

Abstract
Isolated brain mitochondria are a heterogeneous mixture from different cell types and these subsets may have differing sensitivities to Ca2+-induced membrane permeability transition (MPT) and to inhibition of the MPT by cyclosporin A (CsA). This study tested the hypothesis that mitochondria within primary cultures of astrocytes and neurons exhibit different energy-dependent Ca2+ uptake capacities and different degrees to which CsA increases their uptake capacity. Astrocytes and neurons were suspended in a cytosol-like medium containing respiratory substrates, ATP, and Mg2+ in the presence of digitonin to selectively permeabilize the plasma membrane. Uptake of added Ca2+ by mitochondria within the cells was measured by Calcium Green 5N fluorescent monitoring of the medium [Ca2+]. Permeabilized astrocytes had a fourfold higher Ca2+ uptake capacity, relative to neurons and a twofold higher content based on relative contents of mitochondria assessed by measurements of mitochondrial DNA and cytochrome oxidase subunit 1 protein. In astrocytes the Ca2+ uptake capacity was increased twofold by preincubation with 2–5 μM CsA, while in neurons CsA had no effect. Similar results were obtained using measurements of the effects of added Ca2+ on mitochondrial membrane potential. FK506, a drug similar to CsA but without MPT inhibitory activity, had no effect on either cell type.
These results are consistent with the presence of a calcium-induced MPT in astrocytes, even in the presence of ATP, and indicate that the MPT in cerebellar granule neurons is resistant to CsA inhibition. Some of the protective effects of CsA in vivo may therefore be mediated by preservation of mitochondrial functional integrity within astrocytes.
Keywords: Astrocyte, Neuron, Cyclosporin A, Mitochondrial permeability transition, Digitonin, Primary culture, Respiration
Introduction
Supranormal elevation of intracellular Ca2+ occurs during neuronal excitotoxicity and cerebral ischemia, contributing to mitochondrial dysfunction and cell death (Budd, 1998; Fiskum, 2000; Kruman and Mattson, 1999; Nicholls and Budd, 2000; White and Reynolds, 1996). A major cause of Ca2+-induced mitochondrial damage is the opening by Ca2+ of a nonspecific pore in the inner mitochondrial membrane leading to activation of the mitochondrial permeability transition (MPT). Activation of the MPT causes loss of inner mitochondrial membrane potential, osmotic swelling of the matrix, rupture of the outer mitochondrial membrane, and leakage of intermembrane proteins, e.g., cytochrome c, to the cytoplasm where they activate caspases (Bernardi et al., 1994; Crompton, 1999; Halestrap et al., 1998; Ravagnan et al., 2002; Zoratti and Szabo, 1995). The MPT is inhibited by Mg2+, adenine nucleotides and the immunosuppressant drug cyclosporin A (CsA) (Gunter and Pfeiffer, 1990; Halestrap et al., 1997).
In vivo, CsA protects against brain mitochondrial injury and neuronal death following ischemia, suggesting an important role of the MPT in ischemic and other forms of brain injury. Results with isolated brain mitochondria have been mixed- Ca2+ can stimulate cytochrome c release from rat brain mitochondria without inducing swelling, a hallmark of the MPT (Andreyev et al., 1998), and even in the presence of the MPT inhibitor CsA (Andreyev and Fiskum, 1999), while other investigators have found that the MPT in brain mitochondria is only partially- not completely- inhibited by CsA (Heiskanen et al., 1999; Kristal and Dubinsky, 1997). In intact cultured neurons, mitochondrial depolarization in response to calcium ionophore is not blocked by CsA (Dubinsky and Levi, 1998) although CsA pretreatment has been shown to reduce or delay mitochondrial depolarization in response to glutamate (Dubinsky and Levi, 1998; Nieminen et al., 1996).
One factor that needs to be considered is that the intact brain contains astrocytes as well as neurons and that astrocytes contribute to mitochondria isolated from brain. Most studies of ischemic brain damage have focused on neurons, but in recent years it has become clear that astrocytes are also damaged in ischemia (Petito et al., 1998; Liu et al., 1999; Dugan and Kim-Han, 2004; Swanson et al., 2004). There is evidence in vitro that oxidative stress can induce an MPT in astrocytes (Muyderman et al., 2004), however the role of Ca2+ in MPT in astrocytes is less clear. One study imaging mitochondria in intact astrocytes found a CsA-inhibited shape change associated with calcium ionophore (Kahlert and Reiser, 2002), but no quantitative measures of the amount of calcium required to induce this change were made. We have investigated the ability of CsA to protect astrocyte mitochondria from calcium-induced stress by examining Ca2+ uptake in permeabilized cells, where in situ mitochondria can be exposed to known concentrations of Ca2+. For comparison we also examined permeabilized cerebellar granule cells, a neuron that has been extensively studied in models of glutamate-induced mitochondrial depolarization (Nicholls et al., 1999). By using the permeabilized cell approach we were able to study astrocyte and neuron mitochondria in situ in response to similar concentrations of calcium, which is not possible when the calcium stress is presented by treatment of cells with glutamate or calcium ionophore. If neuronal and astrocytic mitochondria differ in their responses to Ca2+, a partial inhibition of an MPT by CsA in isolated brain mitochondria could reflect contributions from CsA-sensitive and CsA-insensitive cells.
Methods
Cortical astrocytes were isolated from E20 Sprague Daley rats (Booher and Sensenbrenner, 1972) and cultured in DMEM/F12 (1/1) 10%FBS with penicillin and streptomycin in 95% air/5% CO2 at 37°C. The culture medium was changed every 3 days and the cells were used at 10 div. The cultures were determined to be >95% astrocytes by immunocytochemistry for glial fibrilary acidic protein (GFAP) in cultures counterstained with the DNA dye propidium iodide (PI). Cerebellar neurons were isolated from P5 Sprague Daley rats (Schousboe et al., 1989) and cultured in BMEM, 10% FBS in 95% air/5% CO2 at 37°C. Cultures were used at 1day in vitro (div). Cultures were >95% neurons by MAP2 immunocytochemistry with PI counterstaining. All cell culture reagents were from Invitrogen. Unless otherwise stated, all chemical reagents were from Sigma-Aldrich, USA.
Measurements of mitochondrial respiration and Ca2+ uptake in permeabilized cells were performed as previously described (Murphy et al., 1996). Astrocytes and neurons were harvested by trypsinization and counted. The cells were then pelleted and resuspended in KCl medium [125 mM KCl, 2 mM K2HPO4, 1 mM MgCl2, 20 mM HEPES pH 7.0]. Respiration was measured polargraphically in KCl medium at 37°C with a Clarke electrode. 1–2 × 107 cells were transferred to the oxygen electrode chamber and permeabilized with digitonin in the presence of 5 mM succinate, 2 μM rotenone and 0.25 mM ADP. Digitonin was titrated in until the maximum rate of respiration was observed (Fiskum et al., 2000). Using this concentration of digitonin (typically 0.005–0.05% digitonin), respiration was determined with 5 mM malate, 5 mM glutamate, 0.25 mM ADP in the absence and presence of oligomycin (2 μg/ml) and the ratio of these two rates—the respiratory control ratio—calculated. Average respiratory control ratios for astrocytes and neurons were 4.9 ± 0.3 (n = 4) and 4.6 ± 0.3 (n = 4) (mean ± SEM), respectively, showing that the mitochondria in the permeabilized astrocytes and neurons were similar and functioning at the beginning of the experiments.
For measurement of mitochondrial Ca2+ uptake 1–2 × 107 cells were resuspended in 1 ml KCl medium with glutamate and malate (5 mM) and 3 mM ATP and the hexapotassium salt of the low affinity Ca2+ indicator Calcium green-5N (0.1 μM, Molecular Probes). The cells were permeabilized with the titrated concentration of digitonin determined from the respiration experiment. Pulses of 25–200 nmoles Ca2+ were added and extramitochondrial Ca2+ was determined by continuous monitoring of Calcium green fluorescence at 506 nm excitation and 531 nm emission in a fluorescence spectrometer equipped with a stirring device. In the present study, the maximal Ca2+ uptake capacity was determined as the cumulative amount of Ca2+ up to the last addition completely taken up by the mitochondria and expressed as μmoles/107/cells.
Mitochondrial membrane potential was monitored in experiments performed in parallel to the Ca2+ uptake measurements but using the membrane potential-sensitive dye safranine orange (5 μM, Molecular Probes). Safranine O was added to the KCl medium and fluorescence measured at 485 nm excitation and 585 nm emission. Pulses of Ca2+ were added as described above. Each addition of Ca2+ caused a transient depolarization of the mitochondrial membrane reflected by a transient increase in Safranine O fluorescence (Fiskum et al., 2000). Sequential additions of Ca2+ were made until mitochondrial membrane potential no longer recovered to baseline.
For Southern blot analysis, total DNA was extracted from neurons and astrocytes using the Qiagen DNA extraction kit. One μg of total DNA was digested with restriction endonuclease PvuII, separated on a 0.8% agarose gel, denatured and transferred to nitrocellulose membrane. The blots were hybridized first with a [α-32P]-labeled 1.7-kb cDNA fragment of mtDNA-encoded COX I gene. The fragment was purified from a plasmid containing the COX I cDNA insert. The COX I cDNA was random primer labeled with [α-32P]-dCTP, purified from unlabeled nucleotide, and hybridized as described (Chandrasekaran et al., 2001). After washing, the membranes were analyzed in a phosphorimager (Molecular Dynamics) and the signal intensity was quantified. Filters were stripped and rehybridized with a 1.8-kb fragment of the nuclear DNA-encoded 18S rRNA (Vu et al., 1998). The [α-32P]-labeled probe anneals to a 12.0-kb chromosomal 18S rRNA PvuII fragment. The ratio of the mitochondrial- to nuclear-encoded bands was calculated.
For western blot analysis, cells, were solubilized in lysis buffer containing 50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.1% SDS, and 2.0% β-mercaptoethanol. After centrifugation at 15, 000 × g, the protein concentration was determined with the Bio-Rad protein assay (Hercules, CA). Proteins (10 μg/lane) were separated by SDS-PAGE on a 4–20% gradient gel, transferred to a nitrocellulose membrane, and probed with anti-COX I antibody (Molecular Probes, OR). Next the membrane was reacted with an appropriate horseradish peroxidase-conjugated secondary antibody (Amersham, Piscataway, NJ), and bands were visualized with enhanced chemiluminescent reagent (Amersham). Cox protein was normalized to the 43 kDa band (corresponding to actin) on a Poinceau stained gel.
Results
When mitochondrial Ca2+ uptake was measured in permeabilized astrocytes, Calcium green fluorescence increased with each Ca2+ addition and returned to baseline as the mitochondria sequestered the Ca2+. Sequential additions of Ca2+ were made until the mitochondria failed to reduce extramitochondrial Ca2+ (Fig. 1A). To test for involvement of the MPT in the loss of further Ca2+ accumulation, permeabilized cells were pretreated with 2 μM CsA. In the representative experiment shown in Fig. 1A, CsA increased the Ca2+ uptake capacity of astrocytes, suggesting that the loss of ability to take up further calcium correlated with the opening of an MPT pore in the astrocyte mitochondria. As a control for non-MPT effects of CsA, astrocyte mitochondria calcium uptake was measured in the presence of FK506. 1 μM FK506 did not increase astrocyte mitochondrial uptake, 0.216 ± 0.021 (n = 11) vs. 0.215 ± 0.034 (n = 5) μmoles calcium/107 cells for control and FK506-treated cells, respectively. To confirm that the loss of calcium uptake correlated with a loss of mitochondrial membrane potential, parallel experiments were performed with the potential-dependent dye safranine O. As anticipated, pulses of calcium caused a series of reversible drops in membrane potential until reaching a point of no return. CsA increased the amount of calcium required for irreversible membrane depolarization from 0.225 μmoles/107 cells to 0.600 μmoles/107 cells (means of two experiments), consistent with the calcium uptake data (Fig. 2).
Fig. 1.
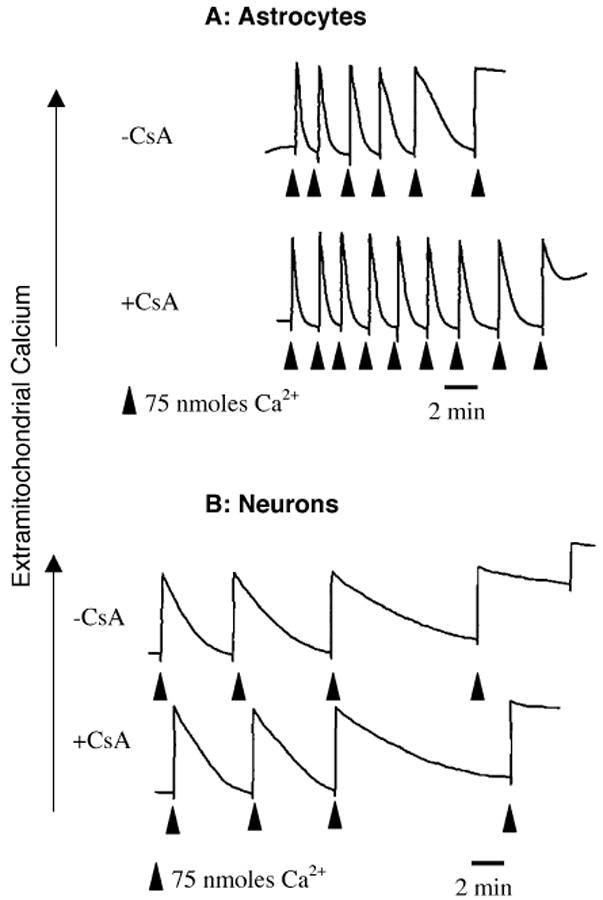
Calcium uptake in permeabilized astrocytes and neurons in the absence (top traces) or presence (bottom traces) of 2 μM CsA. Extramitochondrial calcium was measured by Calcium Green fluorescence. Arrows indicate additions of CaCl2. Calcium Green fluorescence increased with each addition of calcium and decreased as the calcium was sequestered by the mitochondria. 107 cells/experiment, 75 nmoles of calcium/addition. Astrocytes took up more calcium/cell than neurons and the uptake capacity of astrocytes, but not neurons, was increased by CsA
Fig. 2.
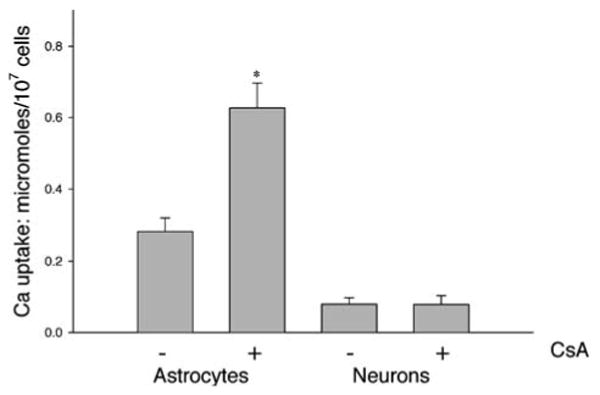
Maximal calcium uptake in astrocytes and neurons in the absence or presence of CsA (2 μM). Bars are Mean ± SEM for 7–9 experiments for astrocytes and 4–5 experiments for neurons. *Significantly different from 0 CsA by t-test, p < 0.01
For comparison, calcium uptake experiments were carried out on cerebellar granule neurons. As with the astrocytes, the neurons were able to take up several pulses of Ca2+ before reaching a threshold beyond which there was no further accumulation of added Ca2+ (Fig. 1B). However, CsA was not able to increase the neuronal calcium uptake capacity. These results are summarized in Fig. 2, showing that CsA significantly increased the maximal mitochondrial Ca2+ uptake of astrocytes by approximately 100%, but had no effect on the uptake of Ca2+ by digitonin-permeabilized cerebellar granule neurons.
We noted that the Ca2+ uptake capacities expressed in Fig. 2 as μmoles/107 cells indicated a greater uptake (approximately fourfold) by astrocytes compared to neurons. To determine if the increased Ca2+ uptake capacity of permeabilized astrocytes was due to the presence of more mitochondria within cortical astrocytes, the relative mitochondrial content of neurons and astrocytes was determined by measuring levels of mtDNA, using nuclear DNA as internal control. We found a significant increase (p < 0.005) in the mtDNA to 18S rRNA ratio in cortical astrocytes compared to cerebellar granule neurons (Table I). We also measured levels of mtDNA-encoded COX I protein in astrocytes and neurons and found a significant increase in the ratio of COX I protein to the 43 kDa actin band in cortical astrocytes compared to cerebellar granule neurons (Table I). Since both protein and DNA content measurements agree, we conclude that astrocytes have a twofold increase in mitochondrial content compared to cerebellar granule neurons. Using the estimate of twice as much mitochondrial mass present in astrocytes compared to neurons, the Ca2+ uptake capacity in mitochondrial equivalents in astrocytes is still 190% greater than that of cerebellar granule neurons.
Table 1.
Mitochondrial DNA and Protein/cell: To Determine the Mitochondrial Content/cell, the Ratios of mtDNA Coding for COX I to Nuclear DNA for 18S rRNA and of COX I Protein to Actin (From Western Blots) were Determined in Extracts from Cultured Astrocytes and Neurons
Note. The mean values of the ratios from the neurons was set as one and used to normalize the ratios obtained with astrocytes. As measured by DNA and protein, astrocytes have double the mitochondrial content of neurons. Data are means ± SEM for 3 experiments.
Significantly different from neurons at p < 0.005, t-test.
Discussion
Qualitatively, the responses of permeabilized neurons and astrocytes to repeated additions of calcium were the same. Sequential additions of calcium were sequestered successfully until a threshold was reached beyond which the mitochondria depolarized and no further calcium uptake occurred. The difference between the two cell types was that the mitochondrial permeability transition inhibitor CsA had a protective effect on astrocytes, increasing their maximal mitochondrial calcium uptake capacity, while the same or higher concentrations of CsA had no effect on the neurons. A second, quantitative difference observed was that the astrocytes had twice the calcium uptake capacity, expressed per mitochondrial protein or DNA, as did the neurons. This is unlikely to reflect more damage to neuronal mitochondria during the isolation as both cells had similar respiratory control ratios, it may reflect either the different brain regions or times in culture of the cells used in the present experiments.
The MPT has been defined by electron microscopy as mitochondrial swelling and pharmacologically on the basis of its inhibition by CsA. CsA is thought to act by binding cyclophilin D, which then interacts with the ANT, a candidate component of the multimeric pore complex. The present data could be explained if mitochondria in cerebellar granule cells can undergo a calcium-dependent MPT that is not CsA-sensitive, either due to differences in cyclophilin D expression or cyclophilin D-ANT-MPT pore interactions.
The differences in CsA-sensitivity for the mitochondrial changes reported here for neurons and astrocytes may underlie the reported heterogeneity of calcium-induced permeability transition in brain mitochondria (Kristian et al., 2002) where some mitochondria appear to have a CsA-dependent and some a CsA-independent calcium-induced swelling. Although neurons are more vulnerable than astrocytes to ischemic injury, brain astrocytes are also subject to increased calcium loading and damage in ischemia (Petito et al., 1998; Liu et al., 1999). Damage to astrocytes could contribute to neuron death in ischemia due to a loss of astrocytic regulation of the extracellular environment and supply of cytokine and metabolic substrates (Montgomery, 1994; Walz, 2000). Reports, for example, of therapeutic benefit of the MPT inhibition component of CsA action in models of stroke (Uchino et al., 1995; Uchino et al., 1998; Friberg et al., 1998; Yoshimoto and Siesjo, 1999) could reflect a direct protection of astrocytes leading indirectly to neuroprotection.
Acknowledgments
The authors thank Steven Russell for his expert technical assistance and acknowledge the support of NIH grants NS34152 and HD16596 to GF.
Contributor Information
Linda L. Bambrick, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD 21201C
Krish Chandrasekaran, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD 21201C.
Zara Mehrabian, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD 21201C.
Christopher Wright, Department of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201.
Bruce K. Krueger, Department of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201
Gary Fiskum, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD 21201C.
Bibliography
- Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM. J Neurochem. 2002;81:207–217. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]
- Andreyev A, Fahy B, Fiskum G. FEBS Lett. 1998;439:373–376. doi: 10.1016/s0014-5793(98)01394-5. [DOI] [PubMed] [Google Scholar]
- Andreyev A, Fiskum G. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Broekemeier KM, Pfeiffer DR. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- Booher J, Sensenbrenner M. Neurobiology. 1972;2:97–105. [PubMed] [Google Scholar]
- Budd SL. Pharmacol Ther. 1998;80:203–229. doi: 10.1016/s0163-7258(98)00029-1. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran K, Liu LI, Hatanpaa K, Shetty U, Mehrabian Z, Murray PD, Fiskum G, Rapoport SI. Mitochondrion. 2001;1:141–150. doi: 10.1016/s1567-7249(01)00010-1. [DOI] [PubMed] [Google Scholar]
- Crompton M. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, Levi Y. J Neurosci Res. 1998;53:728–741. doi: 10.1002/(SICI)1097-4547(19980915)53:6<728::AID-JNR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Kim-Han JS. J Bioenerg Biomembr. 2004;36:317–321. doi: 10.1023/B:JOBB.0000041761.61554.44. [DOI] [PubMed] [Google Scholar]
- Fiskum G. J Neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Kowaltowski AJ, Andreyev A, Kushnareva YE, Starkov AA. Methods Enzymol. 2000;322:222–234. doi: 10.1016/s0076-6879(00)22023-5. [DOI] [PubMed] [Google Scholar]
- Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T. J Neurosci. 1998;18:5151–5159. doi: 10.1523/JNEUROSCI.18-14-05151.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Mol Cell Biochem. 1997;174:167–172. [PubMed] [Google Scholar]
- Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Biochim Biophys Acta. 1998;1366:79–94. doi: 10.1016/s0005-2728(98)00122-4. [DOI] [PubMed] [Google Scholar]
- Heiskanen KM, Bhat MB, Wang HW, Ma J, Nieminen AL. J Biol Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Kahlert S, Reiser G. FEBS Lett. 2002;529:351–355. doi: 10.1016/s0014-5793(02)03394-x. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristal BS, Dubinsky JM. J Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- Kristian T, Weatherby TM, Bates TE, Fiskum G. J Neurochem. 2002;83:1297–1308. doi: 10.1046/j.1471-4159.2002.01238.x. [DOI] [PubMed] [Google Scholar]
- Kruman II, Mattson MP. J Neurochem. 1999;72:529–540. doi: 10.1046/j.1471-4159.1999.0720529.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA. Brain Res Mol Brain Res. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. [DOI] [PubMed] [Google Scholar]
- Montgomery DL. Vet Pathol. 1994;31:145–167. doi: 10.1177/030098589403100201. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyderman H, Nilsson M, Sims NR. J Neurosci. 2004;24:8019–8028. doi: 10.1523/JNEUROSCI.1103-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL, Ward MW, Castilho RF. Biochem Soc Symp. 1999;66:55–67. doi: 10.1042/bss0660055. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Petito CK, Olarte JP, Roberts B, Nowak TSJ, Pulsinelli WA. J Neuropathol Exp Neurol. 1998;57:231–238. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Roumier T, Kroemer G. J Cell Physiol. 2002;192:131–137. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Meier E, Drejer L, Hertz L. A Dissection and Tissue Culture Manual of the Nervous System. Alan R. Liss; New York: 1989. [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Uchino H, Elmer E, Uchino K, Li PA, He QP, Smith ML, Siesjo BK. Brain Res. 1998;812:216–226. doi: 10.1016/s0006-8993(98)00902-0. [DOI] [PubMed] [Google Scholar]
- Uchino H, Elmer E, Uchino K, Lindvall O, Siesjo BK. Acta Physiol Scand. 1995;155:469–471. 0. doi: 10.1111/j.1748-1716.1995.tb09999.x. [DOI] [PubMed] [Google Scholar]
- Vu TH, Tanji K, Valsamis H, DiMauro S, Bonilla E. Neurology. 1998;51:1190–1193. doi: 10.1212/wnl.51.4.1190. [DOI] [PubMed] [Google Scholar]
- Walz W. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto T, Siesjo BK. Brain Res. 1999;839:283–291. doi: 10.1016/s0006-8993(99)01733-3. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]