

Abstract
CC-chemokine ligand 2 (CCL2) is the major chemoattractant protein that recruits monocytes to sites of inflammation and increased expression of CCL2 is associated with numerous inflammatory diseases including human immunodeficiency virus-associated dementia (HIV-D). The −2578 guanine polymorphism in the CCL2 promoter has been associated with increased expression of CCL2 as well as pathogenesis of HIV-D; however, the molecular mechanism of regulation is unknown. We propose a molecular model for −2578G regulated CCL2 expression in astrocytes, which are major producers of CCL2 in the brain. The −2578G polymorphism creates a consensus binding site for the transcriptional regulator Prep1, which along with binding partner Pbx2, preferentially binds the −2578G allele. CCL2 promoters harboring the G allele under unstimulated conditions exhibit a lower basal activity compared to the ancestral A allele. Upon IL-1β stimulation, Prep1/Pbx2 complexes maintain the ability to bind −2578G alleles, yet transcription levels from promoters that harbor the A allele or G allele are equally activated, suggesting that the −2578 region does not influence CCL2 transcription under pro-inflammatory conditions. Therefore, promoters that harbor the −2578G allele undergo a higher fold induction and by extension, individuals homozygous for −2578G would be expected to exhibit hyper-responsive CCL2 phenotypes during periods of inflammation.
Keywords: TALE, CCL2, SNP, Promoter, Prep1, Pbx2
INTRODUCTION
CC-chemokine ligand 2 (CCL2, formally known as monocyte chemoattractant protein 1, MCP-1), a member of the C-C chemokine family, is produced by macrophages, microglia, activated astrocytes and endothelial cells to recruit circulating monocytes to sites of inflammation1. Because of its central role in inflammatory responses, increased levels of CCL2 have been implicated in a variety of disease pathologies including arthersosclerosis, cancer metastisis, multiple sclerosis, Alzheimer's disease and HIV-dementia 2-7. In other settings, CCL2 appears to be beneficial as it protects against initial HIV infection, is neuroprotective against HIV Tat and N-methyl-D-aspartate (NMDA)-induced apoptosis during early HIV infection, and promotes healing following myocardial infarct 8-10. Genetic analysis of the distal CCL2 promoter revealed a polymorphism at −2578 (alternatively designated −251811) with functional consequences in the context of a luciferase reporter assay. Specifically, CCL2 distal promoters that harbor the −2578 G allele exhibit higher levels of activity than those harboring the A allele in IL-1β-stimulated cells 11. Other studies demonstrated that the guanine polymorphism increased the levels of CCL2 produced in cultured PBMCs, monocytes and hepatic stellate cells providing evidence that the polymorphism regulates CCL2 expression in the context of the endogenous promoter 11-16.
Almost five years ago, a provocative study associated the −2578 G allele with protection against initial HIV infection; however, once individuals were infected, they exhibited a more rapid progression to AIDS and a 4.5 fold increased chance of developing HIV-associated dementia compared to individuals carrying the ancestral −2578 A allele 9. Preliminary molecular analysis in this study suggested that the region encompassing the −2578 polymorphism shared homology with an Interferon Stimulated Response Element (ISRE) and that the transactivator Interferon Regulatory Factor (IRF) −1 bound the −2578 A allele with higher affinity than the −2578 G allele.
Presently, numerous reports have associated the −2578 G polymorphism with a wide variety of pathological sequelae such as progression of M. turberculosis infection, earlier age of onset of idiopathic immune-mediated posterior segment uveitis, asthma severity, premature kidney graft failure and HCV-related liver disease severity among others12-14; 17; 18. A number of studies have also correlated the −2578 G polymorphism with increased expression of CCL2 in vivo. For example, plasma levels of CCL2 in unaffected-individuals and patients with tuberculosis, Behcet's disease and Alzheimer's disease were significantly higher in individuals that harbor the −2578 G allele compared to those that harbor the −2578 A allele 3; 9; 13; 16; 19. Further, levels of CCL2 in the CSF of HIV infected individuals were significantly higher in individuals that harbored the G at −2578 compared to those which harbored the A, indicating that this polymorphism influences in vivo central nervous system (CNS) levels of CCL29. Finally, patients with systemic lupus erythematosus (SLE) that carried the −2578 G allele demonstrated an increase in interstitial macrophages in the kidney, which is indicative of increased trafficking of monocytes into tissue, thereby providing biological significance of the −2578 G allele to increased levels of CCL29.
Because the majority of studies have demonstrated that the −2578 G allele confers increased expression of CCL2 in cells that are stimulated with proinflammatory cytokines as compared to the −2578 A allele, we were puzzled by a potential regulatory mechanism involving decreased binding of a classical transactivator protein (IRF-1) and intrigued by the report of a specific complex able to bind the −2578 G allele in EMSA but not the A allele 12. Here we propose a molecular model by which the −2578 G polymorphism regulates CCL2 expression in astrocytes and identify 2 proteins that bind as a complex specifically to the −2578 G CCL2 allele. Neither protein is a member of the IRF family (in fact, no definitive binding of IRF-1 or -2 to either −2578 allele could be demonstrated in our studies); rather, one protein was identified as Prep1 (also known as Pknox1), a member of the three amino acid loop extension (TALE) family of proteins in the subfamily MEINOX (Meis/Pknox1), and the second protein was identified as Pbx2, a second TALE family member known to complex with Prep1. Both proteins are required to obtain binding of the complex to the −2578 G allele. With broader implications perhaps relevant to the wide variety of diseases impacted by the −2578 polymorphism, the identification of this site as a MEINOX consensus sequence suggests that the G allele has the potential to bind any of the MEINOX family members thereby conferring cell/tissue specific effects on expression of CCL2.
RESULTS
Promoters that Harbor the −2578 G Polymorphism Exhibit Lower Basal Transcription in Reporter Assays
Transcriptional activity of the distal promoter region of CCL2 (929 bp) has previously been studied using luciferase assays in the A172 astrocytoma cell line, which demonstrated elevated transcriptional levels for promoters that contained the −2578 G polymorphism upon IL-1β stimulation11. In the present studies, we used U87-MG cells to examine the molecular basis of the −2578 G phenotype in another relevant astrocytic cell line widely used to study CCL2 expression20-22 and since astrocytes are key producers of CCL2 in the brain, which promotes an influx of monocytes that correlates with HIV/SIV encephalitis and dementia2. We utilized the same 929-bp distal human CCL2 promoter region as the previous study inserted into a pGL4.11 firefly luciferase vector in conjunction with a pGL4.74 renilla luciferase vector in co-transfection experiments to control for transfection efficiency. Extracts prepared from transfected U87-MG cells that were either stimulated with IL-1β or left unstimulated for 3 hours were subjected to Promega's Dual-Luciferase Reporter Assay to quantitate both firefly and renilla luciferase activity. A consistent reduction in the basal level of transcription was observed in cells transfected with vectors that contain the −2578 G allele as compared to the −2578 A allele (Figure 1, p = 0.0012). However, transcriptional levels were equivalent after stimulation with IL-1β (Figure 1, p = 0.88), reflecting a greater fold induction for the −2578 G allele (Figure 1, p = 0.016). These data suggested two possibilities: 1) a transcription factor(s) binds preferentially to the −2578 A allele in unstimulated cells providing a higher level of constitutive transcriptional activation and/or 2) a transcription factor(s) with repressor activity binds preferentially to the −2578 G promoter in unstimulated cells.
Figure 1.
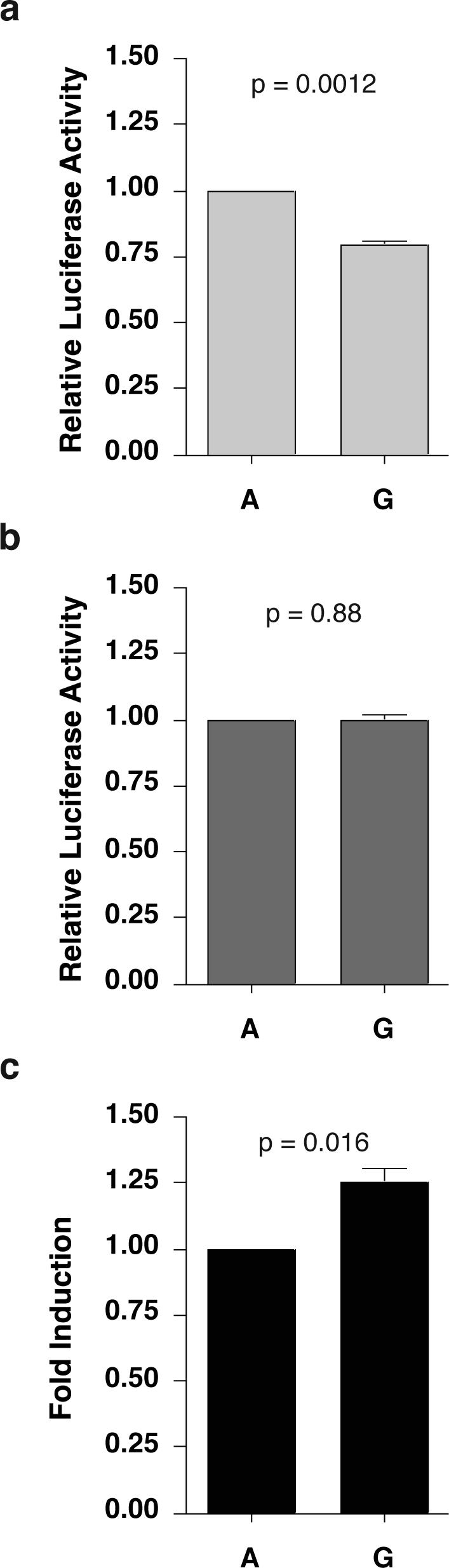
The −2578 G Polymorphism Influences Basal Activity of the CCL2 Distal Promoter. Distal promoters (929 bp) harboring either the −2578 G or A polymorphism were cloned directly upstream of the firefly luciferase gene in the pGL4.11 vector. Constructs were transfected into U87-MG cells (human astrocytic cell line) along with pGL4.74, a renilla luciferase construct that serves to control for transfection efficiency. [pGL4 vectors without inserts resulted in nominal background luciferase activity (not shown).] Transfected cells were either untreated or treated with human recombinant IL-1β (10 ng/mL) for 3 hours. Results are presented relative to the −2578 A construct. (A) Basal activity for the −2578 G construct is significantly lower than the −2578 A construct (p = 0.0012). (B) IL-1β stimulated activity is equal for constructs containing either the −2578 G or A allele (p = 0.88). (C) The fold induction is greater for the −2578 G construct (p = 0.016) as compared to the A construct. The results represent the mean of 4 individual experiments in which samples were processed in triplicate, controlled for transfection efficiency, and normalized to the −2578 A construct.
The −2578 G Region of the Human CCL2 Promoter Exhibits Enhanced Binding of a Specific Nuclear Complex that Does Not Contain IRF-1 or IRF-2
Previous studies suggested that the −2578 region is an ISRE and that the −2578 G polymorphism disrupts the binding of the transcription activator IRF-19. However, aligning the consensus binding elements for IRF-1 and IRF-2 with the canonical ISRE, the CCL2 ISRE, and the probes used in this study (in reverse-complement orientation, Figure 2a), revealed that an A or a G at position −2578 disrupts the ISRE consensus site where ISRE, IRF-1 and IRF-2 binding sites are 100% conserved. Although this alignment suggests that the −2578 region is a poor ISRE, we nonetheless included IRF-1 and IRF-2 (a known repressor of ISREs) in our analyses in an effort to reconcile previously reported studies. To examine the presence of IRF-1 and IRF-2 in U87-MG cells, we performed western blot analysis on nuclear lysates, which demonstrated that IRF-2 is constitutively expressed (Figure 2B) and IRF-1, while expressed at low levels in unstimulated cells, is induced to a great extent by IL-1β (Figure 2C). To determine whether IRF-1 and IRF-2 bind the −2578 region, labeled double-stranded DNA oligonucleotides containing either the −2578 A or G polymorphism were subjected to EMSA analysis in parallel with labeled ISRE consensus double stranded DNA oligonucleotides that provided a control for specific IRF-1 and IRF-2 binding. As expected, IRF-2 bound the ISRE consensus oligonucleotide producing a distinct band (Figure 2D, lane 1) that was shifted by α-IRF-2 antibody (Figure 2D, lane 2) and IRF-1 bound the ISRE consensus oligonucleotide producing one distinct band just below the IRF-2 band (Figure 2E, lane 1) that was shifted by α-IRF-1 antibody (Figure 2E, lane 2). Neither IRF-2 nor IRF-1 bound the −2578 A or G CCL2 oligonucleotide as there was no evidence of a supershift with antibody to either IRF-2 or IRF-1 (Figure 2D, lanes 5−8; Figure 2E, lanes 5− 8). Moreover, while 200× unlabeled ISRE completely competed the IRF-2 band complexed to labeled ISRE oligonucleotides in competition experiments (Figure 2F, lane 2) there was no competition with either the −2578 A or G unlabeled oligonucleotides (Figure 2F, lanes 3 and 4). Of note, 200× −2578 A unlabeled oligonucleotide appeared to compete for IRF-1 binding to the ISRE (Figure 2G, lane 3), albeit very weakly, suggesting that IRF-1 may have some affinity for −2578 A allele.
Figure 2.

The −2578 G allele specifically binds a protein complex that does not contain IRF-1 or IRF-2. (A) Alignment of IRF-1, IRF-2 and ISRE consensus sites with the CCL2 ISRE (reverse-complement orientation) and both the −2578 A and G oligonucleotides, designed and used in EMSA experiments. Italicized nucleotides represent flexibility in the consensus sites (R = G or A; Y = C or T; S = C or G; W = A or T; and N = A, G, C or T). Bold nucleotides represent nucleotides in the CCL2 ISRE that do not match the consensus binding sequences. Bold/Italicized nucleotides highlight the location of the −2578 polymorphism, where neither polymorphism matches the consensus sequence. Western blot analysis to identify IRF-2 (B) and IRF-1 (C) in nuclear extracts from unstimulated U87-MG cells and U87-MG cells stimulated with 10 ng/ml IL-1β (a known activator of IRF-1) for 2.5 hours. EMSAs were performed using the unstimulated (D) and IL-1β stimulated (E) U87-MG nuclear lysates with labeled ISRE consensus oligonucleotide, −2578 A and G oligonucleotides. Antibodies were added to binding reactions as indicated above the gel lanes; ss, antibody complex resulting from supershift analysis using α-IRF-2 and α-IRF-1; IRF-2, indicates band that contains IRF-2 as part off the complex; IRF-1, indicates band that contains IRF-1 as part of the complex; GSC, protein complex that is specific to the −2578 G oligonucleotide. Competition EMSAs were performed using 200× unlabeled ISRE oligonucleotide (lane 2), −2578 A oligonucleotide (lane 3), −2578 G oligonucleotide (lane 4) and scrambled G oligonucleotide (lane 5, to account for nonspecific affects) with either unstimulated (F) or IL-1β stimulated (G) U87-MG nuclear extracts. To determine the specificity of the intense band present in the −2578 G lanes of the ISRE EMSA (D and E, lanes 7 and 8), competition EMSAs were performed using 100x unlabeled oligonucleotide of −2578 A oligonucleotide, −2578 G oligonucleotide, ISRE consensus oligonucleotide and scrambled G oligonucleotide against labeled-2578 A oligonucleotide (H) and labeled-2578 G oligonucleotide (I) using unstimulated U87-MG nuclear lysates. The G specific band is the only region shown for each EMSA because Figure 2D and 2E (lanes 7 and 8) clearly show that lanes containing the G specific band are void of nonspecific bands. Data are representative of at least three independent experiments.
EMSA analysis of nuclear extracts from U87-MG cells clearly revealed a protein complex that binds specifically to the −2578 G oligonucleotide (Figure 2 D and E, lanes 7 and 8) consistent with a previous report12, although weak binding to the −2578 A oligonucleotide was observed (Figure 2 D and E, lanes 5 and 6). Notably, this band is not present in the ISRE consensus lanes and is not affected by the addition of either IRF-2 or IRF-1 antibody. Competition experiments using unstimulated U87-MG nuclear extracts incubated with the −2578 A labeled oligonucleotide again demonstrated weak binding of the complex compared to the -2578 G oligonucleotide (Figure 2H, lanes 2 and 1, respectively) which was competed away by 100× unlabeled −2578 A and G oligonucleotides (Figure 2H, lanes 3 and 4) but not unlabeled ISRE or scrambled G oligonucleotides (Figure 2H, lanes 5 and 6). Competition experiments using labeled −2578 G oligonucleotides demonstrated notable but incomplete competition with 100X unlabeled −2578 A oligonucleotides (Figure 2I, lane 3), complete competition with 100X unlabeled −2578 G oligonucleotides (Figure 2I, lane 4), and no competition with 100X unlabeled ISRE and scrambled −2578 G oligonucleotides (Figure 2I, lanes 5 and 6, respectively). Collectively, these observations suggest that a protein complex exists (not containing IRF-1 or −2) that has a high affinity for the −2578 G allele and binds only weakly to the −2578 A allele.
The −2578 G Polymorphism Creates a Consensus Binding Site (TGACAG) for the MEINOX (Meis Pknox) Proteins in the Three Amino Acid Loop Extension (TALE) Family
To identify factors that potentially bind the −2578 region, a search was conducted using MatInspector (Geomatrix), which identified the G allele but not the A allele as the Meis1 consensus binding site (Figure 3A). The Meis1 consensus site, TGACAG, is shared by the other members of the MEINOX subclass of TALE proteins and exhibits 100% sequence alignment to the −2578 G region. To investigate whether the MEINOX consensus site is required for the G specific band, we first compared the binding of U87-MG nuclear lysates to an oligonucleotide reported by Chang et al. that was used to define the Meis consensus site [herein referred to as the Chang oligonucleotide23] in parallel with the −2578 A and G oligonucleotides by EMSA. The intensity of the band for the −2578 A oligonucleotide was very weak, but the band increased in intensity in the presence of −2578 G oligonucleotides with the highest intensity observed in the presence of Chang oligonucleotides (Figure 3B). Although the Chang oligonucleotide and the −2578 G oligonucleotide share only limited alignment outside of their common TGACAG consensus sites (Figure 3A), the labeled Chang oligonucleotide migrated with the labeled G oligonucleotide, suggesting that the core sequence shared by both oligos is necessary for binding of this complex. To examine whether the −2578 G oligonucleotide and the Chang oligonucleotide bound the same protein complexes, competition experiments were performed with labeled −2578 G or Chang oligonucleotides and 100X of each unlabeled oligonucleotide. Competition of labeled −2578 G complexes with 100X unlabeled −2578 A oligonucleotide reduced the G specific band but not completely as shown above (Figure 3C, lane 3) whereas complete competition was observed with 100x unlabeled −2578 G and Chang oligonucleotides and no competition was observed with 100x unlabeled scrambled −2578 G oligonucleotide (Figure 3C, lanes 4, 5 and 6, respectively). Competition of labeled Chang complexes with 100x unlabeled −2578 A oligonucleotide only slightly decreased the intensity; competition with 100x unlabeled −2578 G oligonucleotide competed better than the −2578 A oligonucleotide but did not completely compete for the Chang complexes (Figure 3D, lanes 3 and 4), which occurred only with 100x unlabeled Chang oligonucleotide (Figure 3D, lanes 5). These results suggest that 1) the protein complex that binds the −2578 G oligonucleotide can bind the Chang oligonucleotide; and 2) the complex has higher affinity for the Chang oligonucleotide, possibly due to residues outside the core TGACAG. The Chang oligonucleotide has a Pbx consensus site (TGAT) adjacent to the MEINOX binding site (TGACAG), which is absent from the −2578 G oligonucleotide. Since many of the MEINOX proteins have to complex to Pbx proteins to enter the nucleus and regulate transcription, it seemed likely that the MEINOX protein that is bound to the −2578 G oligonucleotide and the Chang oligonucleotide may be complexed with its Pbx binding partner.
Figure 3.

The −2578 G polymorphism creates a MEINOX consensus binding site (TGACAG). (A) Alignment of the MEINOX consensus site with the −2578 G CCL2 oligonucleotide (reverse complement) and the Chang oligonucleotide (an oligonucleotide developed to study the Meis1 consensus site here named Chang oligonucleotide 23). MEINOX family members share the same binding site consensus sequence (TGACAG). Bold nucleotides represent nucleotides in the Chang oligonucleotide that do not match the −2578 G CCL2 oligonucleotide sequence, while a bold/italicized nucleotide represents the location of the −2578 polymorphism. (B) EMSA using nuclear extracts from U87-MG cells comparing nuclear complex binding of the −2578 A oligonucleotide (lane 1), the −2578 G oligonucleotide (lane 2) and the Chang oligonucleotide (lane 3). Competition EMSAs using 100X concentration of unlabeled −2578 A oligonucleotide (lane 3),−2578 G oligonucleotide (lane 4). Chang oligonucleotide (lane 5) and scrambled G oligonucleotide (lane 6) against labeled-2578 G oligonucleotide (C) and labeled Chang oligonucleotide (D). High-affinity DNA binding sites for Meis1 (E). The results of binding site selection for in vitro-produced monomeric Meis1 displayed as the percent frequency of each nucleotide at each position. The consensus binding sites are listed below the percentages. (Adapted from Chang et al. 1997) (F) Alignment of the MEINOX consensus site with the −2578 G CCL2 oligonucleotide (reverse complement) and the mutant −2578 G CCL2 oligonucleotides (including −2578 A CCL2 oligonucleotide). EMSAs using the oligonucleotides from the first mutant panel (all transitions mutations) (G) and the second mutant panel (different mutations at positions 3 and 5) (H) bound to complexes from unstimulated U87-MG extracts. The G specific band is the only region shown for each EMSA because Figure 2D and 2E (lanes 7 and 8) clearly show that lanes containing the G specific band are void of nonspecific bands. Data are representative of at least three independent experiments.
To further investigate whether the −2578 G site is a MEINOX consensus site, we mutated the MEINOX consensus site in the −2578 G oligonucleotide one nucleotide at a time and tested mutated oligonucleotides in EMSA experiments. For simplicity, pyrimidines were mutated to pyrimidines and purines were mutated to purines (Figure 3F). Previous analyses defining in vitro monomeric Meis1 high affinity consensus site from binding site selection experiments23 (Figure 3E) suggested that positions 3 and 5 could tolerate an A to G mutation at those sites but not a C; therefore, those sites were also mutated to C as well. EMSA analysis of the mutant panel consisting of all transition changes (Figure 3G) demonstrates that positions 2 (lane 3) and 4 (lane 5, −2578 polymorphic site) could not tolerate mutational changes and binding to the G specific banding pattern was lost, results that are strikingly consistent with previous studies that demonstrate position 2 does not tolerate nucleotide substitution for binding of Meis1 and Prep123; 24. Mutant oligonucleotides 1, 3, 5, 6 bound the protein complex but with reduced band intensity (lanes 2, 4, 6 and 7 respectively) while mutant oligonucleotides 7 and 8 bound the complex with equal intensity as the −2578 G nucleotide (lanes 8 and 9 respectively); this observation is again consistent with a previous study demonstrating the Meis1 high affinity consensus site 23. Moreover, the band observed with the −2578 G oligonucleotide is lost when positions 3 and 5 are mutated to C (Figure 3H, lanes 4 and 6 respectively), exhibiting a direct correlation with the Meis1 binding consensus shared by all MEINOX proteins.
The Complex Binding to the −2578 G Region Is Comprised of Prep-1 and Pbx2
Pbx proteins are regulated by the MEINOX subfamily (Meis1, Meis2, Meis3, Prep1, Prep2) of TALE family proteins 23-25. Western blot analysis was used to examine the expression of MEINOX proteins in unstimulated and IL-1β-stimulated U87-MG cells, which demonstrated that all 5 proteins were expressed (Figure 4 A and B; data not shown). Western blot analysis was also used to examine the expression of Pbx binding partners in unstimulated and IL-1β-stimulated U87-MG cells, which demonstrated constitutive expression of Pbx2 (Figure 4C), but failed to detect Pbx1 (which could not be detected by RT-PCR using mRNA from the U87-MG cells; data not shown). Meis1, Meis2, Meis3, Prep1, Prep2 and Pbx2 expression in U87-MG cells was further confirmed by cloning and sequencing of RT-PCR amplification products from U87-MG mRNA (data not shown).
Figure 4.

Prep1 and Pbx2 complexes bind the −2578 G allele. Western blot analysis of nuclear extracts from unstimulated U87-MG cells and U87-MG cells stimulated with 10 ng/ml IL-1β for 2.5 hours for (A) Prep-1, (B) Pbx-2 (a known binding partner of Prep1) and (C) Meis-2; no protein expression could be visualized for Pbx1 (the other known binding partner for prep1, data not shown). (D) EMSA performed using the unstimulated U87-MG nuclear lysates with labeled −2578 A oligonucleotide (lane 1) or the −2578 G oligonucleotide (lanes 2−6). Antibodies were added to binding reactions as indicated above the gel lanes; ss, antibody complex resulting from supershift analysis using α-Pbx2. U87-MG cells were transfected with siRNA for Prep1, Pbx2 and Meis2 as well as with the Risk Free siGlo and Non-targeting controls. (E) Western blot analysis of whole cell lysates from transfected cells using antibody against Prep1, Pbx2, Meis2 and actin (loading control). (F) Nuclear lysates from U87-MG cells extracted 36 hours after siRNA tranfection were used in EMSA experiments with labeled-2578 G oligonucleotide. Labeled ISRE consensus oligonucleotide was added to each reaction to use the IRF-2 band for quantitation purposes. Figure 4F is representative of three independent experiments. (G) Prep1 (upper gel) and Pbx2 (bottom gel) were coimmunoprecipitated from unstimulated U87-MG nuclear lysates and detected by western blot. The G specific band is the only region shown in some EMSAs because Figure 2D and 2E (lanes 7 and 8) clearly show that lanes containing the G specific band are void of nonspecific bands. Data are representative of at least two independent experiments.
To determine whether any of these proteins are present in the complex that binds the −2578 G oligonucleotide, antibodies against each of the expressed TALE proteins in addition to an isotype control antibody was added to EMSA reactions (Figure 4D). Addition of Prep1 and Meis2 antibodies ablated the G specific band (lanes 3 and 5) whereas addition of Pbx2 antibody supershifted the complex and reduced the intensity of the remaining G specific band (Figure 4D, lane 5). Isotype control antibody as well as antibody against Meis1, Meis3 and Prep2 did not affect the −2578 G oligonucleotide complex (Figure 4D, lane 6; data not shown). EMSAs using IL-1β stimulated U87-MG nuclear extracts and TALE antibodies were identical to the EMSAs using the unstimulated U87-MG cells, further supporting the binding of TALE proteins to the −2578 G oligonucleotide (Data not shown).
Since Prep1 and Meis2 have not been previously demonstrated to exist in a complex, we considered the possibility that one or both antibodies are cross-reactive, a common problem with MEINOX antibodies due to the high homology between all MEINOX family members. Western blots of in vitro-translated Prep1 and Meis2 confirmed that both antibodies recognized both proteins (data not shown). Therefore, to determine which one of these MEINOX proteins potentially bound the −2578 G oligonucleotide with Pbx2, we knocked down the expression of each protein specifically with siRNAs. Western blot analysis of whole cell extracts of U87-MG cells transfected with individual siRNAs, left unstimulated or stimulated with IL-1β, demonstrate significant reduction of Prep1, Pbx2 and Meis2 expression by 48−72 hours after transfection (Figure 4E, lanes 4, 5 and 6, respectively, Data not shown). Nuclear lysates prepared from unstimulated U87-MG cells transfected with the individual siRNAs for 36 hours were used in an EMSA assay. To quantify the effect on binding, labeled ISRE oligonucleotide was added to the −2578 G labeled oligonucleotide prior to addition to the EMSA reactions and the resulting IRF-2/ISRE band (see Figure 2D demonstrating ISRE specificity) was used to normalize the G-specific band in each reaction. The ratio of the G specific band to the IRF-2 band was 3.35 for no siRNA, 3.78 for siGLO risk free control, and 3.66 for the non-targeting siRNA control (Figure 4F, lanes 2, 3 and 4). The siRNA against Prep1 clearly abolishes binding of the protein complex to the −2578 G probe (Figure 4F, lane 5, G/IRF-2 ratio = 0.77) whereas siRNA against Pbx2 reduces the intensity of the band but does not completely abolish the binding of the complex (Figure 4F, lane 6, G/IRF-2 ratio = 2.21). However, siRNA against Meis2 did not reduce the G specific band (Figure 4F, lane 7, G/IRF2 ratio = 3.50) indicating that Meis2 is not present in the protein complex that binds the −2578 G oligonucleotide. The results were identical using siRNA transfected IL-1β stimulated U87-MG cells demonstrating the same binding specificity in stimulated cells (Data not shown).
The Prep1 and Pbx2 siRNA experiments demonstrated that both of these proteins bound the −2578 G oligonucleotide, but did not exclude the possibility that these proteins bind individually to the −2578 G oligonucleotide. However, the G specific band was reduced in intensity with siRNA against either Prep1 or Pbx2 suggesting that neither protein could bind the −2578 G oligonucleotide when not in complex with its partner. To directly examine the potential interaction between Prep1 and Pbx2, nuclear extracts from unstimulated U87-MG cells were subjected to reciprocal-immunoprecipitation and western analysis with antibody against Prep1 or Pbx2, which demonstrated that these proteins do exist in a complex (Figure 4G). In additional experiments neither Prep1 nor Pbx2 could bind the −2578 G oligonucleotide in an EMSA assay when transcribed/translated separately in vitro; however, when Prep1 and Pbx2 were transcribed/translated together, a complex bound the −2578 G oligonucleotide providing additional evidence that both Prep1 and Pbx2 have to be present for binding (Data not shown). Collectively, these observations support the conclusion that Prep1/Pbx2 complexes exist in astrocytes and bind specifically to the −2578 G allele.
DISCUSSION
The −2578 G polymorphism in the CCL2 distal promoter is an example of a functional promoter polymorphism that associates with disease pathogenesis, correlates with protein levels in vivo, and affects reporter gene expression3; 9; 11-16; 19(Figure 1). In the present study, we have investigated the −2578 polymorphism in an astrocytic cell line to model the influence of this site in the brain. Astrocytes are considered to be the major producers of CCL2 in the CNS, and the −2578 G polymorphism has been associated with the development of HIV dementia2; 9. Surprisingly, our data demonstrate that the G allele confers a repressive phenotype on promoters (compared to the A allele) in unstimulated astrocytes, which is overcome by stimulation with IL-1β such that expression from both alleles is equivalent. The subsequent effect on fold induction is consistent with previous studies11 causing the construct with the G allele to have a greater fold induction as compared to the A allele. Our studies also identified the −2578 G polymorphic region as a consensus site for the MEINOX family of transcription factors and demonstrated that the complex binding to the −2578 G region exhibits the same sequence specificity as the MEINOX consensus sequence. Further, we identified Prep1 as the only MEINOX family member that binds specifically to promoters that harbor the −2578 G allele in U87-MG cells and demonstrate that Prep1 is in a complex with its binding partner, Pbx2.
We could not confirm that the −2578 region is an ISRE or that IRF-1 or IRF-2 bind this region. IRF-1 and IRF-2 bind the consensus ISRE oligonucleotides, however these bands and shifts were not detected with the −2578 A and G oligonucleotide lanes. Furthermore, neither the −2578 A or G oligonucleotides competed notably with the consensus ISRE oligonucleotide. Although these results are negative, they challenge the findings of previous work9 and hence must be reported. To date, no study has revealed how this polymorphism regulates transcription of CCL2 or described an alternative transcription regulator that binds this site, although numerous studies continue to investigate the affect of this polymorphism on disease outcome. In the absence of a mechanistic model, reports that explore whether the −2578 G polymorphism affects disease are stymied by the lack of intervention strategies potentially able to alleviate disease symptoms or outcome. It follows then that when the mechanism by which the −2578 G polymorphism regulates disease is understood, the possibility exists for the design of effective therapeutics to treat affected −2578 G individuals.
In this study, we demonstrate that a complex binds specifically to the −2578 G allele yielding a single band in U87-MG cells and identified two complex proteins, Prep1 and Pbx2. Prep1 can modulate the activity of Pbx1a, Pbx1b and Pbx2 depending on their expression profile26 and it has been previously reported that Pbx1 will also bind Prep1 in DNA binding assays, producing a second distinct band26-29. Previous studies have demonstrated that Prep1 is expressed ubiquitously, including all regions examined in the human brain30. The present studies demonstrate that astrocytic cells (U87-MG) express only Pbx2, although both Pbx1 and Pbx2 are expressed in hematopoietic (K562), epithelial (A431) and glial (Hs683) cells23. Cell type specific regulation of Prep1/Pbx-1 or-2 complexes has been well investigated and raises the possibility that the effect of the −2578 G polymorphism may differ depending on which Pbx binding partners are expressed in the cell. Interestingly, Prep1/Pbx2 complexes have been reported to repress promoters31; 32 as well as activate promoters29; 33 containing the TGACAG motif in a cell type-dependent manner 32 whereas Prep1/Pbx1 complexes have only been reported to activate promoters28; 34.
This study has shown that Prep1/Pbx2 nuclear complexes can bind the −2578 G oligonucleotides under unstimulated and IL-1β-stimulated conditions where promoter activity from the G allele is repressed and equivalent respectively, as compared to the A allele. These data suggest that the −2578 site does not influence IL-1β-stimulated CCL2 expression in astrocytes. One reasonable interpretation is that during IL-1β stimulation other potent or dominant transcriptional activators of CCL2 gene expression such as NF-κB, simply overcome the repressive effect of the Prep1/Pbx2 complex. It is also conceivable that the binding of transactivators like NF-κB results in the displacement of Prep1/Pbx2 complexes in the context of an endogenous CCL2 promoter as opposed to oligonucleotides in EMSAs. Regardless, the binding of cell-type specific activator or repressor Prep/Pbx complexes to the −2578 site, which is not known to be a major regulatory region of the CCL2 promoter, coupled with cell-type specific activation or quiescence of the known CCL2 regulators35-41 could account for the conflicting reports regarding the −2578 CCL2 polymorphic alleles and CCL2 levels in different disease models. For example, although several studies have associated the G allele with increased expression of CCL2 in plasma3; 9; 13; 16; 19, other studies have reached the opposite conclusion42; 43 and some studies have failed to find a significant difference in CCL2 levels in individuals harboring either allele42. One study examining a link between this polymorphism and renal survival of Japanese patients with immunoglobulin A nephropathy associated the −2578 A allele with increased incidence of end-stage renal disease and revealed a trend in patients with immunoglobulin A nephropathy to have higher serum CCL2 levels when homozygous for the −2578 A, with a slightly lower level of serum CCL2 for heterozygotes and the lowest levels in patients homozygous for the −2578 G42. A similar finding was described in a study investigating the effect of the −2578 polymorphism on insulin resistance in which the −2578 G allele was associated with decreased plasma CCL2 levels and a decreased prevalence of insulin resistance43. These observations may well reflect the expression of specific Prep/Pbx proteins in different cells/tissues and the activation state of the CCL2 producing cells associated with each disease.
The repressive CCL2 phenotype observed in unstimulated astrocytes described above may well increase our understanding of the association between the −2578 allele and HIV dementia. As discussed, astrocytes are the major producers of CCL2 in the brain and increased levels of CCL2 in the CSF lead to the recruitment of monocytes to the brain, which have been a strong correlate of HIV CNS disease44-48. Under homeostatic conditions, astrocytes from individuals expressing the G allele would be expected to produce lower levels of CCL2 compared to their A allele counterparts based on the studies presented herein. During inflammatory conditions in the brain, a greater fold induction of CCL2 expression would result in astrocytes harboring the −2578 G allele, perhaps reflected in the CSF of HIV positive −2578 G individuals as reported49, and create a steeper increase of CCL2 from basal levels compared to their A allele counterparts, favoring a greater influx of monocytes. We have previously demonstrated that increased levels of CSF CCL2 protein alone, in pigtailed macaques infected with SIV, predict the development of moderate/severe CNS disease demonstrating that it is the level of CCL2 in the CNS that drives monocyte recruitment in this animal model of HIV dementia50. Because CCL2 itself is a pro-inflammatory chemokine, it follows that the hyper-responsiveness of the promoters that contain the −2578 G become amplified in the self-perpetuating inflammatory cycle, resulting in further increases in CCL2 as well as other pro-inflammatory mediators.
MATERIALS AND METHODS
Materials
Antibodies to IRF-1 (sc-497 X), IRF-2 (sc-498 X), Prep1 (sc-6245 X), Pbx1 (sc-889 X), Pbx2 (sc-890 X) and TFIIB (sc-274 X) were obtained from SantaCruz Biotechnologies (Santa Cruz, California). The antibodies for Meis2 (H00004212-M01) and Pbx2 (H00005089-M01) were obtained from Novus Biologicals, Inc. (Littleton, CO). The mouse monoclonal antibody for Prep1 (05−766, referred to as Meis4) was obtained from Upstate (Lake Placid, NY). The antibody for actin (A1978) was obtained from Sigma (St. Louis, MO). The pDONR221, pcDNA3.1/V5 and pEXP1-Dest vectors, the Gateway Vector conversion kit, antibodies to V5 and Xpress epitopes, Lipofectamine2000, Minimal Essential media with Earls salts and siRNA to Prep1 (PKNOX1HSS108055) were obtained from Invitrogen Corporation (Carlsbad, CA). The pGL4.11 and pGL4.74 vectors, as well as the Dual Luciferase Assay system was obtained from Promega Corporation (Madison, WI). ON-TARGETplus siCONTROL non-Targeting pool (D-001810−10), siGlo® Risc-free (D-001600−01), ON-TARGETplus Human PBX2 SMARTpool (L-011746−00), and ON-TARGETplus Human MEIS2 SMARTpool (L-011330−00) siRNAs were obtained from Dharmacon, Inc. (Lafayette, CO). Recombinant Human IL-1β (201-LB) was obtained from R & D Systems (Minneapolis, MN).
DNA Extraction
To clone CCL2 distal promoter regions, DNA was extracted from pelleted U937 and 293T cells (rationale below) using the Qiagen Dneasy tissue kit (Qiagen Inc., Valencia, CA) according to the manufacturer's instructions.
Polymerase Chain Reaction and Plasmid Production
The 929 base pair CCL2 promoter region was amplified by PCR from genomic DNA in 25-μL reactions with Epicentre's Fail-Safe PCR system (Epicentre Biotechnologies, Madison, WI) using the forward primer 5’-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC CGA GAT GTT CCC AGC ACA G-3’ and the reverse primer (DR) 5’-GGG GAC CAC TTT GTA CAA GAA AGC TGG GCT GCT TTG CTT GTG CCT CTT-3’ with the following thermal profile: 35 cycles of 94°C (30-seconds), 55°C (30-seconds) and 72°C (1-minute and 30-seconds) with an initial denaturation step of 94°C (5 min)11. Prep-1 and Pbx2 were amplified from cDNA, created from U87-MG RNA, in 25-μL reactions with Platinum Taq-High Fidelity (Invitrogen) using the forward primer (Prep1) 5’-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTG ATG ATG GCT ACA CAG ACA TT-3’ and reverse primer (Prep1) 5’-GGG GAC CAC TTT GTA CAA GAA AGC TGG GCT ACT GCA GGG AGT CAC TGT-3’, and the forward primer (Pbx2) 5’-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTG ATG GAC GAA CGG CTA CTG GG-3’ and the reverse primer (Pbx2) 5’-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC AGT TGG AGG TAT CAG AGT-3’ with a change in the extension time (72°C for 2-minute and 30-seconds) of the thermal profile above. Bold nucleotides represent the target specific primer sequence, while plain text represent additional nucleotides needed to clone using the BP reaction in the Gateway system. For the distal CCL2 region, two separate PCR reactions were completed, one with DNA from U937 cells (heterozygous at the −2578 site, but contained a SNP at −2408 G to C on the −2578 A allele) to clone the −2578 G allele and one PCR reaction with DNA from 293T cells (homozygous −2578 A) to clone the −2578 A allele. All PCR reactions were resolved on a 1.2% agarose gel, and bands were gel purified (Invitrogen) and subjected to the gateway BP clonase reaction, which transferred the distal promoter regions, Prep1 and Pbx2 coding regions into the pDONR221 vector. The pGL4.11 vector was converted to a Gateway vector using the cassette B and following the protocol from the gateway vector conversion kit. The distal promoter regions were transferred from the pDONR221 vectors to the converted pGL4.11 vectors through the LR clonase reaction. Distal promoter pGL4.11 vectors were sequenced to confirm that the only difference between the two vectors was the −2578 A and G site. Prep1 and Pbx2 inserts were transferred from the pDONR221 into the pcDNA3.1/V5 and pEXP1-Dest vectors through the LR clonase reaction.
Cell Culture and Reagents
U87-MG cells (HTB-14, American Type Culture Collection (Manassas, VA) were maintained in MEM with glutamine supplemented with 10% FBS, 0.1mM nonessential amino acids (MEM NEAA), and 1 mM sodium pyruvate. U87-MG cells are homozygous for the A allele at the −2578 position.
Luciferase Assays
Day 1: One T-75 flask >90% confluent of U87-MG cells was split into 5 6-well plates containing antibiotic free media. Day 2: The media was changed to MEM (serum free, antibiotic free) and 1440 ng of pGL4.11 (no insert, −2578 A or −2578 G) and 480 ng of pGL4.74 (Promega), to control for transfection efficiency, were transfected with Lipofectamine 2000. After 3 hours of incubation at 37°C, the media was decanted and replaced with 2% serum, antibiotic free media. Day 3: 24 hours after transfection, cells were left unstimulated or stimulated with 10 ng/mL IL-1β for 3 hours. After stimulation, cells were washed twice with PBS and lysed with passive lysis buffer (Dual luciferase assay system; Promega). Plates were scraped and the lysates were placed into 1.5 mL microcentrifuge tubes and sonicated for 20 seconds at 95% amplitude in a cup horn. Twenty μL of lysate was subjected to the Dual luciferase assay using the Fluoroskan Ascent FL.
Firefly luciferase activity and renilla luciferase activity were measured for each sample. The firefly luciferase activity was normalized for transfection efficiency to the renilla luciferase values and the mean relative luciferase activity was calculated from triplicate samples in each experiment. The mean relative luciferase activity for −2578 G was normalized to the mean relative luciferase activity for −2578 A in each basal and IL-1β-stimulated experiment. Fold stimulation was calculated by dividing the mean relative luciferase activity for the A or G constructs in IL-1β-stimulated samples by the mean relative luciferase activity for the A or G constructs in basal samples, respectively. Fold stimulation for the −2578 A and −2578 G constructs were normalized to the fold stimulation for the −2578 A construct in each experiment. The normalized values of the −2578 G constructs were compared to the normalized values of the −2578 A constructs. A one-sample T-test was performed for each condition, which tested if the mean of the normalized −2578 G constructs is significantly different from −2578 A (1). All p-values presented are two-tailed.
siRNA Transfection
siRNAs (50nM; none, Risc-free siGlo, Non-targeting siRNA pool, PKNOX1 (Prep1), PBX2 pool and MEIS2 pool) were transfected into T-75 flasks with U87-MG cells (60% confluent) using Lipofectamine 2000. After 4 hours incubation at 37°C, media was changed to fresh MEM 10% FBS with no antibiotics. Nuclear lysates were taken from transfected U87-MG cells at 36 hours after transfection using a nuclear extraction kit and protocol (Marligen Biosciences, Inc., Ijamsville, MD). Whole cell lysates were extracted using RIPA (1% NP40, 0.5% Sodium Desoxycholate, 0.1% SDS, 50mM NaF, 1x protease inhibitor cocktail (Calbiochem, Darmstadt, Germany) in 1x Phosphate buffered saline) buffer with sonication.
Western Blot Analysis
U87-MG nuclear lysates (30 μg) prepared from U87-MG cells that were stimulated or not with IL-1β for 2.5 hours, or whole cell extracts from siRNA treated U87-MG cells (50 μg) were resolved on a 10% Tris-Cl Criterion gel (Bio-Rad Laboratories, Life Science Research, Hercules, CA) and transferred to a nitrocellulose membrane with the criterion wet transfer apparatus. Membranes were washed 5 minutes in TBST, blocked for 20 minutes with 5% milk, and rocked at 4°C overnight with α-Prep1 (1:2000), α -Pbx2 (1:2000), α-Meis2 (1:1000), α-IRF1 (1:2000), α-IRF-2 (1:2000), or α-TFIIB (1:2000) or α-Actin (1:5000). Secondary antibodies were added at 1:5000 and blots were developed with SuperSignal Westpico chemiluminescence substrate or with the more sensitive SuperSignal Westdura extended duration substrate (Pierce Biotechnology, Rockford, IL).
EMSA Analysis
Nuclear extracts were prepared from U87-MG cells (treated or not with IL-1β for 2.5 hr) using a nuclear extraction kit and protocol (Marligen Biosciences, Inc). DNA binding reactions were performed on ice for 30 minutes in a 15 μL reaction volume containing 2 μg of poly[d(I · C)], 75 mM NaCl, 1 mM EDTA, 10 mM Tris-Hcl (pH 7.5), 6% glycerol, and 2 μg of bovine serum albumin (BSA) and subjected to EMSA using 5 % Tris-borate-EDTA (TBE) polyacrylamide gels (Bio-Rad) in 0.25x TBE buffer. DNA probes (50,000 cpm/ binding reaction) were labeled, forward and reverse strand independently, using T4 Polynucleotide kinase and γ-32P-ATP. Labeling reactions for the forward and reverse oligonucleotides were then added together, immediately boiled for 5 minutes, left for 1 hour to equilibrate to room temperature and then passed through the G25 spin columns to separate oligonucleotides from unused γ-32P-ATP. The oligonucleotides for the siRNA EMSA were diluted together with 50,000 cpm of each double stranded oligonucleotide (−2578 A or G with ISRE consensus) per μL; one μL was used in each binding reacting (100,000 cpm/binding reaction). The sequence of the relevant DNA probes are indicated in the figures: ISRE consensus, Figure 2A; −2578 A oligonucleotide, Figure 2A; −2578 G oligonucleotide, Figure 2A, 3A; Chang oligonucleotide, Figure 3A; and the mutant G oligonucleotide panel located in figure 4F. The sequence for the scrambled G oligonucleotide used in competition experiments is 5’ - ATT GCA CAG ACC TCT ACT AGA - 3’ Antibodies and competition oligonucleotides were added before the labeled oligonucleotides and the EMSA reactions were allowed to equilibrate on ice for 15 minutes before the addition of labeled oligonucleotides.
Coimmunoprecipitation
U87-MG nuclear extracts (100 μg) were added to a 1.5 mL tube and the volume was increased to 500 μL with IP buffer (10 mM Tris HCl pH 7.4, 5 mM EDTA pH 8.0, 150 mM NaCl, 0.5% NP40). Prep1 or Pbx2 antibody (3 μg) was added to the samples and rotated at 4°C overnight. Protein G Dynabeads (100 μL) were added to the samples the next day and rotated at 4°C for 2 hours. Beads were washed three times with ice cold TBST for 5 minutes. The beads were boiled in 40 μL western sample buffer, resolved on a 10% Tris-Cl Criterion gel and subjected to western blot analysis.
In Vitro Transcription and Translation
In vitro Luciferase (positive control), Prep1 and Pbx2 was produced using the TNT T7 Coupled Reticulocyte Lysate System (Promega Corporation) according to the manufacturer's instructions.
ACKNOWLEDGMENTS
The authors thank the entire Retrovirus Laboratory at Johns Hopkins University School of Medicine for useful discussions, especially to Susan Follstaedt for her expertise with western blots, and Justyna Dudaronek and Kenneth Witwer for their expertise with siRNAs.
Grant support: This research was supported by NIH grants to J.E.C. (NS050028, NS47984, and MH70306)
REFERENCES
- 1.Leonard EJ, Yoshimura T. Human monocyte chemoattractant protein-1 (MCP-1). Immunol Today. 1990;11(3):97–101. doi: 10.1016/0167-5699(90)90035-8. [DOI] [PubMed] [Google Scholar]
- 2.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95(6):3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fenoglio C, Galimberti D, Lovati C, Guidi I, Gatti A, Fogliarino S, et al. MCP-1 in Alzheimer's disease patients: A-2518G polymorphism and serum levels. Neurobiol Aging. 2004;25(9):1169–1173. doi: 10.1016/j.neurobiolaging.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006;25(4):611–619. doi: 10.1007/s10555-006-9027-x. [DOI] [PubMed] [Google Scholar]
- 5.Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corra B, et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer's disease. Neurobiol Aging. 2006;27(12):1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247(3):349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 7.Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol. 2003;15(1):23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- 8.Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85(5):1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99(21):13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 11.Rovin BH, Lu L, Saxena R. A novel polymorphism in the MCP-1 gene regulatory region that influences MCP-1 expression. Biochem Biophys Res Commun. 1999;259(2):344–348. doi: 10.1006/bbrc.1999.0796. [DOI] [PubMed] [Google Scholar]
- 12.Muhlbauer M, Bosserhoff AK, Hartmann A, Thasler WE, Weiss TS, Herfarth H, et al. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology. 2003;125(4):1085–1093. doi: 10.1016/s0016-5085(03)01213-7. [DOI] [PubMed] [Google Scholar]
- 13.Flores-Villanueva PO, Ruiz-Morales JA, Song CH, Flores LM, Jo EK, Montano M, et al. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J Exp Med. 2005;202(12):1649–1658. doi: 10.1084/jem.20050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kruger B, Schroppel B, Ashkan R, Marder B, Zulke C, Murphy B, et al. A Monocyte chemoattractant protein-1 (MCP-1) polymorphism and outcome after renal transplantation. J Am Soc Nephrol. 2002;13(10):2585–2589. doi: 10.1097/01.asn.0000031701.53792.54. [DOI] [PubMed] [Google Scholar]
- 15.Tucci M, Barnes EV, Sobel ES, Croker BP, Segal MS, Reeves WH, et al. Strong association of a functional polymorphism in the monocyte chemoattractant protein 1 promoter gene with lupus nephritis. Arthritis Rheum. 2004;50(6):1842–1849. doi: 10.1002/art.20266. [DOI] [PubMed] [Google Scholar]
- 16.Cho ML, Kim JY, Ko HJ, Kim YH, Kim WU, Cho CS, et al. The MCP-1 promoter −2518 polymorphism in Behcet's disease: correlation between allele types, MCP-1 production and clinical symptoms among Korean patients. Autoimmunity. 2004;37(1):77–80. doi: 10.1080/08916930310001609446. [DOI] [PubMed] [Google Scholar]
- 17.Szalai C, Kozma GT, Nagy A, Bojszko A, Krikovszky D, Szabo T, et al. Polymorphism in the gene regulatory region of MCP-1 is associated with asthma susceptibility and severity. J Allergy Clin Immunol. 2001;108(3):375–381. doi: 10.1067/mai.2001.117930. [DOI] [PubMed] [Google Scholar]
- 18.Ahad MA, Missotten T, Abdallah A, Lympany PA, Lightman S. Polymorphisms of chemokine and chemokine receptor genes in idiopathic immune-mediated posterior segment uveitis. Mol Vis. 2007;13:388–396. [PMC free article] [PubMed] [Google Scholar]
- 19.Tabara Y, Kohara K, Yamamoto Y, Igase M, Nakura J, Kondo I, et al. Polymorphism of the monocyte chemoattractant protein (MCP-1) gene is associated with the plasma level of MCP-1 but not with carotid intima-media thickness. Hypertens Res. 2003;26(9):677–683. doi: 10.1291/hypres.26.677. [DOI] [PubMed] [Google Scholar]
- 20.Abraham S, Sawaya BE, Safak M, Batuman O, Khalili K, Amini S. Regulation of MCP-1 gene transcription by Smads and HIV-1 Tat in human glial cells. Virology. 2003;309(2):196–202. doi: 10.1016/s0042-6822(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 21.Abraham S, Sweet T, Sawaya BE, Rappaport J, Khalili K, Amini S. Cooperative interaction of C/EBP beta and Tat modulates MCP-1 gene transcription in astrocytes. J Neuroimmunol. 2005;160(1−2):219–227. doi: 10.1016/j.jneuroim.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Lim SP, Garzino-Demo A. The human immunodeficiency virus type 1 Tat protein up-regulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of SP-1, AP-1, and NF-kappaB consensus sites. J Virol. 2000;74(4):1632–1640. doi: 10.1128/jvi.74.4.1632-1640.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang CP, Jacobs Y, Nakamura T, Jenkins NA, Copeland NG, Cleary ML. Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol Cell Biol. 1997;17(10):5679–5687. doi: 10.1128/mcb.17.10.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berthelsen J, Zappavigna V, Mavilio F, Blasi F. Prep1, a novel functional partner of Pbx proteins. Embo J. 1998;17(5):1423–1433. doi: 10.1093/emboj/17.5.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fognani C, Kilstrup-Nielsen C, Berthelsen J, Ferretti E, Zappavigna V, Blasi F. Characterization of PREP2, a paralog of PREP1, which defines a novel sub-family of the MEINOX TALE homeodomain transcription factors. Nucleic Acids Res. 2002;30(9):2043–2051. doi: 10.1093/nar/30.9.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferretti E, Schulz H, Talarico D, Blasi F, Berthelsen J. The PBX-regulating protein PREP1 is present in different PBX-complexed forms in mouse. Mech Dev. 1999;83(1−2):53–64. doi: 10.1016/s0925-4773(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 27.Berthelsen J, Viggiano L, Schulz H, Ferretti E, Consalez GG, Rocchi M, et al. PKNOX1, a gene encoding PREP1, a new regulator of Pbx activity, maps on human chromosome 21q22.3 and murine chromosome 17B/C. Genomics. 1998;47(2):323–324. doi: 10.1006/geno.1997.5086. [DOI] [PubMed] [Google Scholar]
- 28.Berthelsen J, Zappavigna V, Ferretti E, Mavilio F, Blasi F. The novel homeoprotein Prep1 modulates Pbx-Hox protein cooperativity. Embo J. 1998;17(5):1434–1445. doi: 10.1093/emboj/17.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goudet G, Delhalle S, Biemar F, Martial JA, Peers B. Functional and cooperative interactions between the homeodomain PDX1, Pbx, and Prep1 factors on the somatostatin promoter. J Biol Chem. 1999;274(7):4067–4073. doi: 10.1074/jbc.274.7.4067. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Rossier C, Nakamura Y, Lynn A, Chakravarti A, Antonarakis SE. Cloning of a novel homeobox-containing gene, PKNOX1, and mapping to human chromosome 21q22.3. Genomics. 1997;41(2):193–200. doi: 10.1006/geno.1997.4632. [DOI] [PubMed] [Google Scholar]
- 31.Gregory PA, Mackenzie PI. The homeodomain Pbx2-Prep1 complex modulates hepatocyte nuclear factor 1alpha-mediated activation of the UDP-glucuronosyltransferase 2B17 gene. Mol Pharmacol. 2002;62(1):154–161. doi: 10.1124/mol.62.1.154. [DOI] [PubMed] [Google Scholar]
- 32.Herzig S, Fuzesi L, Knepel W. Heterodimeric Pbx-Prep1 homeodomain protein binding to the glucagon gene restricting transcription in a cell type-dependent manner. J Biol Chem. 2000;275(36):27989–27999. doi: 10.1074/jbc.M003345200. [DOI] [PubMed] [Google Scholar]
- 33.Okada Y, Matsuura E, Nagai R, Sato T, Watanabe A, Morita I, et al. PREP1, MEIS1 homolog protein, regulates PF4 gene expression. Biochem Biophys Res Commun. 2003;305(1):155–159. doi: 10.1016/s0006-291x(03)00718-6. [DOI] [PubMed] [Google Scholar]
- 34.Chao SH, Walker JR, Chanda SK, Gray NS, Caldwell JS. Identification of homeodomain proteins, PBX1 and PREP1, involved in the transcription of murine leukemia virus. Mol Cell Biol. 2003;23(3):831–841. doi: 10.1128/MCB.23.3.831-841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rovin BH, Dickerson JA, Tan LC, Hebert CA. Activation of nuclear factor-kappa B correlates with MCP-1 expression by human mesangial cells. Kidney Int. 1995;48(4):1263–1271. doi: 10.1038/ki.1995.410. [DOI] [PubMed] [Google Scholar]
- 36.Zhou ZH, Chaturvedi P, Han YL, Aras S, Li YS, Kolattukudy PE, et al. IFN-gamma induction of the human monocyte chemoattractant protein (hMCP)-1 gene in astrocytoma cells: functional interaction between an IFN-gamma-activated site and a GC-rich element. J Immunol. 1998;160(8):3908–3916. [PubMed] [Google Scholar]
- 37.Zhou ZH, Han Y, Wei T, Aras S, Chaturvedi P, Tyler S, et al. Regulation of monocyte chemoattractant protein (MCP)-1 transcription by interferon-gamma (IFN-gamma) in human astrocytoma cells: postinduction refractory state of the gene, governed by its upstream elements. Faseb J. 2001;15(2):383–392. doi: 10.1096/fj.00-0373com. [DOI] [PubMed] [Google Scholar]
- 38.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3(1):69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 39.Teferedegne B, Green MR, Guo Z, Boss JM. Mechanism of action of a distal NF-kappaB-dependent enhancer. Mol Cell Biol. 2006;26(15):5759–5770. doi: 10.1128/MCB.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ueda A, Ishigatsubo Y, Okubo T, Yoshimura T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J Biol Chem. 1997;272(49):31092–31099. doi: 10.1074/jbc.272.49.31092. [DOI] [PubMed] [Google Scholar]
- 41.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153(5):2052–2063. [PubMed] [Google Scholar]
- 42.Mori H, Kaneko Y, Narita I, Goto S, Saito N, Kondo D, et al. Monocyte chemoattractant protein-1 A-2518G gene polymorphism and renal survival of Japanese patients with immunoglobulin A nephropathy. Clin Exp Nephrol. 2005;9(4):297–303. doi: 10.1007/s10157-005-0375-6. [DOI] [PubMed] [Google Scholar]
- 43.Simeoni E, Hoffmann MM, Winkelmann BR, Ruiz J, Fleury S, Boehm BO, et al. Association between the A-2518G polymorphism in the monocyte chemoattractant protein-1 gene and insulin resistance and Type 2 diabetes mellitus. Diabetologia. 2004;47(9):1574–1580. doi: 10.1007/s00125-004-1494-4. [DOI] [PubMed] [Google Scholar]
- 44.Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38(5):755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 46.Hou J, Major EO. Direct and indirect mechanisms of HIV-1 neuropathogenesis in the human central nervous system. Adv Exp Med Biol. 2001;493:29–34. doi: 10.1007/0-306-47611-8_3. [DOI] [PubMed] [Google Scholar]
- 47.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 48.Zink WE, Zheng J, Persidsky Y, Poluektova L, Gendelman HE. The neuropathogenesis of HIV-1 infection. FEMS Immunol Med Microbiol. 1999;26(3−4):233–241. doi: 10.1111/j.1574-695X.1999.tb01394.x. [DOI] [PubMed] [Google Scholar]
- 49.Letendre S, Marquie-Beck J, Singh KK, de Almeida S, Zimmerman J, Spector SA, et al. The monocyte chemotactic protein-1 −2578G allele is associated with elevated MCP-1 concentrations in cerebrospinal fluid. J Neuroimmunol. 2004;157(1−2):193–196. doi: 10.1016/j.jneuroim.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 50.Wright EK, Jr., Clements JE, Barber SA. Sequence variation in the CC-chemokine ligand 2 promoter of pigtailed macaques is not associated with the incidence or severity of neuropathology in a simian immunodeficiency virus model of human immunodeficiency virus central nervous system disease. J Neurovirol. 2006;12(6):411–419. doi: 10.1080/13550280601009538. [DOI] [PubMed] [Google Scholar]