

Pathobiology of periodontal disease progression
Periodontal disease initiation and progression occurs as a consequence of the host immune inflammatory response to oral pathogens. Periodontal pathogens produce harmful by-products and enzymes (e.g. hyaluronidases, collagenases, proteases) that break down extracellular matrices, such as collagen, as well as host cell membranes, in order to produce nutrients for their growth and possibly subsequent tissue invasion. Many of the microbial surface protein and lipopolysaccharide molecules are responsible for eliciting a host immune response, resulting in local tissue inflammation. Porphyromonas gingivalis, Actinobacillus actinomycetemcomitans and other periodontal pathogens possess multiple virulence factors, such as cytoplasmic membranes, peptidoglycans, outer membrane proteins, lipopolysaccharide, capsules and cell-surface fimbriae (P. gingivalis). Once immune and inflammatory processes are initiated, various inflammatory molecules, such as matrix metalloproteinases, other host enzymes, cytokines and prostaglandins are released from leukocytes, fibroblasts or other tissue-derived cells (59, 60). Proteases can degrade the collagen structure of periodontal tissues and thus create inroads for further leukocyte infiltration. Although the production of collagenase from infiltrating neutrophils and resident periodontal tissue cells is part of the natural host response to infection, in periodontal disease and other chronic inflammatory diseases, there is an imbalance between the level of activated tissue-destroying matrix metalloproteinases and their endogenous inhibitors (173).
The inflammatory infiltrate from the gingival tissue can initiate tissue and alveolar bone destruction through the activation of several pro-inflammatory cytokines, including interleukin-1β, tumor necrosis factor-α and interleukin-6. Within the diseased periodontal tissues, activated osteoclasts comprise an integral component of bone destruction (8, 28). Multiple inflammatory signals can modulate the receptor activator of nuclear factor kappa B (NF-κB) ligand (RANKL), RANK, or osteoprotegerin - three novel members of the tumor necrosis factor ligand and receptor superfamilies. Briefly, a number of hormones and cytokines modulate osteoclastogenesis by enhancing osteoclast differentiation, activation, life span and function (reviewed in 9, 75). These include parathyroid hormone, calcitriol, parathyroid hormone-related protein, prostaglandin E2, interleukin-1β, interleukin-6 and interleukin-11. The formation of active osteoclasts requires macrophage colony-stimulating factor and involves cell-to-cell contact between precursors of the monocyte/macrophage lineage and osteoblasts, marrow stromal cells, or T and B lymphocytes. These cells can all express RANKL, which is essential for this process. For osteoclastogenesis to occur, RANKL must bind to its cognate receptor, RANK, a receptor on the cell surface of osteoclasts and osteoclast precursors, to stimulate proliferation and differentiation of cells from the monocyte/macrophage lineage to form the functional osteoclasts. Osteoprotegerin, a soluble decoy receptor produced by osteoblasts, marrow stromal cells and other cells, profoundly modifies the effects of RANKL by inhibiting RANKL/RANK interaction and has shown promising results for the treatment of bone-related diseases. In the presence of periodontal pathogens (e.g. A. actinomycetencomitans), CD4+ T lymphocytes display increased expression of RANKL, triggering the activation of osteoclasts that leads to bone loss, similar to mechanisms found in rheumatoid arthritis and osteoporotic bone destruction (107, 171). The destructive pattern leads to further accumulation of subgingival plaque biofilm, and the increase in microbial load propagates the destructive periodontal lesion. As the periodontal pocket deepens, the subgingival microflora becomes more anaerobic and the host response becomes more destructive and chronic. The periodontitis lesion may eventually progress to such an extent that the tooth is lost. Approaches to block the progression of inflammatory bone loss observed in periodontitis include host modulation of matrix metalloproteinase or the blockage of inflammatory cytokines. More recently, inflammatory cell signaling pathways that generate these inflammatory and tissue destruction proteins have become promising therapeutic targets. Both host-modulating strategies are the focus of this review.
Cell signaling mechanisms mediating the innate immune response
Chronic periodontal inflammation perpetuates and amplifies itself through numerous autocrine and paracrine loops of cytokines, acting on cells within the periodontium. Central in the process of inflammation-induced bone loss is the host defense against periodontal pathogens. During periodontal disease, the host immune system functions to eliminate microbial pathogens and protect the host. However, an improper or exuberate immune response leads directly towards overproduction of inflammatory cytokines and consequently periodontal attachment loss. This ‘dualedged sword’ is ultimately responsible for perpetuating inflammatory bone loss in periodontitis. Understanding molecular mechanisms governing inflammatory bone loss initiated through the innate immune response in periodontitis is a key aspect towards the development of rational therapeutics that may stop inflammatory bone loss in periodontitis patients.
Unlike adaptive immunity, innate immunity does not recognize every possible antigen. Instead, it is designed to recognize a few highly conserved structures present in many different microorganisms. The structures recognized are pathogen-associated molecular patterns and include lipopolysaccharide from the gram-negative cell wall, peptidoglycan, lipotechoic acids from the gram-positive cell wall, the sugar mannose (common in microbial glycolipids and glycoproteins but rare in humans), bacterial DNA, N-formylmethionine found in bacterial proteins, double-stranded RNA from viruses, and glucans from fungal cell walls. Most body defense cells have pattern-recognition receptors for these common pathogen-associated molecular patterns and so there is an immediate response against the invading microorganism. Pathogen-associated molecular patterns can also be recognized by a series of soluble pattern-recognition receptors in the blood that function as opsonins and initiate the complement pathways. In all, the innate immune system is thought to recognize ≈103 molecular patterns (2, 3).
Upon toll-like receptor recognition of pathogen-associated molecular patterns, a series of orchestrated signaling events occur within host cells, resulting in the production of cytokines that dictate the nature of the host response. Within periodontal tissues, toll-like receptor-2 and toll-like receptor-4 expression is increased in severe disease states, suggesting that these receptors have an increased capacity to signal and influence downstream cytokine expression (120). When considering therapeutic strategies, it is essential to understand the major signaling pathways initiated following toll-like receptor engagement. All toll-like receptors contain a common extracellular leucine-rich domain and a conserved intracellular domain. The intracellular tail of the receptor was shown to be homologous with the intracellular domain of the interleukin-1 receptor type I, currently being designated as the Toll/IL-1R (TIR) domain. The toll-like receptor-pathogen-associated molecular patterns interaction results in the recruitment of specific adaptor molecules, such as MyD88 and Mal, which then bind the interleukin-1R-associated kinase (IRAK). The signal is thereafter transmitted through a chain of signaling molecules, which is apparently common to all toll-like receptors, involving the tumor necrosis factor receptor-associated factor-6 (TRAF6) and mitogen-activated protein kinases (MAPKs) (3). Thereafter, phosphorylation of numerous transcription factors, including NF-κB, activated protein-1, Elk-1 and others, leads towards transcriptional activation of genes involved in the activation of the innate host defense, notably pro-inflammatory cytokines (Fig. 1) (6, 128).
Fig. 1.

Toll-like receptor (TLR) signaling: stimulation of TLRs by periodontal pathogen associated molecular patterns triggers the association of myleloid differentiation primary-response protein 88 (MyD88), which in turn recruits IL-1 receptor associated kinase-4 (IRAK) [which phosphorylates IRAK1 (represented by IRAK)]. Tumor-necrosis factor receptor associated factor-6 (TRAF6) is also recruited to the phosphorylated IRAK complex. IRAK/TRAF6 then dissociate from the receptor complex to a new complex with transforming growth factor β-activated kinase (TAK1) along with TAK-1 and -2 binding protein (TAB1 and -2) (data not shown) which phosphorylate TAK1. TAK1, in turn, phosphorylates both mitogen activated protein kinase kinases-3 and -6 (MKK3, MKK6) and the inhibitor of nuclear factor κB (IκB)-kinase complex (IKK complex) (data not shown). The IKK complex then phosphorylates IκB, which allows nuclear factor-kappa B (NF-κB) transcription factors (p50/p65) to translocate to the nucleus and induce gene expression of cytokine genes. Similarly, MKK3/6 can phosphorylate p38 mitogen-activated protein kinase (MAPK) to activate activator protein-1 transcription factors and initiate gene expression. In addition, p38 can phosphorylate RNA-binding proteins, which can stabilize cytokine mRNA and thus amplify cytokine production. AP1, activating protein-1; ERK, extracellular signal-regulated kinases; MEK, MAPK/ERK kinase; SRE, serum response element.
One major signaling pathway is represented by MAP kinase-kinase-kinase (MAPKKK), such as TAK-1, which is located upstream of MAPK. TAK-1 activation can lead to the activation of either activator protein-1, through MAPK pathways, or NF-κB, through the inhibitor of nuclear factor kappa B (IKK) pathway (12, 34, 85, 128). Regardless of proximal signaling diversity, there are many common pathways, including NF-κB, c-jun N-terminal kinase (JNK) and p38 MAPK pathways, which funnel these diverse signals. These common pathways provide some of the attractive therapeutic targets to block bacterial-induced inflammatory signals to manage chronic periodontitis (123, 178).
Lipopolysaccharide-mediated bone destruction in periodontal disease
P. gingivalis and A. actinomycetemcomitans lipopolysaccharides are considered to be key factors in the development of chronic periodontitis. Lipopolysaccharide induction of disease leads to the initiation of a local host response in gingival tissues that involves recruitment of inflammatory cells, generation of prostanoids and cytokines, elaboration of lytic enzymes and activation of osteoclasts (10, 73). Porphyromonas gingivalis lipopolysaccharide preferentially utilizes toll-like receptor-2 and not toll-like receptor-4 (10, 73, 74). Earlier data indicated that P. gingivalis lipopolysaccharide bound toll-like receptor-4 in gingival fibroblasts (178, 179). Regardless of which toll-like receptor is engaged, lipopolysaccharide increases osteoblastic expression of RANKL, interleukin-1, interleukin-8, prostaglandin E2 and tumor necrosis factor-α, each known to induce osteoclast activity, viability and differentiation (155, 167). An overview of bone resorption/formation and remodeling is shown in Fig. 2.
Fig. 2.
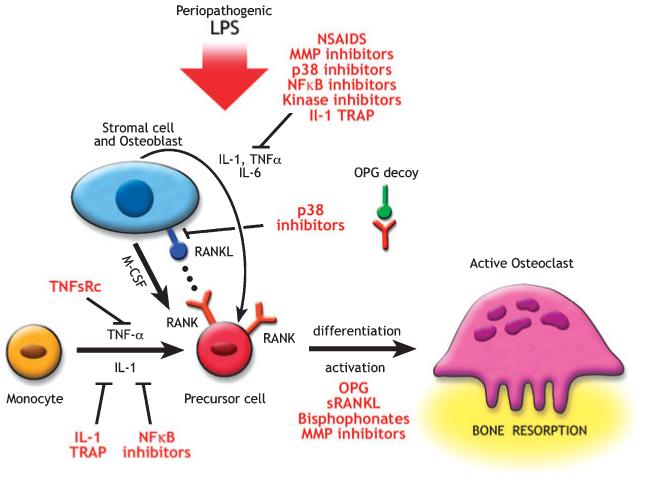
Potential therapeutic strategies to treat bone resorption: agents that block the differentiation or activity of osteoclasts are potential therapeutic agents. Osteoprotegerin (OPG) inhibits the differentiation of osteoclasts through its action as a decoy receptor that blocks receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL) and RANK juxtacrine interaction. Non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory molecules [including p38 mitogen-activated protein kinase (MAPK) inhibitors, c-jun N-terminal kinase (JNK) inhibitors and NF-κB inhibitors] can inhibit the formation of hematoprogenitor cells to pre-osteoclasts. Antibodies to RANKL can also block this interaction. Matrix metalloproteinase (MMP) inhibitors reduce the protease degradation of the organic matrix, and anti-integrins block the initial osteoclast adhesion to the matrix. IL, interleukin; LPS, lipopolysaccharide; M-CSF, macrophage colony-stimulating factor; sRANKL, soluble RANKL; TNF-α, tumor necrosis factor-α; TNFsRC, TNF soluble receptor.
A variety of immune-associated cell populations are responsible for the pathogenesis of periodontal diseases. Activated monocytes, macrophages, and fibroblasts all produce cytokines, such as tumor necrosis factor-α, interleukin-1β and interleukin-6, within periodontal lesions (98, 145). These cytokines orchestrate the cascade of destructive events that occur in the periodontal tissues, and trigger the production of an array of inflammatory enzymes and mediators, including matrix metalloproteinases, prostaglandins and osteoclast recruitment and differentiation through RANKL-dependent and -independent pathways, thus resulting in irreversible hard and soft tissue damage. The pathogenic processes that result in chronic periodontitis are remarkably similar in many ways to those that are observed in rheumatoid arthritis, a destructive bone disease that displays periods of remission and exacerbation (Table 1) (113-115). It is this inherent similarity which provides a common therapeutic basis for disruption of cytokine networks that ultimately result in alveolar bone in periodontitis or joint destruction in rheumatoid arthritis.
Table 1.
Comparison of the pathobiology of periodontal disease and rheumatoid arthritis
| Periodontal disease | Rheumatoid arthritis | |
|---|---|---|
| Pro-inflammatory cytokines IL-1; IL-6; TNF-α | High levels | High levels |
| MMPs | High levels | High levels |
| TIMP (MMP inhibitors) | Low levels | Low levels |
| Prostaglandin E2 | Present | Present |
| Inflammatory infiltrate on lesion site | Present | Present |
| Active osteoclast | Present | Present |
| C-reactive protein | Elevated level | Elevated level |
| NSAIDs effect | Reported | Reported |
IL, interleukin; MMP, matrix metalloproteinase; NSAID, non-steroidal anti-inflammatory drug; TIMP, tissue inhibitor of metalloproteinases; TNF-α, tumor necrosis factor-α. Adapted from Kirkwood et al. (89).
Cytokines and inflammatory diseases
Tumor necrosis factor-α is an inflammatory cytokine that is released by activated monocytes, macrophages and T lymphocytes, and promotes critical inflammatory responses during periodontal disease. Tumor necrosis factor-α binds to two receptors - the type 1 tumor necrosis factor receptor (tumor necrosis factor-R1; p55) and the type 2 receptor (tumor necrosis factor-R2; p75) - that are expressed by a variety of cells (19). Activation of tumor necrosis factor-R1 up-regulates the inflammatory response, while tumor necrosis factor-R2 appears to dampen the response. Tumor necrosis factor-R1 is expressed on multiple cell types, whereas the tumor necrosis factor-R2 is more restricted to expression on endothelial cells and cells of hematopoetic lineage. Blocking tumor necrosis factor-α has been proven to inhibit osteoclast formation effectively (27). Indeed, the blockade of tumor necrosis factor can be used as a probe to understand the molecular basis of osteoclastogenesis and also as a target for therapeutic agent development.
Patients with periodontal disease have high concentrations of tumor necrosis factor in the gingival crevice fluid (98). Studies have shown a very strong association of active bone resorption coincident with high local levels of tumor necrosis factor at the diseased sites. Interleukin-1, interleukin-6 and tumor necrosis factor have all been found to be significantly elevated in diseased periodontal sites compared with healthy or inactive sites (40, 47, 49, 57, 164). Interleukin-1 has also been positively correlated with increased probing depth and attachment loss (43, 47, 57). Other clinical data indicate that elevated interleukin-6 levels are higher in refractory periodontitis, and increased gingival crevice fluid levels correlate with gram-negative fimbriae (72, 149).
Acting in concert with one another, pro-inflammatory cytokines amplify the inflammatory condition. interleukin-1β has synergistic activity with tumor necrosis factor-α or lymphotoxin in stimulating bone resorption (163). Reduction of the bacteria and associated metabolic by-products through periodontal therapy also results in a decrease in both interleukin-1β (4, 136) and tumor necrosis factor-α (47, 81). Although periodontal treatment can successfully reduce inflammation, some individuals have demonstrated aggressive bone destruction and high levels of pro-inflammatory cytokines that cannot be completely explained by the presence of bacteria alone. Based on susceptibility analysis, cells from individuals with immune-mediated diseases exhibit different cytokine-secretion profiles, which may be caused by genetic differences between individuals, causing the hyper-responsiveness (71).
Therapeutic strategies in the treatment of periodontal diseases
Subantimicrobial doxycycline, non-steroidal anti-inflammatory drugs and bisphosphonates
Although the pathogenesis of periodontal disease is not completely understood, it is well established that it is an infectious disease and that the host immune and inflammatory response to the microbial challenge mediates tissue destruction (131). Based on this view, the therapeutic strategies for the treatment of periodontal disease have been directed towards two different and complementary paths: antimicrobial therapy and host modulation. Considering that the primary etiology of the disease are bacteria in the plaque and their products, mechanical and chemical approaches to reduce the presence of periodonto-pathogens in the plaque have been largely used in the treatment of periodontal patients (61). Recently, a better understanding of the participation of host immune-inflammatory mediators in the disease progression has increased the investigation of the use of modulating agents as an adjunctive therapy to the periodontal treatment. Inhibition or blockade of proteolytic enzymes, pro-inflammatory mediators and of osteoclast activity has been the focus of these agents, which has led to encouraging results in preclinical and clinical studies (148). More specifically, three categories of host-modulating agents have been investigated in the periodontal therapy: antiproteinases (represented by tetracyclines); anti-inflammatory drugs; and bone-sparing drugs (represented by antiresorptive agents such as bisphosphonates).
One important group of proteolytic enzymes present in the periodontal tissues is formed by the matrix metalloproteinases, which include collagenases, gelatinases and metalloelastases. Matrix metalloproteinases are produced by fibroblasts, keratinocytes, macrophages, neutrophils and endothelial cells, and are responsible for remodeling the extracellular matrix (15). In pathological conditions, macrophage-derived tumor necrosis factor-α, interleukin-1β and interleukin-6 markedly increase the local production of various matrix metalloproteinases in periodontal tissues (142). In 1985, tetracyclines were discovered to have anticollagenolytic activity and were proposed as a host-modulating agent for periodontal treatment (52). Initial studies demonstrated that doxycycline was the most potent tetracycline in the inhibition of collagenolytic activities (23). This property of doxycycline provided the pharmacological rationale for the use of a low or subantimicrobial dose of doxycycline, which was shown to be efficient in inhibiting mammalian collagenase activity without developing antibiotic resistance (53). Several clinical studies have been conducted, assessing the benefits of the sub-antimicrobial dose of doxycycline as an adjunctive therapy to scaling and root planing in the treatment of the periodontal disease. Reddy et al. (148) recently presented a meta-analysis of six selected clinical studies (7, 25, 54, 55, 126, 141), comparing a long-term systemic subantimicrobial dose of doxycycline (20 mg b.i.d. doxycycline) with placebo control in periodontal patients. A statistically significant adjunctive benefit on clinical attachment level and probing depth was found when a subantimicrobial dose of doxycycline was used in combination with scaling and root planing, in both 4-6 mm and ≤7 mm pocket depth categories. No significant adverse effects were reported in any of the studies. Additional studies published in the last 3 years (42, 48, 63, 100, 143), which evaluated subantimicrobial doses of doxycycline in periodontal treatment, were reviewed and a summary is presented in Table 2.
Table 2.
Subantimicrobial dose of doxycycline adjunctive therapy in periodontal treatment: double-blind, randomized, placebo-controlled, parallel group studies
| Ref. | Subjects/periodontal disease | Groups (n) | Results | ||
|---|---|---|---|---|---|
| Clinical attachment level | Probing depth | Other endpoints | |||
| Gurkan et al. (63) | 26 adults: severe generalized chronic periodontitis (smokers included) | D: SRP + 20 mg doxy b.i.d./3 months (n = 13) C: SRP + placebo b.i.d./3 months (n = 13) |
B to 3/6 months: significant improvement in both groups (no difference between groups) | Similar to clinical attachment level % Sites with PD reduction ≤3 mm: greater in group D (P = 0.01) | GCF TGF-β1 total amount and concentration increased significantly (3 months) in group D |
| Preshaw et al. (143) | 209 adults: moderate to severe chronic periodontitis (smokers included) | D: SRP + 20 mg doxy b.i.d./9 months (n = 107) C: SRP + placebo b.i.d./9 months (n = 102) |
B to 3/6/9 months: significant improvement in both groups. Changes from B at 9 months and % sites with clinical attachment level gain ≤3 mm: greater in group D (P < 0.05) | Similar to clinical attachment level Changes from B at 9 months and % sites with PD reduction ≤3 mm: greater in group D (P < 0.01) |
|
| Emingil et al. (42) | 20 adults: moderate to severe chronic periodontitis [no heavy smokers (>10 cigarettes/day) included] | D: SRP + 20 mg doxy b.i.d./3 months (n = 10) C: SRP + placebo b.i.d./3 months (n = 10) |
B to 3/6/9/12 months: no significant improvement in either group Changes from B at 12 months in 2 study sites/patient: significant improvement (no difference between groups) |
Significant improvement from B at 3/6/9/12 months in both groups Changes from B at 12 months in wholemouth and in 2 study sites/patient: greater in group D (P < 0.05) |
GCF MMP-8 total amount reduced significantly from B at 3/6/9/12 months in both groups. GCF MMP-8 concentr.: significant difference only in group D from B to 3/6 months |
| Lee et al. (100) | 41 adults: chronic periodontitis (no smokers) | D: SRP + 20 mg doxy b.i.d./9 months (n = 24) C: SRP + placebo b.i.d./9 months (n = 17) |
B to 1/3/6/9 months: significant improvement in both groups Change from B at 9 months was greater in D group (P < 0.05) | Similar to clinical attachment level | GCF MMP-8 and 13 levels - decreased significantly (greater reduction in group D at 9 months; P < 0.05) |
| Gapski et al. (48) | 21 adults: severe chronic periodontitis [no heavy smokers (≤2 packs/day) included] | D: SRP + access flap surgery (AFS) + 20 mg doxy b.i.d./6 months (n = 10) C: SRP + AFS + placebo b.i.d./6 months (n = 11) |
B to 3/6/9/12 months: no significant improvement in either group. Stratified data according to baseline PD (4-6 mm and ≤7 mm): significant improvement for both groups (no difference between groups) |
Significant improvement from B at 3/6/9/12 months (P < 0.001) Significant difference between groups only for initially deep PD (P < 0.05) |
GCF ICTP levels - greater reduction in group D during treatment (not significant) BOP - significant reduction in group D. No difference between groups |
B, baseline; BOP, bleeding on probing; doxy, doxycycline; ICTP, cross-linked carboxyterminal telopeptide of type I collagen; MMP, matrix metalloproteinase; PD, probing depth; GCF, gingival crevice fluid; SRP, scaling and root planing; TGF, transforming growth factor.
Overall, these studies confirmed the clinical benefits of a subantimicrobial dose of doxycycline therapy observed by previous studies. Three of these studies failed to find significantly better results, with respect to gain in clinical attachment level, when a subantimicrobial dose of doxycycline therapy was compared with placebo (42, 48, 63). Despite the results from these more recent studies, the pharmacological basis for this host modulation therapy is strong. A subantimicrobial dose of doxycycline was able to reduce gingival crevice fluid levels of matrix metalloproteinase-8, matrix metalloproteinase-13 and cross-linked carboxyterminal telopeptide of type I collagen (ICTP) levels compared with placebo (42, 48, 63, 100). Matrix metalloproteinase-8 is the predominant type of collagenase found in diseased periodontal tissues and initiates the degradation of collagen (162, 182). Although matrix metalloproteinase-8 reduction was observed after mechanical periodontal therapy, a subantimicrobial dose of doxycycline further suppressed matrix metalloproteinase-8 levels, confirming the host-modulation effect of a subantimicrobial dose of doxycycline. Matrix metalloproteinase-13 and cross-linked ICTP are related to bone resorption and their decrease after a subantimicrobial dose of doxycycline therapy, consistent with the ability of a subantimicrobial dose of doxycycline to function as a bone-sparing agent (48).
Another family of pharmacological agents that has been well studied as an inhibitor of the host response in periodontal disease is represented by the non-steroidal anti-inflammatory drugs (NSAIDs) (see Table 3). These agents have been shown to prevent prostanoid formation. In this process, arachidonic acid liberated from membrane phospholipids of cells after tissue damage or stimulus is metabolically transformed, via cyclooxygenase or lipooxygenase pathways, in compounds with potent biological activities (135). The cyclooxygenase enzymes are recognized to have two isoforms: cyclooxygenase 1 (COX-1), which is a constitutive enzyme present in most cells, and cyclooxygenase 2 (COX-2), which is inducible and is present in cells involved in the inflammatory processes (33). The cyclooxygenase pathway produces prostaglandins, prostacyclin and thromboxane, called prostanoids. Some prostanoids have pro-inflammatory properties and have been associated with destructive processes in inflammatory diseases. In periodontal diseases, prostaglandin E2 has been extensively correlated with inflammation and bone resorption (135). Its levels in gingival tissues and in the gingival crevice fluid are significantly elevated in periodontally diseased patients compared with healthy patients (32, 132). Moreover, the gingival crevice fluid prostaglandin E2 concentration was positively associated with the aggressiveness of the periodontal disease (133).
Table 3.
Pharmacological agents with potential host-modulation action
| Agent | Mechanism of action | Periodontal-related effects | References |
|---|---|---|---|
| NSAIDs | Inhibition of cyclooxygenase enzymes that participate in arachidonic acid metabolism Reduction of prostanoid production, specially prostaglandin E2 |
Significant reduction of alveolar bone loss Inconsistent benefits on clinical attachment gain or probing depth reduction |
(14, 20, 26, 84, 124, 146, 183, 184) |
| Bisphosphonates | Inhibition of osteoclast function | Significant attachment gain and probing depth reduction Significant alveolar bone gain |
(41, 96, 147, 151) |
| NO synthase inhibitors | Reduction of nitric oxide production through inhibition of NO synthases | Preclinical studies in rats Significant reduction of bone loss and gingival inflammation |
(101, 105) |
| rhIL-11 | Inhibition of pro-inflammatory cytokines and other mediators Stimulation of TIMP-1 |
Preclinical study in dogs Significant reduction of tissue attachment and bone loss |
(108, 172) |
| Omega-3 fatty acid | Inhibition of cyclooxygenase and lipooxygenase (arachidonic acid cascade) Reduction of prostanoids and leukotrienes, especially leukotriene B4 |
Preclinical study in rats Insignificant reduction of bone loss |
(177) |
| p38 MAPK inhibitors | Inhibit lipopolysaccharide-induced MMP, cytokine (IL-1β, TNF-α, IL-6, IL-8), and prostaglandin expression | Preclinical study in rats Significant reduction in bone loss |
(90) |
| JNK inhibitors | Inhibits TNF-α, IFN-γ, IL-6, COX-2, and MMP expression | None published to date | |
| NF-κB family inhibitors | Inhibits NF-κB-dependent expression (IL-1, TNF-α, IL-6, IL-8), MMPs, IFN-γ, others | None published to date | |
| TNF antagonists | Inhibits TNF-α | Preclinical primate studies Significant reduction of attachment loss and bone loss |
(8, 29, 30, 58) |
| RANKL/RANK/osteoprotegerin disruption therapeutics | Inhibits RANKL/RANK-mediated osteoclastogenesis | Preclinical studies in mice Significant reduction in alveolar bone loss |
(107, 171) |
COX-2, cyclooxygenase 2; JNK, c-jun N-terminal kinase; IFN-γ, interferon-γ; IL, interleukin; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; NO, nitric oxide; NSAID, non-steroidal anti-inflammatory drug; NF-κB, nuclear factor-kappa B; rh, recombinant human; TNF-α, tumor necrosis factor-α; RANK, receptor activator of NF-κB; RANKL, receptor activator of NF-κB ligand.
In vitro (51, 56, 160, 176) and in vivo preclinical (93, 127, 134, 180, 183) studies using NSAIDs have shown the extensive ability of the drugs to reduce prostanoid production by inhibiting cyclooxygenases (see Table 3). Suppression of osteoclast differentiation, as measured by decreased osteoclast numbers and concomitant decreased alveolar bone resorption, is the most frequent sequela following systemic or local delivery of NSAIDs. Recently, selective NSAIDs that are capable of inhibiting COX-2, without affecting the constitutive isoform, COX-1, have indicated sharing of the same bone-sparing effects (13, 78, 79, 160) without inducing adverse effects associated with COX-1 suppression, such as gastroduodenal problems and renal toxicity (67, 103).
Several adjunctive periodontal clinical trials have been conducted with NSAIDs. In a systematic review (148), ten clinical studies, in which the therapeutic outcome of NSAIDs were expressed in clinical attachment level or alveolar crestal height, as measured by subtraction radiography, were selected (14, 20, 26, 45, 64, 70, 84, 124, 146, 184). In these studies, a variety of different NSAIDs, including flurbiprofen, meclofenamate, ibuprofen, ketorolac, naproxen and aspirin, were systemically or locally administered. Although the heterogeneity of data did not allow a meta-analysis, limited quantitative analysis tended to show a significant benefit related to alveolar bone preservation when NSAIDs were associated with conventional therapy (26, 70, 84, 146, 184). Otherwise, superior results were not consistently observed when clinical attachment level was used as the outcome measure.
More recently, a selective COX-2 inhibitor (nimesulide)/scaling and root planing therapy was compared with a non-selective COX inhibitor (naproxen)/scaling and root planing on periodontal clinical parameters and on gingival tissues levels of prostaglandin E2 and prostaglandin F2α. No additional increase was observed in clinical attachment levels and probing depth reduction after both adjunctive therapies when compared with a placebo/scaling and root planing group. Nevertheless, NSAID groups showed significant reduction of prostaglandin F2α and prostaglandin E2 (only naproxen) levels, while the placebo group showed an increase of these levels after 10 days of treatment. Based on the clinical results to date, additional long-term studies are necessary to provide support for the adjunctive use of NSAIDs in the treatment of periodontal disease.
Alveolar bone resorption is the principal sequela and the cause of tooth loss in patients afflicted by periodontal disease. The use of bone-sparing drugs that inhibit alveolar bone resorption is another field in host-modulation therapy. Bisphosphonates are a class of drugs structurally similar to pyrophosphate, a component of human metabolism. It binds to the hydroxyapatite crystals of bone and prevents their dissolution by interfering with osteoclasts function through a variety of direct and indirect mechanisms (153). The antiresorptive properties of bisphosphonates change according to their side chains (153). They are classified from first to third generation as their potency increases. The principal therapeutic purpose of bisphosphonates is in the prevention and treatment of osteoporosis and also in the treatment of Paget’s disease and tumor bone disease (44). In periodontics, their use was proposed for a diagnostic and therapeutic approach. In the latter, radiolabeled bisphosphonates are used to detect changes of metabolic activities at bone (83), but this will not be discussed here. As a therapeutic agent, bisphosphonates were shown to reduce alveolar bone loss and increase mineral density, but not to improve clinical conditions in animal periodontitis models (21, 147). Five studies that assessed the effect of bisphosphonates as an adjunctive agent to scaling and root planing in human periodontal treatment have been found to date (41, 82, 96, 151, 152). Alendronate was the bisphosphonate used in four studies (41, 82, 151, 152), during a period of 6 months. The last study used risedronate during 12 months (96). All the studies presented significant clinical improvement when compared with placebo, including: probing depth reduction (96, 151, 152), clinical attachment gain (96, 151), bleeding on probing reduction (96, 151, 152), alveolar bone gain (82, 151, 152) and increase in bone mineral density (41, 152). These results encourage the use of bisphosphonates as an adjunctive agent to periodontal therapy. Additional studies need to be carried out to confirm the benefits of these drugs.
Recently, the long-term use of bisphosphonates has been related to osteonecrosis of the jaw (109, 157). According to a web-based survey conducted by the International Myeloma Foundation (39), an increased incidence of osteonecrosis of the jaw was observed 36 months after the start of therapy in patients receiving zoledronic acid or pamidronate for the treatment of myeloma or breast cancer. This data also indicated that patients with previous dental problems might have a higher risk of osteonecrosis of the jaw. As the bisphosphonates are potent osteoclast inhibitors, their long-term use may suppress bone turnover and compromise healing of even physiologic micro-injuries within bone (130). Despite the encouraging therapeutic results, further long-term studies are warranted to determine the relative risk-benefit ratio of bisphosphonate therapy.
The improvement of knowledge, and better elucidation of the host mechanisms which participate in the pathogenesis of the periodontal disease, have resulted in the proposal of new agents aimed at host modulation through the inhibition of inflammatory mediators. Preclinical studies in an animal experimental model of periodontitis have shown significant local benefits and provide support for the development of new drugs to periodontal treatment in the future. These studies are briefly discussed below.
Nitric oxide synthase (NOS) inhibitors have demonstrated protective effects against bone resorption and inflammatory process in ligature-induced periodontitis in rats (101, 105). Inducible NOS (iNOS) is responsible for nitric oxide (NO) production by epithelial and inflammatory cells in response to pro-inflammatory cytokines in some inflammatory diseases (118), such as rheumatoid arthritis (166) and periodontal disease (97). Although NO has an anti-microbial protective activity, its elevated concentration in the tissues has a cytotoxic effect towards the host cells. Lohinai et al. (105) and Leitao et al. (101) found a reduction of alveolar bone loss and gingival inflammation after the use of a selective iNOS inhibitor - mercaptoethylguanidine - confirming that NO has a deleterious role in the pathophysiology of periodontitis and that its modulation may prevent tissue destruction.
Interleukin-11 was shown to have anti-inflammatory effects by inhibition of tumor necrosis factor-α and other pro-inflammatory cytokines (172). Moreover, it indirectly minimizes tissue injury through stimulation of tissue inhibitor of metalloproteinases-1 (TIMP-1) (102). Based on these previous studies, Martuscelli et al. (108) investigated the ability of recombinant human interleukin-11 (rhIL-11) to reduce periodontal disease progression in dogs with ligature-induced periodontitis. Significant reduction in the rate of clinical attachment and radiographic bone loss were observed after an 8-week period of rhIL-11 administration, twice a week. The authors suggested that new studies should be conducted towards the therapeutic use of rhIL-11 in periodontal treatment.
Recently, Vardar et al. (177) evaluated the use of omega-3 fatty acids with the purpose of blocking arachidonic acid cascade in induced periodontal disease in rats. This would result in the inhibition of production not only of prostanoids derived from the COX pathway but also of leukotrienes derived from the lipooxygenase pathway. The authors based their therapeutic approach on two facts: first, leukotriene B4, a mediator formed from arachidonic acid via lipooxygenase, plays a significant role in alveolar bone resorption; and, second, inhibition of COX with NSAIDs would result in the accumulation of arachidonic acid, which could be metabolized by the lipooxygenase pathway and result in continuous bone loss. The authors also combined omega-3 fatty acid with celecoxib, looking for a synergism of the anti-inflammatory effects of these two agents. The associated therapy resulted in significant superior reductions on periodontal tissue levels of prostaglandins, leukotriene B4 and platelet-activating factor, which is also a pro-inflammatory mediator. No significant effect was observed on bone loss, which was related to the short period of evaluation.
Hyperproduction of interleukin-6 is observed in the rheumatoid arthritis patients and the serum level of interleukin-6 is closely related to disease activity. Interleukin-6 is a pleiotropic cytokine and its hyperfunctions explain most of the clinical symptoms in rheumatoid arthritis. MRA is a humanized antibody from a mouse anti-human interleukin-6 receptor antibody, which can be administered repeatedly because of its low antigenicity in humans. MRA inhibits interleukin-6 function by blocking interleukin-6 binding to the interleukin-6 receptor, resulting in the prevention of the development of collagen-inducing arthritis in cynomolgus monkeys whose interleukin-6R cross-reacts with MRA. This evidence suggests that MRA has anti-arthritic effects. Treatment of patients with rheumatoid arthritis in a multicenter, double-blind, placebo-controlled studies with MRA for rheumatoid arthritis patients indicates that MRA is as effective as anti-tumor necrosis factor alpha and anti-interleukin-1 therapies (125).
Further studies are necessary to determine the applicability of these agents as therapeutic drugs. Finally, the results of these research studies represent a crescent improvement in the understanding of the immune-inflammatory mechanisms of periodontal disease. It is conceived that new potential agents to modulate host responses will be developed in the future.
Disruption of cell signaling pathways for treating periodontitis
Strategies for preventing cell activation seek to inhibit the intracellular transduction of signals produced when ligands bind to their membrane receptors. Signal transduction pathways are activated not only by cytokines, but also by other factors, such as bacterial proteins, lipopolysaccharide, or environmental stress. These stimuli act on receptors that are coupled to the signal transduction pathways, causing activation of transcription factors and other proteins that control the production of cytokines, proteases and many other compounds involved in the inflammatory process. Inhibition of signal transduction pathways would be expected to abolish both cell activation by cytokines or other stimuli and the production of pro-inflammatory cytokines. Cytokines and bacterial components activate many signal transduction pathways. Signal transduction pathways closely involved in inflammation include the MAPK pathway, phosphatidylinositol-3 protein kinase (PI3) pathway, janus kinase-signal transducer and activator of transcription (Jak-STAT) and NF-κB. In addition, other signal transduction pathways are of fundamental importance in inflammation, such as those involving immunoreceptors (integrins, selectins), G-protein coupled receptors (chemokine receptors) and steroid hormone receptors. Therapeutic strategies have been directed towards many of these major signaling pathways, notably MAPK and NF-κB, which are discussed below.
When a ligand binds to its membrane receptor (such as lipopolysaccharide-binding toll-like receptor-4), the receptor undergoes a conformational change resulting in phosphorylation of the receptor itself or of a receptor-associated enzyme. A complex series of phosphorylated events, mediated by protein kinases, activate transcription factors or other proteins involved in post-transcriptional gene regulation (mRNA stability or translation) to induce protein synthesis. In general, transcription factors become activated upon phosphorylation by protein kinases via enhanced nuclear import or increased transcription factor/DNA affinity. To balance these phosphorylated-mediated events, protein kinase phosphatases (MKPs) can dephosphorylate these kinase-phosphorylated proteins, thereby counteracting the effects of the protein kinase. The importance of this regulation is underscored when MKP-1 isoform knockout mice show enhanced inflammatory responses in macrophages (189). Regulation also occurs at the post-transcriptional stage via RNA-binding proteins (RNABPs, e.g. TIA-1 and TIAR) that bind to specific mRNA sequences called adenosine-uridine-rich elements (AUREs) (31, 62). These cis elements are located in the 3′ untranslated region (UTR) of many inflammatory cytokines (including TNF-α, IL-6 and COX-2), conferring mRNA instability or translational silencing, thereby decreasing protein synthesis. However, in stimulated cells, phosphorylation of TIA-1 and TIAR inhibits mRNA degradation and increases the production of proteins. Thus, RNABPs may serve as a target of cytokine-mediated inflammation.
Other regulatory mechanisms control signal transmission during various steps along cell signaling pathways. Before the ligand binds to the receptor, specific enzymes block the changes needed to activate the ligand. Case in point, interleukin-1β is produced as an inactive molecule, which becomes activated when the interleukin-1β-converting enzyme (ICE, caspase-1) releases a peptide from interleukin-1β. In addition, tumor necrosis factor-α is converted from a membrane form to a soluble form by the enzyme, tumor necrosis factor convertase (TACE). ICE and TACE inhibitors have proved useful in experimental models of inflammation and arthritis (35, 144). The ICE inhibitor pralnacasan, developed by Aventis, showed modest improvements in arthritis until clinical trials were discontinued because of liver toxicity (156).
We will review, below, two major signal transduction pathways, namely NF-κB, and MAPK pathways, directing special attention to the more promising potential therapeutic targets. The transcription factors NF-κB and activator protein-1 control the expression of many mediators involved in periodontal inflammation, such as IL-1, IL-6, tumor necrosis factor-α, collagenases, adhesion molecules and chemokines (99, 110).
The NF-κB pathway
The transcription factors NF-κB are homo- or heterodimers found in the cytoplasm of most human cells. The NF-κB family includes NF-κB1 (p50), NF-κB2 (p52), p65 (RelA), RelB, and c-Rel. In vitro studies have established that both P. gingivalis and other periopathogenic bacteria can activate NF-κB in periodontal tissues (168). Bacterial lipopolysaccharide and the pro-inflammatory cytokines interleukin-1 and tumor necrosis factor, all present in large quantities in periodontal diseased tissues, can also activate NF-κB. More recently, immunohistochemical staining for NF-κB (p50/p65) in human periodontitis indicated an increased expression at sites of periodontal inflammation compared with healthy sites (5). In models of experimental arthritis, activated NF-κB is expressed a few days after immunization with collagen or Freund’s adjuvant and precedes pathosis development in this animal model. NF-κB activation is regulated primarily through IκB and IκB kinase (IKK). In the absence of stimulation, NF-κB is coupled with the inhibitory protein, IκB, which prevents nuclear translocation of NF-κB (110). Stimulation by a pro-inflammatory cytokine induces recruitment of co-stimulatory cytokines, such as TRAF, which leads to activation of the enzyme NF-κB-inducing kinase (NIK). Thus, there are multiple points to inhibit NF-κB signalling. The therapeutic implications are discussed below.
Proteasome inhibitors block NF-κB activation by preventing the degradation of IκB and therefore the release of NF-κB. Phase I and II clinical trials of proteasome inhibitors have been performed in patients with hematological malignancies. Bortezomib (Velcade®) was tested in multiple myeloma with highly promising results and limited side effects (149). The first IKK inhibitor, BMS-345541, was evaluated recently in the collagen-induced arthritis model. When used in a preventive or therapeutic therapy, BMS-345541 improved disease activity scores and decreased both synovial inflammation and joint destruction (111).
Another anti-NF-κB strategy involves increasing the expression of endogenous signal transduction pathway inhibitors. This can be achieved either by administering the recombinant protein or by gene therapy. One or more naturally occurring inhibitors have been identified for each signal transduction pathway. In the classic pathway leading to NF-κB activation, IκB is a natural inhibitor that prevents translocation of NF-κB from the cytosol to the nucleus. Overexpression of IκB within rheumatoid synovial cells decreases the expression of the pro-inflammatory cytokines tumor necrosis factor-α, interleukin-1 and interleukin-6, and of matrix metalloproteinase-1 and matrix metalloproteinase-3 (187).
Inhibition of the protein kinases that activate IκB may also prevent NF-κB activation. IKKβ blockade is a promising anti-inflammatory strategy because NF-κB activation depends largely upon activation of the IKKβ isoform. In an adjuvant arthritis model, gene therapy, ensuring intra-articular administration of the dominant negative form of IKKβ, reduces the number of joints with synovitis (170). However, IKKβ knockout mice have been shown to develop liver failure related to hepatocyte apoptosis, which is exacerbated in the presence of tumor necrosis factor-α (106). Despite the pitfalls of systemic IKKβ inhibition, local administration may be therapeutically and clinically useful.
The MAPK pathway
MAPKs are divided into three families: the extracellular signal-regulated kinases (ERK1/2); JNKs; and p38. Mitogens and growth factors primarily activate ERK1/2, whereas the pro-inflammatory cytokines interleukin-1 and tumor necrosis factor-α and cell stress-inducing factors, such as heat shock, osmotic shock, ultraviolet radiation and oxygen radicals, chiefly activate JNKs and p38. The three MAPKs control the activation of many transcription factors, including activator protein-1 (homo- or heterodimers of the proteins c-fos and c-jun), NF-κB, or C/EBP. MAPKs, most notably p38, can activate NF-κB.
All three MAPK families are assumed to be expressed in diseased periodontal tissues, although the level of expression may differ depending upon the exact cell types activated and the degree of inflammation. Within periodontal resident cell types, including tissue macrophages and other periodontal cells, MAPKs are activated chiefly by lipopolysaccharide, interleukin-1 and tumor necrosis factor-α. The p38 MAPK, most notably its p38α isoform (discussed below), is activated mainly within cells involved in the inflammatory process. Activation of p38 induces synthesis of pro-inflammatory cytokines, such as tumor necrosis factor-α, interleukin-1, interleukin-6 and interleukin-8, either via direct activation of gene transcription or via mRNA stabilization (77, 88, 95, 139). The p38 MAPK stabilizes mRNA via the enzyme substrate, MAP kinase-activated protein kinase 2 (MK2), which may act on one or more proteins capable of binding to mRNA (186). In addition, p38 MAPK controls the synthesis of other compounds, including chemokines, metalloproteinases and prostaglandins (99).
The role for p38 MAPK in the various stages of inflammation has prompted the production of several imidazole compounds capable of inhibiting p38 (SB203580, RWJ 67657, L-167307, VX-745, RPR200765A and others). These pharmacological inhibitors are cytokine-suppressive anti-inflammatory drugs (CSAIDs) responsible for in vitro and in vivo inhibition of lipopolysaccharide-induced tumor necrosis factor-α expression (1). CSAIDs were initially shown to inhibit various inflammatory cytokines before the p38 MAPK was actually discovered. Thus, this class of agents defined the role of p38 well before the activation, regulation and substrates of p38 MAPK were identified.
Most CSAIDs (p38 inhibitors) act through reversible, competitive binding to the ATP-binding pocket (158). This region is often the target of inhibitor drugs because binding of ATP to a protein kinase is critical for phosphotransferase activity (46). Crystallography studies revealed that a Thr106 in the hinge of the p38α ATP-binding pocket interacts with a fluorine atom of SB203580. This places the drug in a preferable orientation for binding. Other structural classes have emerged, namely diphenylimidazoles, pyrimidinylimidazoles and the recent 4-phenyl-5-pyridyl-1,3 thiazole analogues (37, 38, 112). The novel BIRB796 is a pyrazole urea, primarily targeting the site of kinase activity and also interacting with the ATP-binding site, making it concomitantly competitive and non-competitive (46).
A multitude of pharmaceutical companies are developing protein kinase inhibitors, highlighting their enormous therapeutic promise. Few p38 inhibitors have proceeded to clinical trials (36, 46) (Table 3). At the present time, only BIRB-796 and SCIOS-469 are in phase III for the treatment of psoriasis and rheumatoid arthritis, respectively. One p38 inhibitor (VX-745; Vertex) was discontinued from trials, citing that proof of concept information had been obtained. Interestingly, concurrent animal testing revealed adverse neurological effects from high-dose VX-745, although no such effects were reported in human subjects. Studies of VX-702, a newer p38 inhibitor which does not pass through the blood-brain barrier, are currently underway (17).
In experimental arthritis models, p38 inhibitors prevent the development of arthritis and bone erosions. RWJ 67657 has been tested in human volunteers who were given endotoxin. After a single oral dose of RWJ 67657, serum levels of the pro-inflammatory cytokines tumor necrosis factor-α, interleukin-6 and interleukin-8 were decreased by 90% compared with their plasma peak (138). No major side effects were noted, even with the highest doses of p38 inhibitor. Phase II clinical trials of p38 inhibitors are underway in patients with rheumatoid arthritis. Weisman (181) reported preliminary findings from a randomized placebo-controlled study evaluating the efficacy of the oral p38 inhibitor, VX-745, in patients with rheumatoid arthritis. Of the 44 patients included in the Weisman study, 14 were withdrawn because the treatment was ineffective or induced side effects, most notably gastrointestinal symptoms. Although modest improvements were observed, these changes provide promise for future p38 inhibitor trials.
Pharmacological inhibitors of JNK, ERK1/2 and PI3 kinase have shown in vitro and in vivo efficacy in inhibiting the production of pro-inflammatory compounds. The specific JNK inhibitor, SP600125, not only diminishes the production of tumor necrosis factor-α, interferon-γ, interleukin-6, COX-2 and matrix metalloproteinase, but also decreases joint destruction in the adjuvant arthritis model (65). The specific ERK1/2 inhibitor, Ro 09-2210, blocks T-lymphocyte activation and proliferation and induces modest anti-inflammatory effects in several experimental models (185). To date, no human trials have been initiated with these inhibitors. With JNK, it seems that both isoforms (JNK1 and JNK2) must be inhibited to produce an anti-inflammatory effect. In mice lacking the JNK2 gene, expression of the transcription factor activator protein-1 is comparable to that in control mice. In contrast, mice that are missing both the JNK1 and the JNK2 genes have decreased levels of activator protein-1 and collagenase within synovial fibroblasts (119).
In conclusion, the efficacy of anticytokine biotherapies in patients with inflammatory bone diseases is proof that blocking the effects of a cytokine can slow a disease process. In recent years, the identification of pro-inflammatory signal transduction pathways has suggested new therapeutic targets. Because these pathways are shared by several cytokines, their inhibition will probably prove more powerful than current treatment strategies. Pharmacological inhibitors are undergoing evaluation in patients with rheumatoid arthritis. Although promising, the preliminary results were obtained in only small numbers of patients. The main advantages of pharmacological inhibitors are ease of administration and low cost compared with biotherapies or gene therapy. Finally, the well-founded enthusiasm generated by this new therapeutic family should perhaps be tempered by the knowledge that signal transduction pathways contribute to physiological processes and that their inhibition may therefore result in adverse effects.
Novel protein antagonist strategies in the treatment of periodontal diseases
Disruption of the RANKL/RANK/osteoprotegerin axis
Osteotrophic factors such as hormones (vitamin D3, parathyroid hormone, parathyroid hormone-related protein), cytokines (interleukin-1, interleukin-6, interleukin-11 and interleukin-17), growth factors (tumor necrosis factor-α and bone morphogenetic protein-2) and other molecules [prostaglandin E2, the T-cell-activating CD40 ligand (CD40L), and glucocorticoids] all enhance expression of the RANKL gene in osteoblasts/stromal cells (76, 122). Sequentially, RANKL mediates a signal for osteoclastogenesis through RANK on pre-osteoclast cells. In summary, the RANKL/RANK interaction is responsible for differentiation and maturation of osteoclast precursor cells to activate osteoclasts. Osteoprotegerin acts as a decoy receptor, expressed by osteoblastic cells, which binds to RANKL and inhibits osteoclast development. Several studies have shown the opposite effect of RANKL and osteoprotegerin in bone modulation. Osteoprotegerin over-expressing or RANKL-/- mice resulted in osteopetrosis (92, 161). An opposite phenotype was observed in RANKL over-expressing and osteoprotegerin-/- mice which developed osteoporosis (22, 116, 174). Moreover, several stimulators of bone resorption that up-regulate RANKL expression inhibit osteoprotegerin expression in osteoblasts/stromal cells (66, 76).
In pathological bone resorption observed in hormonal disorders, inflammatory diseases and in certain types of cancer, the equilibrium of this interaction is disregulated. In periodontal disease, the role of RANKL in alveolar bone resorption was first investigated by Teng et al. (171). Several previous studies had suggested that T lymphocytes could modulate inflammation and/or alveolar bone resorption, but the mechanism by which host immune responses contribute to alveolar destruction remained unclear. Teng et al. (171) orally inoculated HuPBL-NOD/SCID mice (mice that lack endogenous T and B cells and seeded with human CD4+ cells) with A. actinomycetencomitans, which resulted in the activation of CD4+ T cells, up-regulation of RANKL in these cells and alveolar bone destruction. This result suggests that RANKL expression by CD4+ T cells has a significant role in the bone destruction observed in periodontitis. Liu et al. (104) and Crotti et al. (28), in 2003, demonstrated an over-expression of RANKL in inflamed periodontal tissues, as evidenced by immunohistochemistry, semiquantitative reverse transcription-polymerase chain reaction and in situ hybridization, suggesting expression by lymphocytes and macrophages. Moreover, the RANKL/osteoprotegerin ratio was increased in periodontitis when compared with healthy subjects, suggesting that this molecular interaction may play an important role in modulating local bone loss. The RANKL/osteoprotegerin ratio was found to be significantly increased in the crevicular fluid of patients with periodontitis as compared to healthy patients (117).
The use of osteoprotegerin as a therapeutic agent was first evaluated by Simonet et al. (161) when they treated ovariectomized rats with murine osteoprotegerin-Fc protein and protected them against losses of bone volume associated with deficiencies of estrogen. Other preclinical studies demonstrated a potential therapeutic role of osteoprotegerin in the prevention and reduction of lytic bone lesions associated with skeletal tumor, prostatic carcinoma metastases, hypercalcemia of malignancy and breast cancer (24, 121, 137, 188). Osteoprotegerin blocked increased osteoclast formation responsible for resorptive processes in rheumatoid arthritis patients (69, 91, 154) and periprosthetic bone tissue (50, 68, 87). Gene therapy for the lifelong delivery of osteoprotegerin has also been proposed as a more practical therapy for chronic inflammatory diseases. Osteoprotegerin-expressing adenoviral vectors provided sustained and efficacious levels of circulating osteoprotegerin that enhanced bone mineral density and reduced osteoclast numbers for an extended period of time (18 months) in ovariectomized mice (18). A recombinant adeno-associated virus vector co-expressing osteoprotegerin, and administered in a single injection, demonstrated complete inhibition of osteolysis in a periprosthetic bone resorption model in mice (175) and reversed osteopenia in ovariectomized mice without liver toxicity (94).
Osteoprotegerin administered by single injection to postmenopausal women resulted in a significant decrease in bone collagen degradation products measured in urine, without adverse side effects, suggesting a potential use of osteoprotegerin in osteoporosis treatment (11). The antiresorptive effect of AMGN-0007, a genetically engineered osteoprotegerin-Fc construct, with lytic bone disease associated with breast carcinoma or multiple myeloma was shown to be effective in inhibiting bone resorption (16).
The use of osteoprotegerin as an inhibitor of bone alveolar destruction in periodontal disease was investigated in mice orally infected with A. actinomycemcomitans (107, 171). Inhibition of RANKL function with osteoprotegerin treatment significantly reduced the number of osteoclasts and the alveolar bone destruction in both studies.
In summary, based on pre-clinical animal studies and on preliminary human clinical studies, the osteoprotegerin/RANKL/RANK axis is a new target for the treatment of destructive periodontal disease and other bone resorption-related diseases. Further studies are necessary to determine the most efficacious therapeutic approach on that molecular interaction.
Tumor necrosis factor antagonists to block inflammatory diseases
Tumor necrosis factor-α, an inflammatory cytokine that is released by activated monocytes, macrophages and T lymphocytes, promotes inflammatory responses that are important in the pathogenesis of rheumatoid arthritis and periodontal diseases. Tumor necrosis factor-α binds to two receptors that are expressed by a variety of cells: the type 1 tumor necrosis factor receptor (p55); and the type 2 receptor (p75) (19). Activation of tumor necrosis factor-R1 upregulates the inflammatory response, while tumor necrosis factor-R2 appears to dampen the response (140). Tumor necrosis factor-R1 is expressed on multiple cell types, whereas tumor necrosis factor-R2 is more restricted to expression on endothelial cells and cells of hematopoetic lineage (80).
Patients with rheumatoid arthritis and periodontal disease have high concentrations of tumor necrosis factor in the synovial fluid and gingival crevice fluid, respectively. Studies of experimental models of rheumatoid arthritis and periodontal diseases have shown a very strong association of active bone resorption, coincident with high local levels of tumor necrosis factor at the disease sites. Both interleukin-1 and tumor necrosis factor-α have been found to be significantly elevated in diseased periodontal sites compared with healthy or inactive sites (40, 47, 57, 165). Interleukin-1 has also been positively correlated with increased probing depth and attachment loss (30). In addition, interleukin-1β has synergistic activity with tumor necrosis factor-α or lymphotoxin in stimulating bone resorption. Incubation of interleukin-1 with tumor necrosis factor or lymphotoxin in an in vitro model resulted in a twofold increase in the bone-resorbing activity of interleukin-1, and in a 100-fold increase in the activity of tumor necrosis factor or lymphotoxin (163). Reduction of the bacteria and their metabolic by-products through periodontal therapy also results in a decrease in both interleukin-1 (4, 136) and tumor necrosis factor (47, 81). Thus, improvement in clinical parameters is paralleled by a decrease in these cytokines, suggesting their significance in the pathogenesis of periodontitis.
Blocking the activity of pro-inflammatory cytokines may be a beneficial therapeutic modality for periodontitis. Investigations on the soluble protein delivery of antagonists to interleukin-1 and tumor necrosis factor in a primate model of periodontitis have shown promising results (29, 30, 129). Histological investigations revealed a 51% reduction in connective tissue attachment loss and a 91% reduction in alveolar bone loss (29). To date, the only therapy available that acts as a host modulatory agent for periodontal disease is a low-dose formulation of the antibiotic doxycycline, which requires twice-daily administration (25, 48, 126). However, the harsh enzymatic environment in periodontal lesions may destroy the soluble cytokine antagonists prior to their peak activity, which may necessitate more frequent administration of the active agents to the defects. Thus, gene transfer of TNFR antagonists may offer a more efficient mode of delivery of disease-controlling agents to the periodontal structures (169). Thus, the blocking of tumor necrosis factor pathways offers significant potential in blocking disease progression.
Summary
A variety of treatment strategies have been developed to target the host response to periodontal infection. This review has sought to provide mechanistic overviews and clinical applications on the use of host modulatory therapeutic regimens for periodontal disease management. Matrix metalloproteinase inhibitors, such as low-dose formulations of doxycycline, have been used in combination with scaling and root planing or surgical therapy. In addition, high-risk patient populations, such as patients with diabetes or refractory periodontal disease, have benefited from systemic matrix metalloproteinase administration. Encouraging results have been shown using soluble antagonists of tumor necrosis factor and interleukin-1 delivered locally to periodontal tissues in non-human primates, as well as more recent evidence using gene therapy vectors to provide a longer-term delivery of tumor necrosis factor receptor antagonists at the periodontium. In addition, the use of lipoxins has demonstrated significant potential in the management of the host response to periodontitis (86, 159).
Other therapeutic strategies being explored are aimed at inhibiting signal transduction pathways involved in inflammation. Pharmacological inhibitors of NF-κB and p38 MAPK pathways are actively being developed to manage rheumatoid arthritis and inflammatory bone diseases (1, 95). Using this novel strategy, inflammatory mediators, including pro-inflammatory cytokines (interleukin-1, tumor necrosis factor, interleukin-6), matrix metalloproteinases and others, would be inhibited at the level of cell-signaling pathways required for transcription factor activation necessary for inflammatory gene expression or mRNA stability. These therapies may provide the next wave of disease-specific chemotherapeutics to manage chronic periodontitis.
Acknowledgements
These studies were supported by NIH / NIDCR grants DE 13397 and DE 16619 to WVG and DOD grant W81XWH-05-0075 to KLK.
References
- 1.Adams JL, Badger AM, Kumar S, Lee JC. p38 MAP kinase: molecular target for the inhibition of pro-inflammatory cytokines. Prog Med Chem. 2001;38:1–60. doi: 10.1016/s0079-6468(08)70091-2. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis. 2003;35:555–562. doi: 10.1080/00365540310015683. [DOI] [PubMed] [Google Scholar]
- 4.Al-Shammari KF, Giannobile WV, Aldredge WA, Iacono VJ, Eber RM, Wang HL, Oringer RJ. Effect of non-surgical periodontal therapy on C-telopeptide pyridinoline cross-links (ICTP) and interleukin-1 levels. J Periodontol. 2001;72:1045–1051. doi: 10.1902/jop.2001.72.8.1045. [DOI] [PubMed] [Google Scholar]
- 5.Ambili R, Santhi WS, Janam P, Nandakumar K, Pillai MR. Expression of activated transcription factor nuclear factor-kappaB in periodontally diseased tissues. J Periodontol. 2005;76:1148–1153. doi: 10.1902/jop.2005.76.7.1148. [DOI] [PubMed] [Google Scholar]
- 6.An H, Yu Y, Zhang M, Xu H, Qi R, Yan X, Liu S, Wang W, Guo Z, Guo J, Qin Z, Cao X. Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002;106:38–45. doi: 10.1046/j.1365-2567.2002.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashley RA. Clinical trials of a matrix metalloproteinase inhibitor in human periodontal disease. SDD Clinical Research Team. Ann N Y Acad Sci. 1999;878:335–346. doi: 10.1111/j.1749-6632.1999.tb07693.x. [DOI] [PubMed] [Google Scholar]
- 8.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403–409. [PubMed] [Google Scholar]
- 9.Aubin JE, Bonnelye E. Osteoprotegerin and its ligand: a new paradigm for regulation of osteoclastogenesis and bone resorption. Medscape Womens Health. 2000;5:5. [PubMed] [Google Scholar]
- 10.Bainbridge BW, Darveau RP. Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol Scand. 2001;59:131–138. doi: 10.1080/000163501750266710. [DOI] [PubMed] [Google Scholar]
- 11.Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16:348–360. doi: 10.1359/jbmr.2001.16.2.348. [DOI] [PubMed] [Google Scholar]
- 12.Beutler B. Innate immune sensing of microbial infection: the mechanism and the therapeutic challenge. Wien Med Wochenschr. 2002;152:547–551. doi: 10.1046/j.1563-258x.2002.02097.x. [DOI] [PubMed] [Google Scholar]
- 13.Bezerra MM, de Lima V, Alencar VB, Vieira IB, Brito GA, Ribeiro RA, Rocha FA. Selective cyclooxygenase-2 inhibition prevents alveolar bone loss in experimental periodontitis in rats. J Periodontol. 2000;71:1009–1014. doi: 10.1902/jop.2000.71.6.1009. [DOI] [PubMed] [Google Scholar]
- 14.Bichara J, Greenwell H, Drisko C, Wittwer JW, Vest TM, Yancey J, Goldsmith J, Rebitski G. The effect of postsurgical naproxen and a bioabsorbable membrane on osseous healing in intrabony defects. J Periodontol. 1999;70:869–877. doi: 10.1902/jop.1999.70.8.869. [DOI] [PubMed] [Google Scholar]
- 15.Birkedal-Hansen H. Role of matrix metalloproteinases in human periodontal diseases. J Periodontol. 1993;64:474–484. doi: 10.1902/jop.1993.64.5s.474. [DOI] [PubMed] [Google Scholar]
- 16.Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, Nakanishi A, Holloway D, Martin SW, Dunstan CR, Bekker PJ. A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003;97:887–892. doi: 10.1002/cncr.11138. [DOI] [PubMed] [Google Scholar]
- 17.Boger J. Vertex moves to re-allocate resources from VX-745 in p38 MAP kinase program to accelerate development of second generation drug candidates VX-702 and VX-850. http://www.pr.newswire.com.
- 18.Bolon B, Carter C, Daris M, Morony S, Capparelli C, Hsieh A, Mao M, Kostenuik P, Dunstan CR, Lacey DL, Sheng JZ. Adenoviral delivery of osteoprotegerin ameliorates bone resorption in a mouse ovariectomy model of osteoporosis. Mol Ther. 2001;3:197–205. doi: 10.1006/mthe.2001.0245. [DOI] [PubMed] [Google Scholar]
- 19.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 20.Bragger U, Muhle T, Fourmousis I, Lang NP, Mombelli A. Effect of the NSAID flurbiprofen on remodelling after periodontal surgery. J Periodontal Res. 1997;32:575–582. doi: 10.1111/j.1600-0765.1997.tb00934.x. [DOI] [PubMed] [Google Scholar]
- 21.Brunsvold MA, Chaves ES, Kornman KS, Aufdemorte TB, Wood R. Effects of a bisphosphonate on experimental periodontitis in monkeys. J Periodontol. 1992;63:825–830. doi: 10.1902/jop.1992.63.10.825. [DOI] [PubMed] [Google Scholar]
- 22.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burns FR, Stack MS, Gray RD, Paterson CA. Inhibition of purified collagenase from alkali-burned rabbit corneas. Invest Ophthalmol Vis Sci. 1989;30:1569–1575. [PubMed] [Google Scholar]
- 24.Capparelli C, Kostenuik PJ, Morony S, Starnes C, Weimann B, Van G, Scully S, Qi M, Lacey DL, Dunstan CR. Osteoprotegerin prevents and reverses hypercalcemia in a murine model of humoral hypercalcemia of malignancy. Cancer Res. 2000;60:783–787. [PubMed] [Google Scholar]
- 25.Caton JG, Ciancio SG, Blieden TM, Bradshaw M, Crout RJ, Hefti AF, Massaro JM, Polson AM, Thomas J, Walker C. Treatment with subantimicrobial dose doxycycline improves the efficacy of scaling and root planing in patients with adult periodontitis. J Periodontol. 2000;71:521–532. doi: 10.1902/jop.2000.71.4.521. [DOI] [PubMed] [Google Scholar]
- 26.Cavanaugh PF, Jr, Meredith MP, Buchanan W, Doyle MJ, Reddy MS, Jeffcoat MK. Coordinate production of PGE2 and IL-1 beta in the gingival crevicular fluid of adults with periodontitis: its relationship to alveolar bone loss and disruption by twice daily treatment with ketorolac tromethamine oral rinse. J Periodontal Res. 1998;33:75–82. doi: 10.1111/j.1600-0765.1998.tb02295.x. [DOI] [PubMed] [Google Scholar]
- 27.Cheng X, Kinosaki M, Murali R, Greene MI. The TNF receptor superfamily: role in immune inflammation and bone formation. Immunol Res. 2003;27:287–294. doi: 10.1385/IR:27:2-3:287. [DOI] [PubMed] [Google Scholar]
- 28.Crotti T, Smith MD, Hirsch R, Soukoulis S, Weedon H, Capone M, Ahern MJ, Haynes D. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J Periodontal Res. 2003;38:380–387. doi: 10.1034/j.1600-0765.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 29.Delima AJ, Oates T, Assuma R, Schwartz Z, Cochran D, Amar S, Graves DT. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J Clin Periodontol. 2001;28:233–240. doi: 10.1034/j.1600-051x.2001.028003233.x. [DOI] [PubMed] [Google Scholar]
- 30.Delima AJ, Karatzas S, Amar S, Graves DT. Inflammation and tissue loss caused by periodontal pathogens is reduced by interleukin-1 antagonists. J Infect Dis. 2002;186:511–516. doi: 10.1086/341778. [DOI] [PubMed] [Google Scholar]
- 31.Derrigo M, Cestelli A, Savettieri G, Di Liegro I. RNA-protein interactions in the control of stability and localization of messenger RNA (review) Int J Mol Med. 2000;5:111–123. [PubMed] [Google Scholar]
- 32.Dewhirst FE, Moss DE, Offenbacher S, Goodson JM. Levels of prostaglandin E2, thromboxane, and prostacyclin in periodontal tissues. J Periodontal Res. 1983;18:156–163. doi: 10.1111/j.1600-0765.1983.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 33.DeWitt DL, Meade EA, Smith WL. PGH synthase isoenzyme selectivity: the potential for safer nonsteroidal anti-inflammatory drugs. Am J Med. 1993;95:40S–44S. doi: 10.1016/0002-9343(93)90396-7. [DOI] [PubMed] [Google Scholar]
- 34.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 35.Doggrell SA. TACE inhibition: a new approach to treating inflammation. Expert Opin Investig Drugs. 2002;11:1003–1006. doi: 10.1517/13543784.11.7.1003. [DOI] [PubMed] [Google Scholar]
- 36.Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Curr Opin Drug Discov Devel. 2005;8:421–430. [PubMed] [Google Scholar]
- 37.Dumas J, Hatoum-Mokdad H, Sibley R, Riedl B, Scott WJ, Monahan MK, Lowinger TB, Brennan C, Natero R, Turner T, Johnson JS, Schoenleber R, Bhargava A, Wilhelm SM, Housley TJ, Ranges GE, Shrikhande A. 1-Phenyl-5-pyra-zolyl ureas: potent and selective p38 kinase inhibitors. Bioorg Med Chem Lett. 2000;10:2051–2054. doi: 10.1016/s0960-894x(00)00272-9. [DOI] [PubMed] [Google Scholar]
- 38.Dumas J, Hatoum-Mokdad H, Sibley RN, Smith RA, Scott WJ, Khire U, Lee W, Wood J, Wolanin D, Cooley J, Bankston D, Redman AM, Schoenleber R, Caringal Y, Gunn D, Romero R, Osterhout M, Paulsen H, Housley TJ, Wilhelm SM, Pirro J, Chien du S, Ranges GE, Shrikhande A, Muzsi A, Bortolon E, Wakefield J, Gianpaolo Ostravage C, Bhargava A, Chau T. Synthesis and pharmacological characterization of a potent, orally active p38 kinase inhibitor. Bioorg Med Chem Lett. 2002;12:1559–1562. doi: 10.1016/s0960-894x(02)00238-x. [DOI] [PubMed] [Google Scholar]
- 39.Durie BG, Katz M, Crowley J. Osteonecrosis of the jaw and bisphosphonates. N Engl J Med. 2005;353:99–102. doi: 10.1056/NEJM200507073530120. discussion 99-102. [DOI] [PubMed] [Google Scholar]
- 40.Ejeil AL, Gaultier F, Igondjo-Tchen S, Senni K, Pellat B, Godeau G, Gogly B. Are cytokines linked to collagen breakdown during periodontal disease progression? J Periodontol. 2003;74:196–201. doi: 10.1902/jop.2003.74.2.196. [DOI] [PubMed] [Google Scholar]
- 41.El-Shinnawi UM, El-Tantawy SI. The effect of alendronate sodium on alveolar bone loss in periodontitis (clinical trial) J Int Acad Periodontol. 2003;5:5–10. [PubMed] [Google Scholar]
- 42.Emingil G, Atilla G, Sorsa T, Luoto H, Kirilmaz L, Baylas H. The effect of adjunctive low-dose doxycycline therapy on clinical parameters and gingival crevicular fluid matrix metalloproteinase-8 levels in chronic periodontitis. J Periodontol. 2004;75:106–115. doi: 10.1902/jop.2004.75.1.106. [DOI] [PubMed] [Google Scholar]
- 43.Engebretson SP, Hey-Hadavi J, Ehrhardt FJ, Hsu D, Celenti RS, Grbic JT, Lamster IB. Gingival crevicular fluid levels of interleukin-1beta and glycemic control in patients with chronic periodontitis and type 2 diabetes. J Periodontol. 2004;75:1203–1208. doi: 10.1902/jop.2004.75.9.1203. [DOI] [PubMed] [Google Scholar]
- 44.Fleisch H. Bisphosphonates: mechanisms of action and clinical use in osteoporosis - an update. Horm Metab Res. 1997;29:145–150. doi: 10.1055/s-2007-979008. [DOI] [PubMed] [Google Scholar]
- 45.Flemmig TF, Rumetsch M, Klaiber B. Efficacy of systemically administered acetylsalicylic acid plus scaling on periodontal health and elastase-alpha 1-proteinase inhibitor in gingival crevicular fluid. J Clin Periodontol. 1996;23:153–159. doi: 10.1111/j.1600-051x.1996.tb02070.x. [DOI] [PubMed] [Google Scholar]
- 46.Force T, Kuida K, Namchuk M, Parang K, Kyriakis JM. Inhibitors of protein kinase signaling pathways: emerging therapies for cardiovascular disease. Circulation. 2004;109:1196–1205. doi: 10.1161/01.CIR.0000118538.21306.A9. [DOI] [PubMed] [Google Scholar]
- 47.Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Levels of interleukin-1 beta, -8, and -10 and RANTES in gingival crevicular fluid and cell populations in adult periodontitis patients and the effect of periodontal treatment. J Periodontol. 2000;71:1535–1545. doi: 10.1902/jop.2000.71.10.1535. [DOI] [PubMed] [Google Scholar]
- 48.Gapski R, Barr JL, Sarment DP, Layher MG, Socransky SS, Giannobile WV. Effect of systemic matrix metalloproteinase inhibition on periodontal wound repair: a proof of concept trial. J Periodontol. 2004;75:441–452. doi: 10.1902/jop.2004.75.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geivelis M, Turner DW, Pederson ED, Lamberts BL. Measurements of interleukin-6 in gingival crevicular fluid from adults with destructive periodontal disease. J Periodontol. 1993;64:980–983. doi: 10.1902/jop.1993.64.10.980. [DOI] [PubMed] [Google Scholar]
- 50.Goater JJ, O’Keefe RJ, Rosier RN, Puzas JE, Schwarz EM. Efficacy of ex vivo OPG gene therapy in preventing wear debris induced osteolysis. J Orthop Res. 2002;20:169–173. doi: 10.1016/S0736-0266(01)00083-3. [DOI] [PubMed] [Google Scholar]
- 51.Goldhaber P, Rabadjija L, Beyer WR, Kornhauser A. Bone resorption in tissue culture and its relevance to periodontal disease. J Am Dent Assoc. 1973;87:1027–1033. doi: 10.14219/jada.archive.1973.0010. [DOI] [PubMed] [Google Scholar]
- 52.Golub LM, Wolff M, Lee HM, McNamara TF, Ramamurthy NS, Zambon J, Ciancio S. Further evidence that tetracyclines inhibit collagenase activity in human crevicular fluid and from other mammalian sources. J Periodontal Res. 1985;20:12–23. doi: 10.1111/j.1600-0765.1985.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 53.Golub LM, Ciancio S, Ramamamurthy NS, Leung M, McNamara TF. Low-dose doxycycline therapy: effect on gingival and crevicular fluid collagenase activity in humans. J Periodontal Res. 1990;25:321–330. doi: 10.1111/j.1600-0765.1990.tb00923.x. [DOI] [PubMed] [Google Scholar]
- 54.Golub LM, Lee HM, Greenwald RA, Ryan ME, Sorsa T, Salo T, Giannobile WV. A matrix metalloproteinase inhibitor reduces bone-type collagen degradation fragments and specific collagenases in gingival crevicular fluid during adult periodontitis. Inflamm Res. 1997;46:310–319. doi: 10.1007/s000110050193. [DOI] [PubMed] [Google Scholar]
- 55.Golub LM, McNamara TF, Ryan ME, Kohut B, Blieden T, Payonk G, Sipos T, Baron HJ. Adjunctive treatment with subantimicrobial doses of doxycycline: effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J Clin Periodontol. 2001;28:146–156. doi: 10.1034/j.1600-051x.2001.028002146.x. [DOI] [PubMed] [Google Scholar]
- 56.Gomes BD, Hausmann E, Wienfeld N, De Luca C. Prostaglandins: bone resorption stimulating factors released from monkey gingiva. Calcif Tissue Res. 1976;19:285–293. doi: 10.1007/BF02564011. [DOI] [PubMed] [Google Scholar]
- 57.Gorska R, Gregorek H, Kowalski J, Laskus-Perendyk A, Syczewska M, Madalinski K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J Clin Periodontol. 2003;30:1046–1052. doi: 10.1046/j.0303-6979.2003.00425.x. [DOI] [PubMed] [Google Scholar]
- 58.Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 59.Graves DT, Jiang Y, Valente AJ. The expression of monocyte chemoattractant protein-1 and other chemokines by osteoblasts. Front Biosci. 1999;4:D571–D580. doi: 10.2741/graves. [DOI] [PubMed] [Google Scholar]
- 60.Graves DT, Oskoui M, Volejnikova S, Naguib G, Cai S, Desta T, Kakouras A, Jiang Y. Tumor necrosis factor modulates fibroblast apoptosis, PMN recruitment, and osteoclast formation in response to P. gingivalis infection. J Dent Res. 2001;80:1875–1879. doi: 10.1177/00220345010800100301. [DOI] [PubMed] [Google Scholar]
- 61.Greenwell H. Position paper: Guidelines for periodontal therapy. J Periodontol. 2001;72:1624–1628. doi: 10.1902/jop.2001.72.11.1624. [DOI] [PubMed] [Google Scholar]
- 62.Grzybowska EA, Wilczynska A, Siedlecki JA. Regulatory functions of 3′UTRs. Biochem Biophys Res Commun. 2001;288:291–295. doi: 10.1006/bbrc.2001.5738. [DOI] [PubMed] [Google Scholar]
- 63.Gurkan A, Cinarcik S, Huseyinov A. Adjunctive subantimicrobial dose doxycycline: effect on clinical parameters and gingival crevicular fluid transforming growth factorbeta levels in severe, generalized chronic periodontitis. J Clin Periodontol. 2005;32:244–253. doi: 10.1111/j.1600-051X.2005.00663.x. [DOI] [PubMed] [Google Scholar]
- 64.Haffajee AD, Dibart S, Kent RL, Jr, Socransky SS. Clinical and microbiological changes associated with the use of 4 adjunctive systemically administered agents in the treatment of periodontal infections. J Clin Periodontol. 1995;22:618–627. doi: 10.1111/j.1600-051x.1995.tb00815.x. [DOI] [PubMed] [Google Scholar]
- 65.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasegawa T, Yoshimura Y, Kikuiri T, Yawaka Y, Takeyama S, Matsumoto A, Oguchi H, Shirakawa T. Expression of receptor activator of NF-kappa B ligand and osteoprotegerin in culture of human periodontal ligament cells. J Periodontal Res. 2002;37:405–411. doi: 10.1034/j.1600-0765.2002.01603.x. [DOI] [PubMed] [Google Scholar]
- 67.Hawkey CJ. Gastroduodenal problems associated with non-steroidal, anti-inflammatory drugs (NSAIDs) Scand J Gastroenterol Suppl. 1993;200:94–95. doi: 10.3109/00365529309101583. [DOI] [PubMed] [Google Scholar]
- 68.Haynes DR, Crotti TN, Potter AE, Loric M, Atkins GJ, Howie DW, Findlay DM. The osteoclastogenic molecules RANKL and RANK are associated with periprosthetic osteolysis. J Bone Joint Surg Br. 2001;83:902–911. doi: 10.1302/0301-620x.83b6.10905. [DOI] [PubMed] [Google Scholar]
- 69.Haynes DR, Crotti TN, Loric M, Bain GI, Atkins GJ, Findlay DM. Osteoprotegerin and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclast formation by cells in the human rheumatoid arthritic joint. Rheumatology (Oxford) 2001;40:623–630. doi: 10.1093/rheumatology/40.6.623. [DOI] [PubMed] [Google Scholar]
- 70.Heasman PA, Benn DK, Kelly PJ, Seymour RA, Aitken D. The use of topical flurbiprofen as an adjunct to non-surgical management of periodontal disease. J Clin Periodontol. 1993;20:457–464. doi: 10.1111/j.1600-051x.1993.tb00389.x. [DOI] [PubMed] [Google Scholar]
- 71.Hill AV. Immunogenetics. Defence by diversity. Nature. 1999;398:668–669. doi: 10.1038/19431. [DOI] [PubMed] [Google Scholar]
- 72.Hirose K, Isogai E, Miura H, Ueda I. Levels of Porphyromonas gingivalis Fimbriae and inflammatory cytokines in gingival crevicular fluid from adult human subjects. Microbiol Immunol. 1997;41:21–26. doi: 10.1111/j.1348-0421.1997.tb01168.x. [DOI] [PubMed] [Google Scholar]
- 73.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 74.Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM, Vogel SN. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun. 2001;69:1477–1482. doi: 10.1128/IAI.69.3.1477-1482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001;79:243–253. doi: 10.1007/s001090100226. [DOI] [PubMed] [Google Scholar]
- 76.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- 77.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- 78.Holzhausen M, Rossa Junior C, Marcantonio Junior E, Nassar PO, Spolidorio DM, Spolidorio LC. Effect of selective cyclooxygenase-2 inhibition on the development of ligature-induced periodontitis in rats. J Periodontol. 2002;73:1030–1036. doi: 10.1902/jop.2002.73.9.1030. [DOI] [PubMed] [Google Scholar]
- 79.Holzhausen M, Spolidorio DM, Muscara MN, Hebling J, Spolidorio LC. Protective effects of etoricoxib, a selective inhibitor of cyclooxygenase-2, in experimental periodontitis in rats. J Periodontal Res. 2005;40:208–211. doi: 10.1111/j.1600-0765.2005.00787.x. [DOI] [PubMed] [Google Scholar]
- 80.Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily: structure-function relationship(s) Microsc Res Tech. 2000;50:184–195. doi: 10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 81.Iwamoto Y, Nishimura F, Soga Y, Takeuchi K, Kurihara M, Takashiba S, Murayama Y. Antimicrobial periodontal treatment decreases serum C-reactive protein, tumor necrosis factor-alpha, but not adiponectin levels in patients with chronic periodontitis. J Periodontol. 2003;74:1231–1236. doi: 10.1902/jop.2003.74.8.1231. [DOI] [PubMed] [Google Scholar]
- 82.Jeffcoat MR, Reddy MS. Alveolar bone loss and osteoporosis: Evidence for a common mode of therapy using the bisphosphonate alendronate. In: Davidovitch ZN, Norton LA, editors. Biological mechanisms of tooth movement and craniofacial adaptation. Vol. 1. Harvard Society for the Advancement of Orthodontics; Boston: 1996. pp. 365–373. [Google Scholar]
- 83.Jeffcoat MK, Williams RC, Holman BL, English R, Goldhaber P. Detection of active alveolar bone destruction in human periodontal disease by analysis of radiopharmaceutical uptake after a single injection of 99 m-Tc-methylene diphosphonate. J Periodontal Res. 1986;21:677–684. doi: 10.1111/j.1600-0765.1986.tb01505.x. [DOI] [PubMed] [Google Scholar]
- 84.Jeffcoat MK, Reddy MS, Haigh S, Buchanan W, Doyle MJ, Meredith MP, Nelson SL, Goodale MB, Wehmeyer KR. A comparison of topical ketorolac, systemic flurbiprofen, and placebo for the inhibition of bone loss in adult periodontitis. J Periodontol. 1995;66:329–338. doi: 10.1902/jop.1995.66.5.329. [DOI] [PubMed] [Google Scholar]
- 85.Jones BW, Heldwein KA, Means TK, Saukkonen JJ, Fenton MJ. Differential roles of Toll-like receptors in the elicitation of pro-inflammatory responses by macrophages. Ann Rheum Dis. 2001;60(Suppl 3):iii6–iii12. doi: 10.1136/ard.60.90003.iii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kantarci A, Van Dyke TE. Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins Leukot Essent Fatty Acids. 2005;73:289–299. doi: 10.1016/j.plefa.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 87.Kim KJ, Kotake S, Udagawa N, Ida H, Ishii M, Takei I, Kubo T, Takagi M. Osteoprotegerin inhibits in vitro mouse osteoclast formation induced by joint fluid from failed total hip arthroplasty. J Biomed Mater Res. 2001;58:393–400. doi: 10.1002/jbm.1033. [DOI] [PubMed] [Google Scholar]
- 88.Kirkwood K, Martin T, Andreadis ST, Kim YJ. Chemically modified tetracyclines selectively inhibit IL-6 expression in osteoblasts by decreasing mRNA stability. Biochem Pharmacol. 2003;66:1809–1819. doi: 10.1016/s0006-2952(03)00450-7. [DOI] [PubMed] [Google Scholar]
- 89.Kirkwood KL, Taba M, Jr., Rossa C, Giannobile WV. Comparison of pathology of periodontal disease and rheumatoid arthritis. In: Newman MG, Takei H, Carranza FA, Klokkevold YJ, editors. Carranza’s Clinical Periodontology. 10th Ed. Elseveir; St. Louis: 2006. Chapter 12. [Google Scholar]
- 90.Kirkwood KL, Li F, Rogers JE, Otremba J, Coatney DD, Kreider JM, D’Silva NJ, Chakravarty S, Dugar S, Higgins LS, Protter AA, Medicherla S. A. p38alpha selective mitogen activated protein kinase inhibitor prevents periodontal bone loss. J Pharmacol Exp Ther. 2007 doi: 10.1124/jpet.106.112466. in press. [DOI] [PubMed] [Google Scholar]
- 91.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 92.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 93.Kornman KS, Blodgett RF, Brunsvold M, Holt SC. Effects of topical applications of meclofenamic acid and ibuprofen on bone loss, subgingival microbiota and gingival PMN response in the primate Macaca fascicularis. J Periodontal Res. 1990;25:300–307. doi: 10.1111/j.1600-0765.1990.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 94.Kostenuik PJ, Bolon B, Morony S, Daris M, Geng Z, Carter C, Sheng J. Gene therapy with human recombinant osteoprotegerin reverses established osteopenia in ovariectomized mice. Bone. 2004;34:656–664. doi: 10.1016/j.bone.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 95.Kumar S, Votta BJ, Rieman DJ, Badger AM, Gowen M, Lee JC. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J Cell Physiol. 2001;187:294–303. doi: 10.1002/jcp.1082. [DOI] [PubMed] [Google Scholar]
- 96.Lane N, Armitage GC, Loomer P, Hsieh S, Majumdar S, Wang HY, Jeffcoat M, Munoz T. Bisphosphonate therapy improves the outcome of conventional periodontal treatment: results of a 12-month, randomized, placebo-controlled study. J Periodontol. 2005;76:1113–1122. doi: 10.1902/jop.2005.76.7.1113. [DOI] [PubMed] [Google Scholar]
- 97.Lappin DF, Kjeldsen M, Sander L, Kinane DF. Inducible nitric oxide synthase expression in periodontitis. J Periodontal Res. 2000;35:369–373. doi: 10.1034/j.1600-0765.2000.035006369.x. [DOI] [PubMed] [Google Scholar]
- 98.Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. J Clin Periodontol. 1995;22:885–890. doi: 10.1111/j.1600-051x.1995.tb01788.x. [DOI] [PubMed] [Google Scholar]
- 99.Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 100.Lee JY, Lee YM, Shin SY, Seol YJ, Ku Y, Rhyu IC, Chung CP, Han SB. Effect of subantimicrobial dose doxycycline as an effective adjunct to scaling and root planing. J Periodontol. 2004;75:1500–1508. doi: 10.1902/jop.2004.75.11.1500. [DOI] [PubMed] [Google Scholar]
- 101.Leitao RF, Ribeiro RA, Chaves HV, Rocha FA, Lima V, Brito GA. Nitric oxide synthase inhibition prevents alveolar bone resorption in experimental periodontitis in rats. J Periodontol. 2005;76:956–963. doi: 10.1902/jop.2005.76.6.956. [DOI] [PubMed] [Google Scholar]
- 102.Leng SX, Elias JA. Interleukin-11 inhibits macrophage interleukin-12 production. J Immunol. 1997;159:2161–2168. [PubMed] [Google Scholar]
- 103.Lindsley CB, Warady BA. Nonsteroidal antiinflammatory drugs. Renal toxicity. Review of pediatric issues. Clin Pediatr (Phila) 1990;29:10–13. doi: 10.1177/000992289002900101. [DOI] [PubMed] [Google Scholar]
- 104.Liu D, Xu JK, Figliomeni L, Huang L, Pavlos NJ, Rogers M, Tan A, Price P, Zheng MH. Expression of RANKL and OPG mRNA in periodontal disease: possible involvement in bone destruction. Int J Mol Med. 2003;11:17–21. doi: 10.3892/ijmm.11.1.17. [DOI] [PubMed] [Google Scholar]
- 105.Lohinai Z, Benedek P, Feher E, Gyorfi A, Rosivall L, Fazekas A, Salzman AL, Szabo C. Protective effects of mercaptoethylguanidine, a selective inhibitor of inducible nitric oxide synthase, in ligature-induced periodontitis in the rat. Br J Pharmacol. 1998;123:353–360. doi: 10.1038/sj.bjp.0701604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 107.Mahamed DA, Marleau A, Alnaeeli M, Singh B, Zhang X, Penninger JM, Teng YT. G(-) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes. 2005;54:1477–1486. doi: 10.2337/diabetes.54.5.1477. [DOI] [PubMed] [Google Scholar]
- 108.Martuscelli G, Fiorellini JP, Crohin CC, Howell TH. The effect of interleukin-11 on the progression of ligature-induced periodontal disease in the beagle dog. J Periodontol. 2000;71:573–578. doi: 10.1902/jop.2000.71.4.573. [DOI] [PubMed] [Google Scholar]
- 109.Marx RE. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 2003;61:1115–1117. doi: 10.1016/s0278-2391(03)00720-1. [DOI] [PubMed] [Google Scholar]
- 110.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 111.McIntyre KW, Shuster DJ, Gillooly KM, Dambach DM, Pattoli MA, Lu P, Zhou XD, Qiu Y, Zusi FC, Burke JR. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652–2659. doi: 10.1002/art.11131. [DOI] [PubMed] [Google Scholar]
- 112.McLay LM, Halley F, Souness JE, McKenna J, Benning V, Birrell M, Burton B, Belvisi M, Collis A, Constan A, Foster M, Hele D, Jayyosi Z, Kelley M, Maslen C, Miller G, Ouldelhkim MC, Page K, Phipps S, Pollock K, Porter B, Ratcliffe AJ, Redford EJ, Webber S, Slater B, Thybaud V, Wilsher N. The discovery of RPR 200765A, a p38 MAP kinase inhibitor displaying a good oral anti-arthritic efficacy. Bioorg Med Chem. 2001;9:537–554. doi: 10.1016/s0968-0896(00)00331-x. [DOI] [PubMed] [Google Scholar]
- 113.Mercado F, Marshall RI, Klestov AC, Bartold PM. Is there a relationship between rheumatoid arthritis and periodontal disease? J Clin Periodontol. 2000;27:267–272. doi: 10.1034/j.1600-051x.2000.027004267.x. [DOI] [PubMed] [Google Scholar]
- 114.Mercado FB, Marshall RI, Klestov AC, Bartold PM. Relationship between rheumatoid arthritis and periodontitis. J Periodontol. 2001;72:779–787. doi: 10.1902/jop.2001.72.6.779. [DOI] [PubMed] [Google Scholar]
- 115.Mercado FB, Marshall RI, Bartold PM. Inter-relationships between rheumatoid arthritis and periodontal disease. A review. J Clin Periodontol. 2003;30:761–772. doi: 10.1034/j.1600-051x.2003.00371.x. [DOI] [PubMed] [Google Scholar]
- 116.Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- 117.Mogi M, Otogoto J, Ota N, Togari A. Differential expression of RANKL and osteoprotegerin in gingival crevicular fluid of patients with periodontitis. J Dent Res. 2004;83:166–169. doi: 10.1177/154405910408300216. [DOI] [PubMed] [Google Scholar]
- 118.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 119.Morel J, Berenbaum F. Signal transduction pathways: new targets for treating rheumatoid arthritis. Joint Bone Spine. 2004;71:503–510. doi: 10.1016/j.jbspin.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 120.Mori Y, Yoshimura A, Ukai T, Lien E, Espevik T, Hara Y. Immunohistochemical localization of Toll-like receptors 2 and 4 in gingival tissue from patients with periodontitis. Oral Microbiol Immunol. 2003;18:54–58. doi: 10.1034/j.1399-302x.2003.180109.x. [DOI] [PubMed] [Google Scholar]
- 121.Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR, Kostenuik PJ. Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 2001;61:4432–4436. [PubMed] [Google Scholar]
- 122.Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- 123.Netea MG, van der Graaf C, Van der Meer JW, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004;75:749–755. doi: 10.1189/jlb.1103543. [DOI] [PubMed] [Google Scholar]
- 124.Ng VW, Bissada NF. Clinical evaluation of systemic doxycycline and ibuprofen administration as an adjunctive treatment for adult periodontitis. J Periodontol. 1998;69:772–776. doi: 10.1902/jop.1998.69.7.772. [DOI] [PubMed] [Google Scholar]
- 125.Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Hashimoto J, Azuma J, Kishimoto T. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:1761–1769. doi: 10.1002/art.20303. [DOI] [PubMed] [Google Scholar]
- 126.Novak MJ, Johns LP, Miller RC, Bradshaw MH. Adjunctive benefits of subantimicrobial dose doxycycline in the management of severe, generalized, chronic periodontitis. J Periodontol. 2002;73:762–769. doi: 10.1902/jop.2002.73.7.762. [DOI] [PubMed] [Google Scholar]
- 127.Nyman S, Schroeder HE, Lindhe J. Suppression of inflammation and bone resorption by indomethacin during experimental periodontitis in dogs. J Periodontol. 1979;50:450–461. doi: 10.1902/jop.1979.50.9.450. [DOI] [PubMed] [Google Scholar]
- 128.O’Neill LA, Greene C. Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects, and plants. J Leukoc Biol. 1998;63:650–657. [PubMed] [Google Scholar]
- 129.Oates TW, Graves DT, Cochran DL. Clinical, radiographic and biochemical assessment of IL-1/TNF-alpha antagonist inhibition of bone loss in experimental periodontitis. J Clin Periodontol. 2002;29:137–143. doi: 10.1034/j.1600-051x.2002.290208.x. [DOI] [PubMed] [Google Scholar]
- 130.Odvina CV, Zerwekh JE, Rao DS, Maalouf N, Gottschalk FA, Pak CY. Severely suppressed bone turnover: a potential complication of alendronate therapy. J Clin Endocrinol Metab. 2005;90:1294–1301. doi: 10.1210/jc.2004-0952. [DOI] [PubMed] [Google Scholar]
- 131.Offenbacher S. Periodontal diseases: pathogenesis. Ann Periodontol. 1996;1:821–878. doi: 10.1902/annals.1996.1.1.821. [DOI] [PubMed] [Google Scholar]
- 132.Offenbacher S, Farr DH, Goodson JM. Measurement of prostaglandin E in crevicular fluid. J Clin Periodontol. 1981;8:359–367. doi: 10.1111/j.1600-051x.1981.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 133.Offenbacher S, Odle BM, Gray RC, Van Dyke TE. Crevicular fluid prostaglandin E levels as a measure of the periodontal disease status of adult and juvenile periodontitis patients. J Periodontal Res. 1984;19:1–13. doi: 10.1111/j.1600-0765.1984.tb01190.x. [DOI] [PubMed] [Google Scholar]
- 134.Offenbacher S, Williams RC, Jeffcoat MK, Howell TH, Odle BM, Smith MA, Hall CM, Johnson HG, Goldhaber P. Effects of NSAIDs on beagle crevicular cyclooxygenase metabolites and periodontal bone loss. J Periodontal Res. 1992;27:207–213. doi: 10.1111/j.1600-0765.1992.tb01670.x. [DOI] [PubMed] [Google Scholar]
- 135.Offenbacher S, Heasman PA, Collins JG. Modulation of host PGE2 secretion as a determinant of periodontal disease expression. J Periodontol. 1993;64:432–444. doi: 10.1902/jop.1993.64.5s.432. [DOI] [PubMed] [Google Scholar]
- 136.Oringer RJ, Al-Shammari KF, Aldredge WA, Iacono VJ, Eber RM, Wang HL, Berwald B, Nejat R, Giannobile WV. Effect of locally delivered minocycline microspheres on markers of bone resorption. J Periodontol. 2002;73:835–842. doi: 10.1902/jop.2002.73.8.835. [DOI] [PubMed] [Google Scholar]
- 137.Oyajobi BO, Anderson DM, Traianedes K, Williams PJ, Yoneda T, Mundy GR. Therapeutic efficacy of a soluble receptor activator of nuclear factor kappaB-IgG Fc fusion protein in suppressing bone resorption and hypercalcemia in a model of humoral hypercalcemia of malignancy. Cancer Res. 2001;61:2572–2578. [PubMed] [Google Scholar]
- 138.Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J Clin Pharmacol. 2003;43:406–413. doi: 10.1177/0091270002250615. [DOI] [PubMed] [Google Scholar]
- 139.Patil C, Zhu X, Rossa C, Jr, Kim YJ, Kirkwood KL. p38 MAPK regulates IL-1beta induced IL-6 expression through mRNA stability in osteoblasts. Immunol Invest. 2004;33:213–233. doi: 10.1081/imm-120034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 141.Preshaw PM, Hefti AF, Novak MJ, et al. Subantimicrobial dose doxycycline enhances scaling and root planing. J Dent Res. 2002;81:A–127. (Abstr. 0841) [Google Scholar]
- 142.Preshaw PM, Hefti AF, Jepsen S, Etienne D, Walker C, Bradshaw MH. Subantimicrobial dose doxycycline as adjunctive treatment for periodontitis. A review. J Clin Periodontol. 2004;31:697–707. doi: 10.1111/j.1600-051X.2004.00558.x. [DOI] [PubMed] [Google Scholar]
- 143.Preshaw PM, Hefti AF, Novak MJ, Michalowicz BS, Pihlstrom BL, Schoor R, Trummel CL, Dean J, Van Dyke TE, Walker CB, Bradshaw MH. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: a multicenter trial. J Periodontol. 2004;75:1068–1076. doi: 10.1902/jop.2004.75.8.1068. [DOI] [PubMed] [Google Scholar]
- 144.Randle JC, Harding MW, Ku G, Schonharting M, Kurrle R. ICE/Caspase-1 inhibitors as novel anti-inflammatory drugs. Expert Opin Investig Drugs. 2001;10:1207–1209. doi: 10.1517/13543784.10.7.1207. [DOI] [PubMed] [Google Scholar]
- 145.Reddi K, Wilson M, Nair S, Poole S, Henderson B. Comparison of the pro-inflammatory cytokine-stimulating activity of the surface-associated proteins of periodontopathic bacteria. J Periodontal Res. 1996;31:120–130. doi: 10.1111/j.1600-0765.1996.tb00473.x. [DOI] [PubMed] [Google Scholar]
- 146.Reddy MS, Palcanis KG, Barnett ML, Haigh S, Charles CH, Jeffcoat MK. Efficacy of meclofenamate sodium (Meclomen) in the treatment of rapidly progressive periodontitis. J Clin Periodontol. 1993;20:635–640. doi: 10.1111/j.1600-051x.1993.tb00708.x. [DOI] [PubMed] [Google Scholar]
- 147.Reddy MS, Weatherford TW, 3rd, Smith CA, West BD, Jeffcoat MK, Jacks TM. Alendronate treatment of naturally-occurring periodontitis in beagle dogs. J Periodontol. 1995;66:211–217. doi: 10.1902/jop.1995.66.3.211. [DOI] [PubMed] [Google Scholar]
- 148.Reddy MS, Geurs NC, Gunsolley JC. Periodontal host modulation with antiproteinase, anti-inflammatory, and bone-sparing agents. A systematic review. Ann Periodontol. 2003;8:12–37. doi: 10.1902/annals.2003.8.1.12. [DOI] [PubMed] [Google Scholar]
- 149.Reinhardt RA, Masada MP, Kaldahl WB, DuBois LM, Kornman KS, Choi JI, Kalkwarf KL, Allison AC. Gingival fluid IL-1 and IL-6 levels in refractory periodontitis. J Clin Periodontol. 1993;20:225–231. doi: 10.1111/j.1600-051x.1993.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 150.Richardson P. Clinical update: proteasome inhibitors in hematologic malignancies. Cancer Treat Rev. 2003;29(Suppl 1):33–39. doi: 10.1016/s0305-7372(03)00080-x. [DOI] [PubMed] [Google Scholar]
- 151.Rocha M, Nava LE, Vazquez de la Torre C, Sanchez-Marin F, Garay-Sevilla ME, Malacara JM. Clinical and radiological improvement of periodontal disease in patients with type 2 diabetes mellitus treated with alendronate: a randomized, placebo-controlled trial. J Periodontol. 2001;72:204–209. doi: 10.1902/jop.2001.72.2.204. [DOI] [PubMed] [Google Scholar]
- 152.Rocha ML, Malacara JM, Sanchez-Marin FJ, Vazquez de la Torre CJ, Fajardo ME. Effect of alendronate on periodontal disease in postmenopausal women: a randomized placebo-controlled trial. J Periodontol. 2004;75:1579–1585. doi: 10.1902/jop.2004.75.12.1579. [DOI] [PubMed] [Google Scholar]
- 153.Rogers MJ, Gordon S, Benford HL, Coxon FP, Luckman SP, Monkkonen J, Frith JC. Cellular and molecular mechanisms of action of bisphosphonates. Cancer. 2000;88:2961–2978. doi: 10.1002/1097-0142(20000615)88:12+<2961::aid-cncr12>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 154.Romas E, Sims NA, Hards DK, Lindsay M, Quinn JW, Ryan PF, Dunstan CR, Martin TJ, Gillespie MT. Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol. 2002;161:1419–1427. doi: 10.1016/S0002-9440(10)64417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Roux S, Orcel P. Bone loss. Factors that regulate osteoclast differentiation: an update. Arthritis Res. 2000;2:451–456. doi: 10.1186/ar127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage. 2003;11:738–746. doi: 10.1016/s1063-4584(03)00153-5. [DOI] [PubMed] [Google Scholar]
- 157.Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 2004;62:527–534. doi: 10.1016/j.joms.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 158.Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4:372–377. doi: 10.1016/j.coph.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 159.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, Colgan SP, Stahl GL, Merched A, Petasis NA, Chan L, Van Dyke TE. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 160.Shimizu N, Ozawa Y, Yamaguchi M, Goseki T, Ohzeki K, Abiko Y. Induction of COX-2 expression by mechanical tension force in human periodontal ligament cells. J Periodontol. 1998;69:670–677. doi: 10.1902/jop.1998.69.6.670. [DOI] [PubMed] [Google Scholar]
- 161.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 162.Sorsa T, Uitto VJ, Suomalainen K, Vauhkonen M, Lindy S. Comparison of interstitial collagenases from human gingiva, sulcular fluid and polymorphonuclear leukocytes. J Periodontal Res. 1988;23:386–393. doi: 10.1111/j.1600-0765.1988.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 163.Stashenko P, Dewhirst FE, Peros WJ, Kent RL, Ago JM. Synergistic interactions between interleukin 1, tumor necrosis factor, and lymphotoxin in bone resorption. J Immunol. 1987;138:1464–1468. [PubMed] [Google Scholar]
- 164.Stashenko P, Jandinski JJ, Fujiyoshi P, Rynar J, Socransky SS. Tissue levels of bone resorptive cytokines in periodontal disease. J Periodontol. 1991;62:504–509. doi: 10.1902/jop.1991.62.8.504. [DOI] [PubMed] [Google Scholar]
- 165.Stashenko P, Fujiyoshi P, Obernesser MS, Prostak L, Haffajee AD, Socransky SS. Levels of interleukin 1 beta in tissue from sites of active periodontal disease. J Clin Periodontol. 1991;18:548–554. doi: 10.1111/j.1600-051x.1991.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 166.Stefanovic-Racic M, Stadler J, Evans CH. Nitric oxide and arthritis. Arthritis Rheum. 1993;36:1036–1044. doi: 10.1002/art.1780360803. [DOI] [PubMed] [Google Scholar]
- 167.Suda T, Kobayashi K, Jimi E, Udagawa N, Takahashi N. The molecular basis of osteoclast differentiation and activation. Novartis Found Symp. 2001;232:235–247. doi: 10.1002/0470846658.ch16. discussion 247-250. [DOI] [PubMed] [Google Scholar]
- 168.Sugita N, Kimura A, Matsuki Y, Yamamoto T, Yoshie H, Hara K. Activation of transcription factors and IL-8 expression in neutrophils stimulated with lipopolysaccharide from Porphyromonas gingivalis. Inflammation. 1998;22:253–267. doi: 10.1023/a:1022344031223. [DOI] [PubMed] [Google Scholar]
- 169.Taba M, Jr, Huffer H, Shelburne CE, Kriegl J, Goldstein SA, Lustig KA, Burstein H, Giannobile WV. Gene delivery of TNFR:Fc by adeno-associated virus vector blocks progression of periodontitits. Mol Ther. 2005;10(Suppl):527. [Google Scholar]
- 170.Tak PP, Gerlag DM, Aupperle KR, van de Geest A, Overbeek M, Bennett BL, Boyle DL, Manning AM, Firestein GS. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001;44:1897–1907. doi: 10.1002/1529-0131(200108)44:8<1897::AID-ART328>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 171.Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, Ellen RP, Penninger JM. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106:R59–R67. doi: 10.1172/jci10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Trepicchio WL, Bozza M, Pedneault G, Dorner AJ. Recombinant human IL-11 attenuates the inflammatory response through down-regulation of pro-inflammatory cytokine release and nitric oxide production. J Immunol. 1996;157:3627–3634. [PubMed] [Google Scholar]
- 173.Uchida M, Shima M, Shimoaka T, Fujieda A, Obara K, Suzuki H, Nagai Y, Ikeda T, Yamato H, Kawaguchi H. Regulation of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) by bone resorptive factors in osteoblastic cells. J Cell Physiol. 2000;185:207–214. doi: 10.1002/1097-4652(200011)185:2<207::AID-JCP5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 174.Udagawa N, Takahashi N, Yasuda H, Mizuno A, Itoh K, Ueno Y, Shinki T, Gillespie MT, Martin TJ, Higashio K, Suda T. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology. 2000;141:3478–3484. doi: 10.1210/endo.141.9.7634. [DOI] [PubMed] [Google Scholar]
- 175.Ulrich-Vinther M, Carmody EE, Goater JJ, O’Keefe S, Schwarz EM. Recombinant adeno-associated virus-mediated osteoprotegerin gene therapy inhibits wear debrisinduced osteolysis. J Bone Joint Surg Am. 2002;84-A:1405–1412. doi: 10.2106/00004623-200208000-00016. [DOI] [PubMed] [Google Scholar]
- 176.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 177.Vardar S, Buduneli E, Baylas H, Berdeli AH, Buduneli N, Atilla G. Individual and combined effects of selective cyclooxygenase-2 inhibitor and omega-3 fatty acid on endotoxin-induced periodontitis in rats. J Periodontol. 2005;76:99–106. doi: 10.1902/jop.2005.76.1.99. [DOI] [PubMed] [Google Scholar]
- 178.Wang PL, Ohura K. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med. 2002;13:132–142. doi: 10.1177/154411130201300204. [DOI] [PubMed] [Google Scholar]
- 179.Wang PL, Azuma Y, Shinohara M, Ohura K. Toll-like receptor 4-mediated signal pathway induced by Porphyromonas gingivalis lipopolysaccharide in human gingival fibroblasts. Biochem Biophys Res Commun. 2000;273:1161–1167. doi: 10.1006/bbrc.2000.3060. [DOI] [PubMed] [Google Scholar]
- 180.Weaks-Dybvig M, Sanavi F, Zander H, Rifkin BR. The effect of indomethacin on alveolar bone loss in experimental periodontitis. J Periodontal Res. 1982;17:90–100. doi: 10.1111/j.1600-0765.1982.tb01134.x. [DOI] [PubMed] [Google Scholar]
- 181.Weisman MH. What are the risks of biologic therapy in rheumatoid arthritis? An update on safety. J Rheumatol Suppl. 2002;65:33–38. [PubMed] [Google Scholar]
- 182.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 183.Williams RC, Jeffcoat MK, Howell TH, Reddy MS, Johnson HG, Hall CM, Goldhaber P. Ibuprofen: an inhibitor of alveolar bone resorption in beagles. J Periodontal Res. 1988;23:225–229. doi: 10.1111/j.1600-0765.1988.tb01363.x. [DOI] [PubMed] [Google Scholar]
- 184.Williams RC, Jeffcoat MK, Howell TH, Rolla A, Stubbs D, Teoh KW, Reddy MS, Goldhaber P. Altering the progression of human alveolar bone loss with the non-steroidal anti-inflammatory drug flurbiprofen. J Periodontol. 1989;60:485–490. doi: 10.1902/jop.1989.60.9.485. [DOI] [PubMed] [Google Scholar]
- 185.Williams DH, Wilkinson SE, Purton T, Lamont A, Flotow H, Murray EJ. Ro 09-2210 exhibits potent anti-proliferative effects on activated T cells by selectively blocking MKK activity. Biochemistry. 1998;37:9579–9585. doi: 10.1021/bi972914c. [DOI] [PubMed] [Google Scholar]
- 186.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang HG, Huang N, Liu D, Bilbao L, Zhang X, Yang P, Zhou T, Curiel DT, Mountz JD. Gene therapy that inhibits nuclear translocation of nuclear factor kappaB results in tumor necrosis factor alpha-induced apoptosis of human synovial fibroblasts. Arthritis Rheum. 2000;43:1094–1105. doi: 10.1002/1529-0131(200005)43:5<1094::AID-ANR20>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 188.Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, Mizokami A, Fu Z, Westman J, Keller ET. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–1244. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of pro-inflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem. 2005;280:8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]