chart for BiFC analysis of protein interactions in C. elegans. If the goal is to validate two interacting proteins in worms, use the hsp-16.41 promoter in both pCEBiFC-VN173 and pCE-BiFC-VC155 cloning vectors (22), and subclone the cDNAs encoding protein A and B into the multiple cloning site and follow the procedures on the left side to perform BiFC analysis. If the goal is to visualize temporal and spatial interactions, replace the heat shock promoter with gene specific promoters (PA and PB) and follow the procedures on the right side to perform BiFC analysis. Gene A or Gene B: cDNA encoding protein A or B. VN173: cDNA encoding residues 1−173 of Venus. VC155: cDNA encoding residues 155−238 of Venus.
Temporal and spatial protein interactions are mechanisms that are often utilized for regulation of developmental processes in animals. C. elegans has a transparent body and offers the unique opportunity to visualize temporal and spatial interactions throughout the entire developmental processes. If visualization of temporal and spatial interactions in living worms is the experimental goal, native promoters and full-length cDNAs should be used. This is especially important when two genes are temporally and spatially expressed throughout the development and they are likely involved in the regulation of C. elegans development via their temporal and spatial interactions. For this purpose, replace the heat shock promoter in pCE-BiFC-VN173 and pCE-BiFC-VC155 with native promoters of the two genes under study. Because the use of native promoters allows expression of BiFC fusion proteins throughout the development and because BiFC complexes are essentially irreversible (5), formation of stable complexes could prevent the establishment of transgenic lines. If this happens, it is necessary to establish two transgenic lines separately that allow expression of individual BiFC fusion proteins. However, analysis of temporal and spatial interactions requires crossing of two lines. To facilitate identification of worms that co-express two fusion proteins after crossing, it is necessary to use different co-injection markers (26) for the establishment of each line so that worms can be easily identified for visualization of temporal and spatial interactions.
3. Controls
BiFC assays measure the proximity of two interacting proteins. The reconstitution of fluorescence does not necessarily require physical interaction of two proteins. Because non-fluorescent fragments from all fluorescent proteins tested so far have a tendency to reconstitute fluorescence to a certain degree in the absence of protein interactions, overexpressed BiFC fusion proteins could randomly collide with each other and produce fluorescent signals. Although we did not observe fluorescence when VN173 and VC155 were co-expressed in worms (Fig. 2a), non-specific assembly could potentially occur when other BiFC fusion proteins are co-expressed. It is therefore essential to include negative controls in the BiFC analysis (5, 10, 11, 15, 28). If the interaction interface of the protein of interest is known, introduce substitutions into the interaction interface or make small deletions. Otherwise, interaction mapping using biochemical approaches is necessary in order to identify the interaction interfaces. The control plasmids should be similarly constructed by subcloning cDNAs encoding the mutant proteins into either pCE-BiFC-VN173 or pCE-BiFC-VC155, and follow the same procedures as shown in Figure 1 to establish transgenic lines. Transgenic worms that carry pCE-bJUN-VN173 and pCE-bFOS-VC155 and transgenic worms that carry pCE-bJUN-VN173 and pCE-bFOSΔZIP-VC155 can be used as both positive and negative controls, respectively, to ensure that the system works appropriately (e.g. heat shock conditions, microscope setup) (Fig. 2a). These two transgenic lines (CDH1 and CDH2) have been deposited in the Caenorhabditis Genetics Center (CGC).
Figure 2. BiFC analysis of FOS-1-JUN-1 interactions.

(a). Shown are representative phase and fluorescent images (YFP) acquired from transgenic worms expressing the indicated proteins. All transgenic worms were heat shocked at 33°C for 3 h and recovered at 20°C for 3 h before image acquisition. bFOSΔZIP is a dimerization deficient mutant of FOS-1 and used as a negative control. The lower panel shows images acquired from the transgenic worms that only carry the two BiFC cloning vectors pCE-BiFC-VN173 and pCE-BiFC-VC155. (b). Immunoblotting analysis of expressed fusion proteins from experiments in a. Thirty worms from each group were lysed and the total lysate was used for immunoblotting of bJUN-VN173 (lane 1 and 2) or VN173 (lane 3) with anti-Myc antibody (top panel), or for immunoblotting of bFOS-VC155 (lane 1), bFOSΔZIP-VC155 (lane 2), or VC155 (lane 3) with anti-HA antibody (bottom panel).
4. Microscope setup
The Venus-based BiFC complexes have an excitation peak at 515 nm and an emission peak at 528 nm (11). The fluorescence is detectable with GFP, FITC, and YFP filters that are available on many dissecting fluorescence microscopes or inverted fluorescence microscopes. However, the JP4 CFP/YFP filters from Chroma is the most suitable filter setup for visualization of Venus-based fluorescent signals and has been used for our mammalian and worm BiFC analysis as well as quantification of BiFC signals (11, 22). For quantification of BiFC signals and for high resolution imaging analysis, it is necessary to use high magnification objective lenses (60X or higher) for the acquisition of fluorescent images. In addition, it is necessary to have imaging analysis software (e.g. MetaMorph from Universal Imaging) for quantification of fluorescence intensity.
Validation of interacting proteins in living worms by BiFC analysis
BiFC analysis of interacting proteins using the heat shock promoter is relatively straightforward. Once the transgenic lines are established, pick young adult rollers and heat shock worms at 33°C for 2 h, followed by recovery at 20°C for 30 min. This is usually sufficient to induce expression of fusion proteins and allows direct visualization of fluorescent signals under a dissecting fluorescence microscope or under a low magnification lens of an inverted fluorescence microscope. The distribution of fluorescent signals is ubiquitous and is consistent with the expression patterns of the heat shock promoter, with the highest expression in intestinal cells and late embryos (29) (Fig. 2a). If a 30 min recovery is not enough to see fluorescent signals, extend the incubation at 20°C and examine at a later time. We have observed that following a 2 h heat shock, the peak fluorescence can be seen between 6 h and 12 h (Fig. 3a, Fig. 4). The BiFC complexes composed of the basic region leucine zipper (bZIP) domains of FOS-1 (bFOS) and JUN-1 (bJUN) have a half life of about 6−12 h (Fig. 3b). We have also noticed that a heat shock as short as 30 min produced BiFC signals when bFOS-bJUN interactions were examined (unpublished observations). Heat shock can be performed from 28°C to 33°C, and multiple heat shocks with a 3−6 h interval may be performed. However, heat shock of worms for longer than 6 h or heat shock at high temperatures (>33°C) may kill worms (unpublished observations).
Figure 3. Fluorescence and expression profiles of the bFOS-bJUN interaction.

(a). Ten synchronized young adult worms carrying the plasmids pCE-bFOS-VC155 and pCE-bJUN-VN173 were heat-shocked at 33°C for 3 h, followed by incubation at 20°C. At the indicated time points after the heat shock, fluorescent images were acquired using the YFP filter setup (20X objective lens). The fluorescence intensity in the nucleus of five intestinal cells from each worm was quantified, and the average fluorescence intensity from 10 worms was calculated and presented as a function of time. Error bars indicate standard deviation. (b). Immunoblotting analysis of expressed HA-bFOS-VC155 and Myc-bJUN-VN173 fusion proteins at the indicated time after heat shock.
Figure 4. Comparison of BiFC signals in transgenic worms established by two different transformation methods.

Shown are representative phase (60X) and the corresponding fluorescent images (YFP) acquired at the indicated recovery time at 20°C after heat shock at 33°C for 2 h. Transgenic worms carrying the same BiFC constructs encoding the bZIP domains of two C. elegans bZIP proteins fused to VN173 and VC155, respectively, were established by either microinjection (Injection) or biolistic transformation (Bombardment). The exposure time for the acquisition of each fluorescent image is shown in the bottom right corner.
The introduction of mutations into the interaction interfaces of proteins should abolish or significantly decrease the BiFC signal if the BiFC signal reflects the specific interaction between two proteins. As shown in Figure 2a, deletion of the ZIP region from FOS-1 dramatically decreases the BiFC signal under the same experimental conditions as did for wild-type proteins (Fig. 2a, middle panel). Unlike the mammalian BiFC system in which VN173 and VC155 can readily self-assemble in COS-1 cells (11), co-expression of VN173 and VC155 in worms did not produce any detectable fluorescent signal under the same experimental conditions although these fragments were comparably expressed (Fig. 2a, 2b). If the fluorescence intensity in worms that carry wild-type or mutant fusion proteins is not obviously distinguishable, it is necessary to acquire fluorescent images and quantify the difference in fluorescence intensity between transgenic worms that express wild-type fusion proteins and those that express mutant fusion proteins. To do this, place at least 10 worms on agarose slides and acquire fluorescent images using appropriate objectives. If entire worms will be used as a region of interest (ROI), a low magnification objective lens (e.g. 10X or 20X) should be used. If individual regions or cells are used as ROIs, acquire fluorescent images from corresponding regions of both experimental and control worms using high magnification (60X or above). It is important to use the same exposure time for the acquisition of fluorescent images from experimental and control groups. In addition, select worms at the same developmental stages for quantification because expression levels may vary depending on the developmental stages. It is recommended to run a pilot test to identify appropriate exposure time that not only allows adequate quantification of fluorescence intensity but also avoids acquisition of saturated images. Because intestinal cells show higher expression of proteins driven under the heat shock promoter hsp-16.41, it is convenient to quantify fluorescence intensity in intestinal cells when a high magnification objective lens is used. To determine the statistical significance between experimental and control worms, quantify at least 50 ROIs for comparison between experimental and controls worms using software such as MetaMorph. This can be achieved by selecting 5−10 ROIs from each worm, quantifying the average fluorescence intensity of each ROI, and determining the average of all ROIs from 10−20 worms. Alternatively, quantify total fluorescence intensity in each worm and determine the average total fluorescence intensity from 50 worms if the entire worm is used as ROI.
When using transgenic lines for BiFC analysis, plasmid copy number can vary greatly, affecting fluorescent output. It is therefore critical to determine expression levels of each fusion protein by Western blotting as discussed below in section three of the special considerations. This may aid in normalization of expression levels, allowing comparison of fluorescent signals derived from multiple transgenic lines.
Visualization of temporal and spatial interactions of proteins
To study temporal and spatial interactions of two proteins, transgenic worms that are established through the co-injection of two BiFC plasmids can be directly analyzed if co-expression of two fusion proteins is not lethal. The analysis is analogous to the temporal and spatial study of expression patterns except that the fluorescent signals represent the interaction of two proteins under study. Because expression levels of BiFC fusion proteins driven under native promoters may be low, fluorescent signals derived from the formation of BiFC complexes may not be strong enough to easily differentiate from autofluorescence. To verify if the observed fluorescent signals are from reconstituted Venus, switch the YFP filters to different filter setups such as CFP or DAPI. If the ratio of YFP to CFP or DAPI is higher in experimental lines as compared to negative controls, this indicates a specific interaction. Otherwise, the fluorescent signals most likely come from autofluorescence. To avoid the potential problem that higher levels of expression of two fusion proteins at different developmental stages or in different tissues may produce fluorescent signals independently of specific interactions, transgenic lines that co-express a mutant BiFC fusion protein with another wild-type BiFC fusion protein should be similarly analyzed as described above for the validation of interacting proteins. If necessary, quantification of BiFC signals should be applied using the fluorescent images acquired from the same tissues and at the same developmental stages.
To analyze temporal and spatial interactions using two transgenic lines that express a single BiFC fusion protein, first cross homozygous hermaphrodites of an integrated transgenic line expressing a BiFC fusion protein (e.g. Protein A-VN173) with N2 males, and then cross the heterozygous F1 males with homozygous hermaphrodites of the second integrated transgenic line expressing the second BiFC fusion protein (e.g. Protein B-VC155). Then, identify homozygous F2 progeny for visualization of temporal and spatial interactions. Compared to the direct visualization of protein interactions using the heat shock promoter, study of temporal and spatial interactions require analysis of homozygous F2 progeny. If fluorescent signals are observed at specific developmental stages and/or in specific tissues, similar analysis should be performed by crossing the mutant control lines to establish the specificity of the interactions. However, if formation of stable BiFC complexes is lethal, or causes developmental arrest, further analysis at later developmental stages will not be possible. This may require development of transgenic lines that integrate inducible expression systems as discussed below.
Special considerations for BiFC analysis in C. elegans
1. Optional designs for BiFC plasmids
Because the reconstitution of fluorescence in BiFC analysis depends on the proximity of two non-fluorescent fragments, the position of fusions and the structure of target proteins have a major impact on the successful application of BiFC assay. In most cases, the structural information of proteins of interest may not be available. It is therefore necessary to try different designs of BiFC plasmids if the C-terminal fusions described in this method do not work. In addition, C-terminal fusions may not allow expression of a fusion protein if the C-terminal sequence undergoes post-translational modifications, such as lipid modifications of small GTPase proteins (30). It is therefore necessary to construct two other BiFC plasmids by expressing VN173 or VC155 as an N-terminal fusion. Although we do not provide generic BiFC cloning vectors for tagging VN173 or VC155 to the N-terminus of protein of interest, this can be easily achieved by subcloning the sequences encoding VN173 and VC155 into the multiple cloning sites using the most upstream restriction enzyme sites, and then subclone the cDNAs encoding the proteins of interest downstream of VN173 or VC155. Note that a flexible linker is necessary between the protein of interest and VN173 or VC155. A linker that integrates one to four repeats of the glycine spacer (GGGGS) or uses the residues encoded by the sequences in the multiple cloning sites is appropriate provided that five to 20 residues are used (28). The availability of four BiFC plasmids allows establishment of transgenic lines that co-express fusion proteins in eight different combinations (28). If these efforts do not establish positive interactions, additional optimizations may be tried. For example, vary the length of linkers, use partial structures (e.g. interaction domains) of proteins, or insert VN173 or VC155 into the loops of proteins (e.g. intracellular loops of membrane-bound proteins). The ultimate goal of these efforts is to increase the proximity of VN173 and VC155 if a specific interaction does exist. However, it is important to note that the lack of reconstituted fluorescence does not suggest the lack of interactions because the inappropriate design of BiFC constructs may not allow two fragments to be close enough for complementation, despite the fact that two proteins under study may specifically interact with each other. Similar strategies discussed in details for mammalian BiFC analysis (15, 28) also apply to BiFC analysis in C. elegans.
2. Heat shock conditions
For transgenic lines established by microinjection of BiFC plasmids (10−15 ng/μl BiFC plasmids and 100 ng/μl pRF4) (22), the heat shock time can vary from 30 min to 3 h. A longer heat shock time coincides with a shorter recovery time. However, longer recovery time is necessary if transgenic lines are established by biolistic transformation. In addition, multiple heat shocks may be necessary if one 2 h heat shock is not sufficient to visualize fluorescent signals. This is due to the fact that integrated transgenic lines established by biolistic transformation often have a single or low copy numbers of transgenes (27). We have established a transgenic line expressing the bZIP domains of two C. elegans bZIP proteins as BiFC fusion proteins by biolistic transformation. Although heat shock of worms at 33°C for 2 h with a 3 h recovery did not generate any detectable fluorescent signal, incubation at 20°C for 6 h did show detectable fluorescence, albeit a longer exposure time was used (Fig. 4). By contrast, transgenic lines established by microinjection using the same constructs all produced bright fluorescent signals with the recovering time from 30 min to 24 h (Fig. 4). In fact, we have established 18 different transgenic lines that express different combinations of bZIP domains, and heat shock at 33°C for 2 h followed by a 2 h recovery is sufficient to produce bright fluorescence in lines that express proteins which do interact.
3. Impact of protein expression levels on BiFC analysis
The reconstitution of fluorescence in BiFC analysis can be affected by many factors. These include the design of BiFC constructs described above, number and abundance of endogenous interacting proteins, and the expression levels of fusion proteins. The final BiFC signals only represent those that are structurally favorable and free of binding to endogenous interacting proteins. Under the same genetic background and the experimental conditions, the expression levels of fusion proteins have direct impact on the final signal output. Because wild-type and mutant fusion proteins may have different half-lives, it is necessary to ensure that wild-type and mutant fusion proteins are comparably expressed. For this purpose, we have engineered the sequences encoding HA or Myc tags fused to the N-terminal end of fusion proteins in the two BiFC cloning vectors. This allows detection of expression levels of fusion proteins by immunoblotting and the comparison of wild-type and mutant fusion proteins. While expression levels of β-actin appear to vary at different developmental stages, histone 3 expression levels are relatively consistent and can be used as a loading control if the same number of worms is used for preparation of total lysate for immunoblotting (22) (Fig. 3b). If expression levels of wild-type and mutant fusion proteins are different at the same developmental stages, it is necessary to induce expression of fusion proteins to different levels by varying the heat shock conditions or by incubating worms to different times after the heat shock. Alternatively, multiple heat shocks can be performed. Then, determine the expression levels of fusion proteins under different conditions and identify the conditions that show comparable expression of both wild-type and mutant fusion proteins. The quantified fluorescence intensity from worms that express comparable levels of fusion proteins can then be compared and analyzed for statistical significance. For example, if transgenic worms expressing a wild-type fusion protein 2 h after heat shock are comparable to that from worms that express mutant fusion protein 3 h after the heat shock, then the quantified fluorescence intensity from these two samples can be compared. Contrary to the heat shock-inducible expression system, this approach cannot be used for the quantification of temporal and spatial interactions because of the lack of control of protein expression levels. Selection of different transgenic lines that have different copy number of plasmids integrated can be used for quantitative analysis. However, qualitative analysis of temporal and spatial interactions should satisfy most of the experimental goals.
4. Effect of stable BiFC complexes on visualization of temporal and spatial interactions
Temporal and spatial interactions can readily be analyzed if formation of stable BiFC complexes does not interfere with the function of endogenous signaling and if worms co-expressing two BiFC fusion proteins are viable and healthy. However, stable formation of BiFC complexes could function in a dominant active or dominant negative manner and cause lethality. Although this may still represent valid temporal and spatial interactions, further analysis at later developmental stages is prohibited. If this is the case, on alternative is to use the temperature-sensitive mutant smg-1(cc546ts), a strain that has been successfully used to control the expression of transgenes (31, 32). In C. elegans, the products of the smg genes are required for the degradation of mRNAs that carry a premature stop codon or unusually long 3'UTR. The advantage of smg systems is that transgenes are not expressed at the permissive temperature (16°C) because of the degradation of mRNAs by SMG proteins. However, the SMG-induced degradation of mRNAs is inactivated when worms are cultured at restrictive temperature (23−26°C). This temperature-inducible system allows the establishment of transgenic lines by microinjection under cultivation of worms at 16°C and enables the analysis of temporal and spatial interactions at 23−26°C. The induction of fusion proteins may require incubation of transgenic worms at 23−26°C for 24−48 h. Compared to the heat shock inducible system, the only drawback is that the entire process, including creation of transgenic lines and induction of protein expression, may take a longer time. However, incubation of worms at 23−26°C starting from different developmental stages should allow the analysis of temporal and spatial interactions throughout the development. We have used the 3'UTR from plasmid L3809 (1997 Fire Kit) to construct our BiFC expression plasmids for temporal and spatial interactions of two bZIP proteins (Fig. 5). This construct contains an unusually long 3' UTR which consists of out-of-frame portions of the let-858 coding sequence (1997 Fire Kit). The two BiFC plasmids were co-injected into smg-1(cc546ts) strain (http://www.cbs.umn.edu/CGC/) along with pRF4 that encodes the rol-6(su1006) morphological marker. Although we have failed to establish any transgenic lines by co-injecting the plasmids encoding FOS-1 and JUN-1 fused with VN173 and VC155 under the control of their native promoters into N2 worms, we have recently succeeded in establishing several transgenic lines using the smg system. Incubation of these transgenic worms at 26°C did allow visualization of temporal and spatial interactions of FOS-1 and JUN-1 (unpublished observations).
Figure 5. Strategy for BiFC analysis of temporal and spatial interactions in C. elegans using a smg inducible system.

Shown is a flow chart for BiFC analysis of temporal and spatial interactions using a smg inducible expression system. Note that the 3'UTR is from plasmid L3809 from the 1997 Fire Kit and that smg-1(cc546) I is from CGC. The promoter regions, gene of interests, and the sequences encoding VN173 and VC155 along with the linker sequences can be released or amplified from the constructs in Figure 1 and cloned into L3809.
Concluding remarks
Analysis of protein interactions for C. elegans proteins has been hampered by the lack of available cell lines. Given the transparent nature of C. elegans, BiFC analysis of interacting proteins in living C. elegans offers a unique opportunity to not only identify and validate interacting proteins but also allow visualization of temporal and spatial interactions of interacting proteins throughout development. This is especially important given the fact that many interacting proteins have been identified through the efforts of genome-wide analysis in C. elegans. Although the analysis of a single pair of interacting proteins is presented here, it is also possible to implement a multicolor BiFC analysis to simultaneously visualize multiple interactions throughout development (10, 11). This is particularly useful to study how a protein selectively interacts with different partners throughout development. Although further developments are necessary to facilitate the BiFC analysis of temporal and spatial interactions, we hope the design and general strategies discussed here and the detailed protocols described elsewhere (22) will facilitate the BiFC analysis of protein interactions in living C. elegans.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Atsushi Miyawaki for kindly providing the cDNA of Venus. We also thank Ron E. Ellis and Tom K. Kerppola for helpful discussions and support during the course of this work. This work was supported by a grant from National Science Foundation (0420634-MCB) and the support from the Purdue Cancer Center (NCI-PI30CA23168).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chan FK, Siegel RM, Zacharias D, Swofford R, Holmes KL, Tsien RY, Lenardo MJ. Cytometry. 2001;44:361–8. doi: 10.1002/1097-0320(20010801)44:4<361::aid-cyto1128>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Majoul I, Straub M, Duden R, Hell SW, Soling HD. J. Biotech. 2002;82:267–77. doi: 10.1016/s1389-0352(01)00042-3. [DOI] [PubMed] [Google Scholar]
- 3.Pollok BA, Heim R. Trends Cell Biol. 1999;9:57–60. doi: 10.1016/s0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- 4.Periasamy A, Day RN. Methods Cell Biol. 1999;58:293–314. doi: 10.1016/s0091-679x(08)61962-7. [DOI] [PubMed] [Google Scholar]
- 5.Hu CD, Chinenov Y, Kerppola TK. Mol. Cell. 2002;9:789–98. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 6.Kerppola TK. Nat. Rev. Mol. Cell. Biol. 2006;7:449–56. doi: 10.1038/nrm1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Nat. Rev. Drug Discov. 2007;6:569–82. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 8.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN. Methods Enzymol. 2000;328:208–30. doi: 10.1016/s0076-6879(00)28399-7. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh I, Hamilton AD, Regan LJ. Am. Chem. Soc. 2000;122:5658–59. [Google Scholar]
- 10.Hu CD, Kerppola TK. Nat. Biotech. 2003;21:539–45. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shyu YJ, Liu H, Deng X, Hu CD. Biotechniques. 2006;40:61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- 12.Jach G, Pesch M, Richter K, Frings S, Uhrig JF. Nat. Methods. 2006;3:597–600. doi: 10.1038/nmeth901. [DOI] [PubMed] [Google Scholar]
- 13.Benton R, Sachse S, Michnick SW, Vosshall LB. PLoS Biol. 2006;4:e20. doi: 10.1371/journal.pbio.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saka Y, Hagemann AI, Piepenburg O, Smith JC. Development. 2007;134:4209–18. doi: 10.1242/dev.010645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerppola TK. Nat. Protoc. 2006;1:1278–86. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson CG, Magliery TJ, Regan L. Nat. Method. 2004;1:255–62. doi: 10.1038/nmeth1204-255. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Han M. Bioessays. 2000;22:503–6. doi: 10.1002/(SICI)1521-1878(200006)22:6<503::AID-BIES2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 18.Boulton SJ, Gartner A, Reboul J, Vaglio P, Dyson N, Hill DE, Vidal M. Science. 2002;295:127–31. doi: 10.1126/science.1065986. [DOI] [PubMed] [Google Scholar]
- 19.Walhout AJ, Reboul J, Shtanko O, Bertin N, Vaglio P, Ge H, Lee H, Doucette-Stamm L, Gunsalus KC, Schetter AJ, Morton DG, Kemphues KJ, Reinke V, Kim SK, Piano F, Vidal M. Curr. Biol. 2002;12:1952–8. doi: 10.1016/s0960-9822(02)01279-4. [DOI] [PubMed] [Google Scholar]
- 20.Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Science. 2000;287:116–22. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 21.Zhong W, Sternberg PW. Science. 2006;311:1481–4. doi: 10.1126/science.1123287. [DOI] [PubMed] [Google Scholar]
- 22.Shyu YJ, Hiatt SM, Duren HD, Ellis RE, Kerppola TK, Hu C-D. Nat. Protoc. 2008;3:588–96. doi: 10.1038/nprot.2008.16. [DOI] [PubMed] [Google Scholar]
- 23.Chen B, Liu Q, Ge Q, Xie J, Wang ZW. Curr. Biol. 2007;17:1334–9. doi: 10.1016/j.cub.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. Nat. Biotech. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 25.Mello C, Fire A. Methods Cell Biol. 1995;48:451–82. [PubMed] [Google Scholar]
- 26.Evans TC, editor. The C. elegans Research Community, WormBook. (WormBook) 2006 April 6; doi/10.1895/wormbook.1.108.1, http://www.wormbook.org.
- 27.Praitis V, Casey E, Collar D, Austin J. Genetics. 2001;157:1217–26. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu CD, Grinberg A, Kerppola T. Curr. Prot. Cell Biol. 2005:21.3.1–21.3.21. [Google Scholar]
- 29.Stringham EG, Dixon DK, Jones D, Candido EP. Mol. Biol. Cell. 1992;3:221–33. doi: 10.1091/mbc.3.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang FL, Casey PJ. Annu. Rev. Biochem. 1996;65:241–69. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 31.Mango SE. Trends Genet. 2001;17:646–53. doi: 10.1016/s0168-9525(01)02479-9. [DOI] [PubMed] [Google Scholar]
- 32.Gaudet J, Mango SE. Science. 2002;295:821–5. doi: 10.1126/science.1065175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.