

Abstract
Isolated or nonsyndromic cleft lip and palate (NS CLP) is a complex disorder resulting from multiple genetic and environmental factors. NS CLP has a birth prevalence of 1 per 500 in the Philippines where large families provide an opportunity for gene localization. Genotyping of 392 microsatellite repeat markers at 10 cM intervals over the genome was performed by the Center for Inherited Disease Research (CIDR) on 220 Filipino families with 567 affected and 1,109 unaffected family members genotyped. Among the most statistically significant results from analysis of the genome-wide scan data was a 20 cM region at 8p11-23 in which markers had LODs ≥1.0. This region on 8p11-23 has not been found in any previous genome wide scan nor does it contain any of the candidate genes widely studied in CLP. Fine mapping in 8p11-23 was done in the 220 families plus an additional 51 families, using SNP markers from 10 known genes (FGFR1, NRG1, FZD3, SLC8A1, PPP3CC, EPHX2, BNIP3L, EGR3, PPP2R2A, and NAT1) within the 20 cM region of 8p11-23. Linkage and association analyses of these SNPs yield suggestive results for markers in FGFR1 (recessive multipoint HLOD 1.07) and BAG4 (recessive multipoint HLOD 1.31).
Keywords: cleft lip and palate, linkage analysis, association analysis, fine mapping, SNP markers, FGFR1
Introduction
Nonsyndromic cleft lip and cleft palate (NS CLP) is a common birth defect, and the source of substantial morbidity and mortality worldwide [Murray, 2002]. With an average birth prevalence of 1/700 live births, there is remarkable population to population variation [Mossey and Little, 2002]. In general, Asian populations have a higher birth prevalence of clefting (1/500 births), Caucasians are intermediate (1/1,000 births), and African populations have the lowest (1/2,500 births). A long-term follow-up study of 5,331 Danish individuals with NS CLP found a standardized mortality ratio of 1.4 for males and 1.8 for females. This increased mortality ratio was present in infancy, childhood, and adulthood [Christensen et al., 2004]. Individuals with NS CLP had an increased rate of death from cardiovascular disease, suicide, and epilepsy, as well as a marginally increased rate of death from cancer. Infants with NS CLP also had increased deaths from prematurity, pneumonia, aspiration, and sepsis [Christensen et al., 2004].
An examination of familial recurrence patterns in NS CLP indicated that there may be anywhere from 2 to 14 interacting loci involved in clefting [Schliekelman and Slatkin, 2002]. The upper limit on this range represents more loci than was previously theorized, and indicates that very large sample sizes may be necessary to detect the loci involved in NS CLP [Mitchell and Risch, 1992]. For a complex genetic disorder such as NS CLP, several experimental techniques may be used. These include breakpoint mapping, deletion mapping, direct sequencing of candidate genes, linkage analysis, and linkage disequilibrium analysis [Lidral and Murray, 2004]. Genome wide linkage scans of complex traits can best succeed when heterogeneity is minimized and sample sizes are maximized. The Philippines provides an opportunity to base such a study with the common occurrence of isolated clefting, large average family sizes, and a motivated public health enterprise [Murray et al., 1997].
A study of van der Woude syndrome, a single-gene Mendelian disorder with cleft lip and/or cleft palate as part of the phenotype, identified disease-causing mutations in IRF6 [Kondo et al., 2002]. An association study in Filipino, Caucasian, and South American NS CLP samples found a strong association of a particular V274I mutation with NS CLP [Zucchero et al., 2004], that has since been confirmed in multiple other populations (Italy [Scapoli et al., 2005]; Belgium [Ghassibe et al., 2005]; US [Blanton et al., 2005]; Thailand [Srichomthong et al., 2005]). IRF6 maps to 1q32, and there was also evidence supporting linkage to this region in a meta-analysis of CLP genome scans [Marazita et al., 2004].
In order to extend these earlier studies by identifying possible Filipino-population-specific clefting loci and to identify genes in addition to IRF6 that contribute to NS CLP, we performed a genome scan in 220 extended Filipino families with at least two affected members. Fine mapping and mutation searches were carried out in a region on 8p11-23 where suggestive linkage results were obtained from the genome scan.
Materials and Methods
Sample Collection
Under the auspices of Operation Smile and Operation Smile-Philippines, 271 extended Filipino families were collected. Families had two or more affected members; unaffected family members were collected as well. Clinical aspects of sample collection have been previously described [Murray et al., 1997; Schultz et al., 2004]. The University of Iowa IRB gave approval for sample collection (approval numbers 9701068, 199804081, and 200003065) in conjunction with local approval in the Philippines. In addition, 90 DNA samples from cases in the Philippines and another 90 from cases born in Iowa were utilized to extend the sequencing studies. Whole blood samples were collected by venipuncture. Subjects were reviewed by JCM or SDH to exclude any with syndromic features. Sample and family characteristics are shown in Table I.
Table I.
Sample Characteristics for 271 Filipino Families
| Affected (NS CLP) | 744 |
| Unaffected | 1,464 |
| Phenotype unknown | 28 |
| Total | 2,236 |
| Number of family members for whom samples were unavailable | 2,729 |
DNA Extraction
Stabilization buffer (AS1; Qiagen, Valencia, CA) was added to the whole blood samples for transport back to the United States. Samples were processed using QiaAmp DNA Blood Maxi or Midi kits, depending on sample volume. The protocol was obtained from Qiagen, modified for AS1 use.
Genotyping
DNA samples from the initial 220 extended Filipino kindreds were sent to the Center for Inherited Disease Research (CIDR) for genotyping. Genotyping was completed on 392 markers from the Marshfield Genetics screening set 8. Further information on CIDR is available from their website http://www.cidr.jhmi.edu/.
Fine Mapping
Fine mapping was carried out using TaqMan™ Assays-on-Demand probes (Applied Biosystems, Foster City, CA) and done on an Applied Biosystems 7900 HT Sequence Detection System machine. Single-nucleotide polymorphism (SNP) markers were selected within candidate genes and/or at 1–4 Mb intervals across 8p.
Statistical Analyses
Preliminary steps
Each genome-scan and fine-mapping marker was assessed with PedCheck [O'Connell and Weeks, 1998] to test for inconsistencies due to non-paternity or other errors. For the parametric linkage analyses, allele frequencies were estimated from the unaffected founders in the study families and are available on our laboratory website at genetics.uiowa.edu. The genetic model parameters were taken from segregation analysis results in a sample of the Filipino families (unpublished results). The dominant model was a disease allele frequency of 0.002 with a penetrance of 0.30. The recessive model was a disease allele frequency of 0.04 with a penetrance 0.45.
Linkage calculations
Two-point and multipoint LOD scores
We calculated two-point LOD scores in the extended kindreds employing the LINKAGE program with recent updates to speed calculations (VITESSE and FAST-LINK) [Cottingham, 1993; Terwilliger and Ott, 1994; O'Connell and Weeks, 1995]. For multipoint LOD and heterogeneity LOD (HLOD)[Smith, 1963] calculations, the descent graph method [Sobel and Lange, 1996; Sobel et al., 1996; Lange, 2002] implemented in computer program SIMWALK2 was used. Given the probable complexities in the genetic model for NS CLP (e.g., heterogeneity), we also calculated model-free linkage statistics (both two-point and multipoint) using MERLIN [Abecasis et al., 2002].
Allelic association
Alleles at each genome-scan and fine-mapping marker were tested for association with CLP using the Family Based Association Test (FBAT) [Laird et al., 2000; Rabinowitz and Laird, 2000; Horvath et al., 2001].
Results
Three regions yielded suggestive positive results in the initial 220 multiplex extended Filipino kindreds: 2p21, 6q23, and 8p21. Figure 1 shows the results from the genome scan markers in the 8p11-23 region that was chosen for fine-mapping, the peak of significance ranges from approximately 45 cM to 55 cM. Power calculations using SIMLINK predict the mean maximum LODs for these families assuming 5% of families linked is 2.0 ± 0.10 under a dominant model, and 3.1 ± 0.13 under a recessive model. For 2% of families linked the mean max LODs are 1.3 ± 0.07 and 1.8 ± 0.09 respectively. Therefore, the HLODs for FUT10, BAG4, and FGFR1 are close to the maximum expected under models of heterogeneity.
Fig. 1.
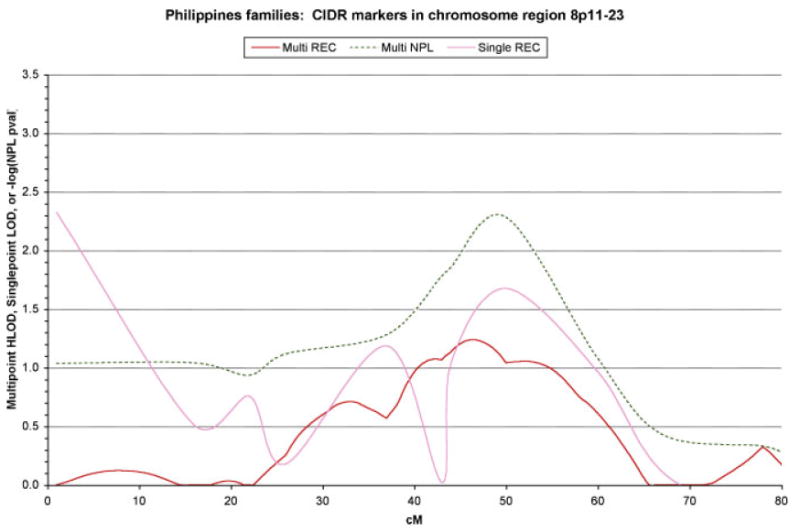
Plot of the results from the microsatellite markers in the 8p11-23 region genotyped by the Center for Inherited Disease Research for 220 Filipino extended multiplex kindreds (i.e., with two or more family members affected with NS CLP).
Figure 2 summarizes the genes genotyped in the region as well as the genome scan markers genotyped by CIDR. Table II summarizes the linkage and association results for the fine-mapping markers genotyped in the 271 multiplex extended Filipino kindreds (the 220 kindreds used for the genome scan plus an additional 54 extended kindreds). Both linkage and association results were positive for markers in FGFR1 and BAG4, while either linkage or association showed positive results for markers in SLC18A1, EPHX2, and FZD3.
Fig. 2.

Map of 8p11-23. Above the chromosome are markers genotyped by the Center for Inherited Disease Research in the genome-wide linkage scan. D8S1771 (in bold) had the largest single-point LOD score and most significant NPL result. Below the chromosome are the SNP markers used for fine-mapping. In bold are those fine-mapping genes with the most significant FBAT P-values (FGFR1, BAG4, EPHX2). Distances in centimorgans between genes are shown in the intervals.
Table II.
Results of 8p11-23 Fine-Mapping From 271 Filipino Multiplex NS CLP Families
| Gene | Marker | Distance to next Marker | Singlepoint LOD | Multipoint HLOD | Multipoint NPL P-value | FBAT multiallele P-value |
|---|---|---|---|---|---|---|
| NAT1 | rs3850751 | 1.97 Mb | No positives | Rec 0.64 α = 0.05 | P = 0.22 | P = 0.19 |
| SLC18A1 | rs227970 | 2.34 Mb | Dom 1.23 θ = 0.2 | Dom 0.42 α = 0.05 | P = 0.21 | P = 0.36 |
| PPP3CC | rs2461491 | 199 kb | Rec 0.53 θ = 0.3 | Dom 0.42 α = 0.05 | P = 0.11 | P = 0.95 |
| EGR3 | rs12541654 | 3.6 Mb | Dom 0.60 θ = 0.3 | Rec 0.41 α = 0.05 | P = 0.14 | P = 0.38 |
| PPP2R2A | rs17055106 | 93 kb | Rec 0.68 θ = 0.3 | Rec 0.37 α = 0.05 | P = 0.11 | P = 0.91 |
| BNIP3L | rs3808579 | 1.13 Mb | No positives | Rec 0.29 α = 0.05 | P = 0.11 | P = 0.73 |
| EPHX2 | rs721619 | 1 Mb | Rec 0.31 θ = 0.4 | Dom 0.45 α = 0.05 | P = 0.10 | P = 0.003 |
| FZD3 | rs2241802 | 2.65 Mb | Dom 1.28 θ = 0.3 | Dom 0.66 α = 0.05 | P = 0.13 | P = 0.94 |
| WRN | rs2725362 | 1.52 Mb | Rec 0.69 θ = 0.3 | Dom 0.66 α = 0.05 | P = 0.09 | P = 0.85 |
| NRG1 | rs2466048 | 768 kb | Rec 0.81 θ = 0.3 | Rec 0.63 α = 0.05 | P = 0.11 | P = 0.25 |
| FUT10 | rs2459516 | 4.66 Mb | Dom 0.15 θ = 0.4 | Rec 1.31 α = 0.10 | P = 0.11 | P = 0.37 |
| BAG4 | rs7836805 | 2.23 Mb | Dom 0.34 θ = 0.4 | Rec 1.31 α = 0.10 | P = 0.07 | P = 0.03 |
| FGFR1 (1) | rs13317 | 36 kb | Dom 0.41 θ = 0.4 | Rec 1.07 α = 0.05 | P = 0.06 | P = 0.01 |
| FGFR1 (2) | rs6996321 | 5.9 kb | Rec 0.09 θ = 0.4 | Rec 1.06 α = 0.05 | P = 0.07 | P = 0.40 |
| FGFR1 (3) | rs10958704 | 4 kb | Rec 0.01 θ = 0.4 | Rec 1.04 α = 0.05 | P = 0.07 | P = 0.36 |
| FGFR1 (4) | rs881301 | N/A | No positives | N/A | P = 0.07 | P = 0.15 |
Mb, megabases; Dom, dominant; Rec, recessive; NPL, non-parametric linkage; FBAT, family based association test; N/A, not applicable; θ, recombination fraction; α, proportion of linked families.
Discussion
In our genome-scan of 220 extended multiplex Filipino families, a 20 cM region on 8p11-23 had evidence for linkage with NS CLP. This region has not previously been identified in any of the five published genome-scans for NS CLP; therefore we targeted that region for fine-mapping studies in 271 extended multiplex Filipino families (the 220 genome-scan kindreds, plus an additional 54 extended kindreds). The association approach can be more powerful than linkage if there is a large degree of heterogeneity. Thirteen CLP candidate gene loci within the 8p11-23 region were assessed yielding positive results for five genes. As this study was exploratory in nature, we did not do any adjustment for multiple comparisons, instead using the usual alpha level of 0.05 as potentially statistically significant. Of these five genes with positive, only FGFR1 has literature supporting its involvement in craniofacial development.
Fibroblast growth factor receptor 1 (FGFR1) is a transmembrane receptor tyrosine kinase involved in signal transduction. Signaling from FGFR1 follows homodimerization, which requires the binding of two FGF molecules to two FGFR molecules in the presence of heparan sulfate proteoglycans [Mohammadi et al., 2005]. In mice, FGFR1 is widely expressed in the branchial arches during development. Mice homozygous for the hypomorphic allele Fgfr1n7/n7 have clefts of the palate in 80% of cases, which appears to be secondary to a defect in neural crest migration into the second branchial arch during development [Trokovic et al., 2003].
A role for FGFR1 in CLP has also been suggested by the report that loss-of-function mutations in FGFR1 cause Kallmann syndrome with clefting as part of the phenotype [Dode et al., 2003; Kim et al., 2005]. In total, 25 novel nonsense, missense, splice-site, frameshift, deletion, and translocation mutations in FGFR1 have been reported in Kallmann syndrome cases [Vermeulen et al., 2002; Dode et al., 2003; Sato et al., 2004; Albuisson et al., 2005; Kim et al., 2005; Pitteloud et al., 2005; Sato et al., 2005]. Seventeen familial and sporadic Kallmann cases have been reported with CLP or CPO, of those 9 have identified mutations in FGFR1 [Lieblich et al., 1982; White et al., 1983; Tompach and Zeitler, 1995; Molsted et al., 1997; Dode et al., 2003; Albuisson et al., 2005; Jonklaas, 2005; Kim et al., 2005; Zenaty et al., 2006]. Interestingly, these families included a mixed clefting pattern, with some family members having cleft palate only (CPO) while other family members have CLP. These two types of clefts are not commonly seen together in families except in cases of MSX1 mutations [van den Boogaard et al., 2000] and Van der Woude Syndrome IRF6 mutations [Kondo et al., 2002]. Kallmann Syndrome is autosomal dominant when found with FGFR1 mutations; the phenotype includes anosmia and hypogonadotrophic infertility as primary features, with CLP or CPO as part of the phenotype in about 5% of cases (M McCray and Jeff Murray, unpublished data).
In humans, a gain of function mutation in FGFR1, P252R, results in the craniosynostoses syndrome features, which can also be caused by mutations at the homologous position in FGFR2 (P253S = Pfieffer Syndrome; P253R = Apert Syndrome) [Slaney et al., 1996; Wilkie et al., 2002]. Three mutations in FGFR2 (S252W, S252F, and P253R) account for most cases of Apert syndrome. Overall, about 44% of Apert syndrome cases have cleft palate, cases whose phenotype includes a cleft are more commonly associated with the S252W mutation (59%) rather than the P253R mutation (17%) [Slaney et al., 1996].
The genome scan data presented in this article were included in the summary results reported in the meta-analysis of 13 total genome scans for CLP [Marazita et al., 2004]. We report here the detailed analysis of one of these populations from the Philippines. This provides insights into a single, mostly homogeneous South Asian population with a high birth prevalence of clefting [Murray et al., 1997] and where the effects of specific genes and/or alleles are likely to be enriched. Notably, 8p21 had a nominally significant P-value of 0.027 in the meta-analysis. The meta-analysis identified multiple genome-wide significant regions that we are now pursuing with additional fine-mapping studies [Marazita et al., 2004].
In summary, we have identified a novel region (8p11-23) that is likely to be involved in nonsyndromic clefting; a search for disease-causing mutations in one of the genes within this region (FGFR1) is underway. Recent work has identified a significant role for the FGF and FGFR genes in NS CLP, including FGFR1 [Riley et al., in press]. If the search for mutations within FGFR1 is successful, then phenotypic expression due to FGFR1 may be similar to MSX1 and IRF6, genes in which multiple mutations and/or polymorphic variants lead to variable phenotypic expression, including both clinically defined syndromes and isolated cleft phenotypes. For example, Witkop syndrome [Jumlongras et al., 2001], hypodontia [van den Boogaard et al., 2000], and isolated CLP [Jezewski et al., 2003; Vieira et al., 2005] are all found with mutations in MSX1. Van der Woude and Popliteal Pterygium syndromes are both found with mutations in IRF6 [Kondo et al., 2002], while NS CLP is associated with polymorphic variants in the same gene [Zucchero et al., 2004]. Previous work has shown that genes that play a role in syndromic clefts may also have presumed hypomorphic variants that play a role in isolated clefting, therefore genetic variation in the FGFR1 gene may contribute to syndromes as well as NS CLP. Finding mutations, despite the lack of complete penetrance for these mutations [Suzuki et al., 2004], can substantially alter recurrence risks and genetic counseling. These results demonstrate the power of a large study sample to further clarify the complex inheritance and etiology of NS CLP.
Acknowledgments
We thank all of the families who participated, without whom this study would not have been possible and the many Operation Smile and HOPE volunteers. Special thanks to Bill Magee, Edith Villanueva, Buena Nepomucena, and Henrietta Gamboa. We thank Sarah O'Brien for sample organization and management and Katie Graf and Erin O'Brien for assisting in genotyping. John Allaman provided computer support. Maggie McCray did a literature search looking for the contributions of Kallmann to clefts.
Grant sponsor: NIH; Grant numbers: R37-DE08559, P50-DE016215, 5T32GM008629; Grant sponsor: Center for Inherited Disease Research (CIDR); Grant number: N01-HG-65403.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Albuisson J, Pecheux C, Carel JC, Lacombe D, Leheup B, Lapuzina P, Bouchard P, Legius E, Matthijs G, Wasniewska M, Delpech M, Young J, Hardelin JP, Dode C. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2) Hum Mutat. 2005;25:98–99. doi: 10.1002/humu.9298. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Cortez A, Stal S, Mulliken JB, Finnell RH, Hecht JT. Variation in IRF6 contributes to nonsyndromic cleft lip and palate. Am J Med Genet Part A. 2005;137A:259–262. doi: 10.1002/ajmg.a.30887. [DOI] [PubMed] [Google Scholar]
- Christensen K, Juel K, Herskind AM, Murray JC. Long term follow up study of survival associated with cleft lip and palate at birth. BMJ. 2004;328:1405. doi: 10.1136/bmj.38106.559120.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham RW, Jr, Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Ghassibe M, Bayet B, Revencu N, Verellen-Dumoulin C, Gillerot Y, Vanwijck R, Vikkula M. Interferon regulatory factor-6: A gene predisposing to isolated cleft lip with or without cleft palate in the Belgian population. Eur J Hum Genet. 2005;13:1239–1242. doi: 10.1038/sj.ejhg.5201486. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. The family based association test method: Strategies for studying general genotype–phenotype associations. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Jezewski PA, Vieira AR, Nishimura C, Ludwig B, Johnson M, O'Brien SE, Daack-Hirsch S, Schultz RE, Weber A, Nepomucena B, Romitti PA, Christensen K, Orioli IM, Castilla EE, Machida J, Natsume N, Murray JC. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40:399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonklaas J. Atypical presentation of a patient with both kallmann syndrome and a craniopharyngioma: Case report and literature review. Endocr Pract. 2005;11:30–36. doi: 10.4158/EP.11.1.30. [DOI] [PubMed] [Google Scholar]
- Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, Seidman CE, Felbor U, Maas R, Seidman JG, Olsen BR. A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet. 2001;69:67–74. doi: 10.1086/321271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Herrick SR, Lemyre E, Kishikawa S, Salisz JA, Seminara S, MacDonald ME, Bruns GA, Morton CC, Quade BJ, Gusella JF. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FG FR1. J Med Genet. 2005;42:666–672. doi: 10.1136/jmg.2004.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19:S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lange K. Mathematical and Statistical Methods for Genetic Analysis. New York, NY: Springer; 2002. Descent graph methods; pp. 167–198. [Google Scholar]
- Lidral AC, Murray JC. Genetic approaches to identify disease genes for birth defects with cleft lip/palate as a model. Birth Defects Res A Clin Mol Teratol. 2004;70:893–901. doi: 10.1002/bdra.20096. [DOI] [PubMed] [Google Scholar]
- Lieblich JM, Rogol AD, White BJ, Rosen SW. Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann syndrome): Clinical and laboratory studies in 23 cases. Am J Med. 1982;73:506–519. doi: 10.1016/0002-9343(82)90329-1. [DOI] [PubMed] [Google Scholar]
- Marazita ML, Murray JC, Lidral AC, Arcos-Burgos M, Cooper ME, Goldstein T, Maher BS, Daack-Hirsch S, Schultz R, Mansilla MA, Field LL, Liu YE, Prescott N, Malcolm S, Winter R, Ray A, Moreno L, Valencia C, Neiswanger K, Wyszynski DF, Bailey-Wilson JE, Albacha-Hejazi H, Beaty TH, McIntosh I, Hetmanski JB, Tuncbilek G, Edwards M, Harkin L, Scott R, Roddick LG. Meta-analysis of 13 genome scans reveals multiple cleft lip/palate genes with novel loci on 9q21 and 2q 32-35. Am J Hum Genet. 2004;75:161–173. doi: 10.1086/422475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LE, Risch N. Mode of inheritance of nonsyndromic cleft lip with or without cleft palate: A reanalysis. Am J Hum Genet. 1992;51:323–332. [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Molsted K, Kjaer I, Giwercman A, Vesterhauge S, Skakkebaek NE. Craniofacial morphology in patients with Kallmann's syndrome with and without cleft lip and palate. Cleft Palate Craniofac J. 1997;34:417–424. doi: 10.1597/1545-1569_1997_034_0417_cmipwk_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Mossey PA, Little J. Epidemiology of oral clefts: An international perspective. In: Wyszynski DF, editor. Cleft Lip and Palate. New York: Oxford University Press; 2002. pp. 127–144. [Google Scholar]
- Murray JC. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- Murray JC, Daack-Hirsch S, Buetow KH, Munger R, Espina L, Paglinawan N, Villanueva E, Rary J, Magee K, Magee W. Clinical and epidemiologic studies of cleft lip and palate in the Philippines. Cleft Palate Craniofac J. 1997;34:7–10. doi: 10.1597/1545-1569_1997_034_0007_caesoc_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE. The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet. 1995;11:402–408. doi: 10.1038/ng1295-402. [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF., Jr Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005;90:1317–1322. doi: 10.1210/jc.2004-1361. [DOI] [PubMed] [Google Scholar]
- Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dodè C, Mohammadi M, Marazita ML, Murray JC. Impaired FGF signaling contributes to cleft lip and palate. PNAS. 2007 doi: 10.1073/pnas.0607956104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, Minowada S, Shimotsuka A, Shishiba Y, Yokozawa M, Yasuda T, Nagasaki K, Hasegawa D, Hasegawa Y, Tachibana K, Naiki Y, Horikawa R, Tanaka T, Ogata T. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab. 2004;89:1079–1088. doi: 10.1210/jc.2003-030476. [DOI] [PubMed] [Google Scholar]
- Sato N, Hasegawa T, Hori N, Fukami M, Yoshimura Y, Ogata T. Gonadotrophin therapy in Kallmann syndrome caused by heterozygous mutations of the gene for fibroblast growth factor receptor 1: Report of three families: Case report. Hum Reprod. 2005;20:2173–2178. doi: 10.1093/humrep/dei052. [DOI] [PubMed] [Google Scholar]
- Scapoli L, Palmieri A, Martinelli M, Pezzetti F, Carinci P, Tognon M, Carinci F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am J Hum Genet. 2005;76:180–183. doi: 10.1086/427344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliekelman P, Slatkin M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet. 2002;71:1369–1385. doi: 10.1086/344779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RE, Cooper ME, Daack-Hirsch S, Shi M, Nepomucena B, Graf KA, O'Brien EK, O'Brien SE, Marazita ML, Murray JC. Targeted scan of fifteen regions for nonsyndromic cleft lip and palate in Filipino families. Am J Med Genet Part A. 2004;125A:17–22. doi: 10.1002/ajmg.a.20424. [DOI] [PubMed] [Google Scholar]
- Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- Smith CA. Testing for heterogeneity of recombination fraction values in human genetics. Ann Hum Genet. 1963;27:175–182. doi: 10.1111/j.1469-1809.1963.tb00210.x. [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K. Descent graphs in pedigree analysis: Applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Lange K, O'Connell JR, Weeks DE. Haplotyping algorithms. In: Speed TP, Waterman MS, editors. Genetic Mapping and DNA Sequencing. New York, NY: Springer-Verlag; 1996. pp. 89–110. [Google Scholar]
- Srichomthong C, Siriwan P, Shotelersuk V. Significant association between IRF6 820G→A and non-syndromic cleft lip with or without cleft palate in the Thai population. J Med Genet. 2005;42:e46. doi: 10.1136/jmg.2005.032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Jezewski PA, Machida J, Watanabe Y, Shi M, Cooper ME, Viet le T, Nguyen TD, Hai H, Natsume N, Shimozato K, Marazita ML, Murray JC. In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med. 2004;6:117–125. doi: 10.1097/01.gim.0000127275.52925.05. [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J. Handbook of Human Genetic Linkage. Baltimore: Johns Hopkins University Press; 1994. [Google Scholar]
- Tompach PC, Zeitler DL. Kallmann syndrome with associated cleft lip and palate: Case report and review of the literature. J Oral Maxillofac Surg. 1995;53:85–87. doi: 10.1016/0278-2391(95)90511-1. [DOI] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Mai P, Partanen J. Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 2003;17:141–153. doi: 10.1101/gad.250703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24:342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- Vermeulen S, Messiaen L, Scheir P, De Bie S, Speleman F, De Paepe A. Kallmann syndrome in a patient with congenital spherocytosis and an interstitial 8p11.2 deletion. Am J Med Genet. 2002;108:315–318. doi: 10.1002/ajmg.10295. [DOI] [PubMed] [Google Scholar]
- Vieira AR, Avila JR, Daack-Hirsch S, Dragan E, Felix TM, Rahimov F, Harrington J, Schultz RR, Watanabe Y, Johnson M, Fang J, O'Brien SE, Orioli IM, Castilla EE, Fitzpatrick DR, Jiang R, Marazita ML, Murray JC. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 2005;1:e64. doi: 10.1371/journal.pgen.0010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BJ, Rogol AD, Brown KS, Lieblich JM, Rosen SW. The syndrome of anosmia with hypogonadotropic hypogonadism: A genetic study of 18 new families and a review. Am J Med Genet. 1983;15:417–435. doi: 10.1002/ajmg.1320150307. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Patey SJ, Kan SH, van den Ouweland AM, Hamel BC. FGFs, their receptors, and human limb malformations: Clinical and molecular correlations. Am J Med Genet. 2002;112:266–278. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]
- Zenaty D, Bretones P, Lambe C, Guemas I, David M, Leger J, de Roux N. Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1) Mol Cell Endocrinol. 2006;254-255:78–83. doi: 10.1016/j.mce.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, Natsume N, Yoshiura K, Vieira AR, Orioli IM, Castilla EE, Moreno L, Arcos-Burgos M, Lidral AC, Field LL, Liu YE, Ray A, Goldstein TH, Schultz RE, Shi M, Johnson MK, Kondo S, Schutte BC, Marazita ML, Murray JC. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]