

Abstract
The initial synapse in the olfactory system is from olfactory nerve (ON) terminals to postsynaptic targets in olfactory bulb glomeruli. Recent studies have disclosed multiple presynaptic factors that regulate this important linkage, but less is known about the contribution of postsynaptic intrinsic conductances to integration at these synapses. The present study demonstrates voltage-dependent amplification of EPSPs in external tufted (ET) cells in response to monosynaptic (ON) inputs. This amplification is mainly exerted by persistent Na+ conductance. Larger EPSPs, which bring the membrane potential to a relatively depolarized level, are further boosted by the low-voltage-activated Ca2+ conductance. In contrast, the hyperpolarization-activated nonselective cation conductance (Ih) attenuates EPSPs mainly by reducing EPSP duration; this also reduces temporal summation of multiple EPSPs. Regulation of EPSPs by these subthreshold, voltage-dependent conductances can enhance both the signal-to-noise ratio and the temporal summation of multiple synaptic inputs and thus help ET cells differentiate high- and low-frequency synaptic inputs. Ih can also transform inhibitory inputs to postsynaptic excitation. When the ET cell membrane potential is relatively depolarized, as during a burst of action potentials, IPSPs produce classic inhibition. However, near resting membrane potentials where Ih is engaged, IPSPs produce rebound bursts of action potentials. ET cells excite GABAergic PG cells. Thus, the transformation of inhibitory inputs to postsynaptic excitation in ET cells may enhance intraglomerular inhibition of mitral/tufted cells, the main output neurons in the olfactory bulb, and hence shape signaling to olfactory cortex.
Keywords: synaptic integration, persistent Na+ current, hyperpolarization-activated nonselective cation current, EPSP, IPSP, rebound depolarization
Introduction
The olfactory system transmits signals directly from peripheral olfactory receptor neurons (ORNs) to the main olfactory bulb (MOB) and then to olfactory cortex. Thus, the MOB is the initial site of integration for olfactory information. The glomerular layer of the MOB contains numerous discrete, spherical structures: glomeruli in which axonal terminals of ORNs synapse directly on mitral/tufted cells, the major output neurons of the bulb, and on local interneurons called juxtaglomerular (JG) cells. Each glomerulus receives input from ORNs that express the same odorant receptor. Recent studies have significantly advanced our understanding of the complexity of the glomerular synaptic network (Aungst et al., 2003; Hayar et al., 2004a,b, 2005; Murphy et al., 2005; Wachowiak and Shipley, 2006). JG cells form local circuits that make dendrodendritic synaptic connections with each other and with mitral-cell apical dendrites to regulate the transfer of sensory information from ORNs to output neurons, i.e., the glomerular input–output function.
External tufted (ET) cells, a subpopulation of JG cells, receive monosynaptic input from ORNs, and provide monosynaptic glutamatergic input to and receive inhibitory synaptic feedback from their postsynaptic targets in the glomerular layer, including most GABAergic periglomerular (PG) cells (Hayar et al., 2004a, 2005; Murphy et al., 2005; Wachowiak and Shipley, 2006). Because ET cells play such a pivotal role in driving the glomerular local inhibitory circuits, their synaptic input–output properties are of importance for sensory encoding. The probability of glutamate release from ORN terminals is unusually high. Feedback inhibition of glutamate release from ORN terminals is mediated by activation of presynaptic GABAB and D2 receptors (Aroniadou-Anderjaska et al., 2000; Ennis et al., 2001). However, little is known about the contribution of postsynaptic intrinsic excitability to synaptic integration in ET cells.
We recently reported that ET cells have multiple subthreshold conductances that cooperatively regulate spontaneous intrinsic bursting (Liu and Shipley, 2008). Because these intrinsic conductances, including the persistent sodium current (INaP), hyperpolarization-activated nonselective cation current (Ih), and L- and/or T-type calcium-channel-mediated low-voltage-activated (LVA) calcium current (IL/T), operate at membrane potentials below action potential threshold, they may actively shape synaptic integration in ET cells. Therefore, the aim of the present study was to determine the roles of these voltage-gated intrinsic conductances in shaping subthreshold and suprathreshold responses of ET cells to excitatory olfactory nerve (ON) inputs and inhibitory feedback inputs from glomerular local circuits.
Materials and Methods
Olfactory bulb slices from 5- to 6-week-old male C57BL/6J mice were prepared as described previously (Liu and Shipley, 2008). Briefly, horizontal slices (350 μm) were cut with a Leica VT1000 s vibratome in an ice-cold and oxygenated (95% O2–5% CO2) sucrose-based artificial CSF (sucrose-aCSF) containing (in mm) 220 sucrose, 3 KCl, 1.25 NaH2PO4, 2.6 MgSO4, 26 NaHCO3, and 10 glucose. After 30 min incubation in normal aCSF at 30°C, slices were then transferred to aCSF at room temperature until they were used for experiments. Normal aCSF was continuously bubbled with 95% O2–5% CO2 and had the following composition (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 1.3 CaCl2, 26 NaHCO3, 10 glucose. During experiments, slices were perfused at 3 ml/min with aCSF equilibrated with 95% O2–5% CO2 and warmed to 30°C.
Electrophysiological recordings were made from olfactory bulb ET cells visualized using a BX50WI (Olympus) fixed-stage upright microscope equipped with near-infrared differential interference contrast optics. ET cells were visually preselected by their somatic location within the deep half of the glomerular layer (GL) or at the GL–external plexiform layer border, having relatively large and pear-shaped cell bodies with one single apical dendrite toward a single glomerulus. Physiologically, they were identified by their distinctive and characteristic bursting firing of two to five action potentials superimposed on a slow depolarizing envelope. In some cases, ET cell identity was confirmed by biocytin filling and staining.
Current or voltage signals were recorded with a MultiClamp 700B amplifier (Molecular Devices) and low-pass filtered at 4 kHz, and sampled at 10 kHz with a Digidata 1322A 16-bit analog-to-digital converter (Molecular Devices) using Clampex 9.2 (Molecular Devices). Patch-recording electrodes were pulled from standard-wall glass capillary tubes without filament (Sutter Instrument). Patch pipettes (4–7 MΩ) contained (in mm) 5.5 EGTA, 0.5 CaCl2, 120 K-gluconate, 2 MgCl2, 10 Na-HEPES, 3 Mg-ATP, 0.3 Na3-GTP, and 10 Tris-phosphocreatine, and 0.1% biocytin. Osmolarity of the pipette solution for whole-cell recording was adjusted to 285–295 mOsm, pH to 7.3, with KOH. Electrical stimulation of the ON was delivered by bipolar glass electrodes made from theta pipettes (Sutter Instrument). The isolated and constant current stimulation pulses (100 μs) were triggered by a PG4000A digital stimulator (Cygnus Technology). Data were analyzed with Clampfit 9.2 (Molecular Devices) and Origin 7.5 (OriginLab)
The liquid junction potential (11–13 mV) was not corrected. Access resistance was typically <30 MΩ and not compensated.
The biocytin-filled cells were developed as described previously (Karnup and Stelzer, 1999). Briefly, the paraformaldehyde-fixed slices were embedded in 10% gelatin and sectioned at 50 μm thickness. The sections were subsequently reacted with 0.3% H2O2, 0.3% Triton X-100, ABC complex, and Ni-DAB chromagen. After dehydration, the sections were mounted in DPX (a mixture of distyrene, tricresyl phosphate, and xylene). Two-dimensional reconstructions of filled neurons were made with Neurolucida software (MicroBrightField).
2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) disodium salt (10 μm), dl-2-amino-5-phosphonovaleric acid (APV; 50 μm), gabazine (10 μm), 3-[[4-(4-chlorophenyl)-4-hydroxypiperidin-l-yl]methyl-1H-indole (l-741626; 5 μm), tetrodotoxin (TTX; 1 μm), 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288; 10 μm), and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845; 10 μm) were purchased from Tocris Bioscience. (1S,2S)-2-(2-(N-[(3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro-1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride (NNC55-0396; 50 μm), nimodipine (20 μm), 1,4-dihydro-2,6-dimethyl-5-nitro-4-(2-[trifluoromethyl]phenyl)pyridine-3-carboxylic acid methyl ester (Bay K8644; 5 μm), and all other chemicals were purchased from Sigma-Aldrich. All drugs were bath applied by diluting in aCSF at the above indicated doses unless otherwise stated.
Results
ON stimulation consistently evokes stable burst-firing response in ET cells
ET cells were selected on the basis of their morphological features including somatic location near the deep half of the glomerular layer and a single primary dendrite extensively ramifying in a single glomerulus (Fig. 1A), by their ability to spontaneously generate rhythmic bursts of action potentials, and by the fact that all ET cells receive excitatory monosynaptic inputs from the ON (Hayar et al., 2004a,b; Liu and Shipley, 2008). In voltage clamp with the holding potential at −60 mV, ON stimulation evoked EPSCs in ET cells with a constant latency (1.9 ± 0.1 ms, n = 20) (Fig. 1B). In current clamp, EPSPs or burst of action potentials were evoked by subthreshold or suprathreshold ON stimulation, respectively (Fig. 1C). These synaptic responses were completely abolished by NBQX (10 μm), indicating that they are mainly mediated by AMPA receptors.
Figure 1.

Stable burst-firing response to ON stimulation. A, Two-dimensional reconstruction of a typical biocytin-filled ET cell. EPL, External plexiform layer; ONL, olfactory nerve layer. B, Typical voltage-clamp recording (holding potential, −60 mV) showing ON-evoked and AMPA/kainate receptor-mediated EPSCs with constant and short latency. C, Current-clamp recording of the same cell showing ON-evoked subthreshold EPSPs and suprathreshold burst-firing responses that were blocked by NBQX (10 μm). D, Plotting EPSP amplitude against stimulus intensity shows the step-like input–output relationship of subthreshold responses from a typical cell. Inset, Current-clamp recording of EPSPs in response to ON stimuli with incremental intensities corresponding to colored solid points. E, Input–output relationship from the same cell in voltage clamp. F, Bar graph showing the number of cells with the number of EPSP steps evoked by incremental stimulus intensities (n = 19). G, Input–output relationship of suprathreshold responses from four cells showing that spike number per response does not continue to increase with incremental intensity of ON stimulation. H, Latency of the first spike within each burst-firing response decreases with incremental ON stimulation intensity. I, Plot of the number of spikes per response to ON stimulation with an intensity of 100 μA remains very stable over 20 min.
In assessing the subthreshold synaptic input–output relationship, we found that the EPSP amplitude increased with intensity of stimulation in a highly nonlinear manner (n = 19 cells). As shown by a representative recording in Figure 1D and inset, no EPSPs were detected until stimulus intensity reached 9 μA, above which EPSPs of two distinct amplitudes were recorded before suprathreshold burst-firing synaptic responses. EPSPs with stable amplitude of ∼6 mV were evoked by stimuli with intensity between 9 and 14 μA. At ≥15 μA, the EPSP amplitude jumped to ∼15 mV, and this amplitude was maintained until the stimulus intensity reached ≥20 μA, at which point spike firing was triggered. EPSPs of two discrete amplitudes were evoked by increased stimulus intensity in 11 of 19 cells (Fig. 1F). A similar input–output relationship was obtained in voltage clamp (Fig. 1E). We interpret this as attributable to presynaptic recruitment of discrete olfactory nerve fascicles (Feinstein and Mombaerts, 2004; Mombaerts, 2006) with increased stimulus intensity.
In contrast, the input–output relationship of suprathreshold responses was relatively linear and stable in terms of spikes/response versus stimulus intensity. Once the stimulus intensity reached threshold for burst firing, increasing shock strength decreased the latency of the initial spike but did not significantly increase the number of spikes per response (Fig. 1G,H). In all 24 cells tested, suprathreshold responses evoked by stimuli of constant intensity were stable (Fig. 1I), demonstrating reliable transmission at the ON→ET cell synapse.
Persistent sodium conductance boosts synaptic response and enhances summation
ET cells have an INaP that is active at resting membrane potential, approximately −55 mV (without correction of junction potential), and increases with membrane depolarization (Hayar et al., 2004b; Liu and Shipley, 2008). Thus, INaP might be engaged by excitatory synaptic ON inputs. TTX blocks INaP, but because it also blocks the transient sodium current (INaT) that is required for action potentials, TTX completely eliminates ON-evoked synaptic responses in ET cells. Thus, to investigate the influence of INaP on excitatory synaptic inputs, we devised an alternative strategy, viz., we examined the voltage changes of ET cells in response to simulated EPSPs (simEPSPs) before and after application of TTX and in the presence of NBQX and APV to eliminate spontaneous excitatory synaptic events. EPSPs were simulated by applying a current command derived from an inverted averaged EPSC recorded from a typical ET cell in response to ON stimulation (Fig. 2A–C). This current command had amplitude of 32.4 pA and a rise time (20–80%) of 0.3 ms, and its decay is well fitted by a single exponential with time constant of 3.5 ms.
Figure 2.

Persistent Na+ conductance amplifies subthreshold postsynaptic excitatory responses and produces temporal summation. A, Typical recording showing that EPSC is completely blocked by NBQX (10 μm). B, Inverted NBQX-sensitive EPSC from A. C, Comparison of two simEPSPs evoked by injecting the inverted EPSC in B from the same cell with holding potential at either −55 (black) or −75 mV (green). D, Pooled data from five cells showing that amplitude (left) and decay time constant (right) of simEPSP at −55 mV is significantly greater than at −75 mV (n = 5). E, Comparison of two simEPSPs from the same cell at −55 mV before (black) and after (red) TTX (1 μm). F, Plots showing both amplitude (left) and decay (right) of simEPSP from five cells at −55 mV are reduced by TTX (1 μm; n = 5). G, Comparison of responses to injection of a train of five inverted EPSCs at 40 Hz before (black) and after (red) TTX (1 μm). H, Plots showing that TTX significantly reduces the peak amplitude (left; n = 5) of the second to the fifth simEPSPs and the integrated area (right; n = 5) under five simEPSPs evoked by the train of inverted EPSCs. Error bars indicate SEM. *p < 0.05; **p < 0.01.
We first compared simEPSPs at both −75 mV, a membrane potential at which INaP is inactive, and at −55 mV, at which it is active. As shown in Figure 2C, the simulated EPSP at −55 mV exhibited a larger amplitude and slower decay time constant than at −75 mV. Pooled data from five cells showed that simEPSPs had an amplitude of 1.5 ± 0.16 mV at −75 mV compared with 2.1 ± 0.18 mV (Fig. 2D, left) (n = 5, p < 0.01) at −55 mV, and a decay time constant of 37.3 ± 6.1 ms at −55 mV compared with 9.6 ± 1.2 ms (Fig. 2D, right) (n = 5, p < 0.01) at −75 mV. This suggests that there is voltage-dependent amplification of synaptic responses. Because INaP activates at approximately −60 mV (without adjustment for the average junction potential of 11–13 mV) (Hayar et al., 2004b; Liu and Shipley, 2008), INaP might contribute to synaptic amplification. To test this, simEPSPs were measured before and after application of TTX. As shown in Figure 2, E and F, at −55 mV TTX significantly reduced both amplitude (Fig. 2F, left) (n = 5, p < 0.05) and decay time constant (Fig. 2F, right) (n = 5, p < 0.01), respectively. In contrast, neither the amplitude nor decay of simEPSPs was affected by TTX when the membrane potential was held at −75 mV (Fig. 2E, inset). These results support the conclusion that INaP amplifies excitatory synaptic transmission from ONs to ET cells.
In addition to increasing the amplitude of EPSPs, INaP also prolonged their decay time, suggesting that this conductance plays a role in the summation of multiple synaptic inputs. Therefore, we investigated the voltage responses to injected trains of five inverted EPSCs at 40 Hz (Fig. 2G). Temporal summation was measured in two ways: (1) the increase in peak amplitude of the second to fifth simEPSPs relative to that of the first; and (2) the integrated area under the train of simEPSPs. In the control condition, the relative peak amplitude of the second and fifth simEPSPs were 135.4 ± 5.2% (n = 5, p < 0.001) and 165.4 ± 7.6% (n = 5, p < 0.001), respectively, indicating a significant temporal summation. TTX (5 min) significantly reduced the peak of all simEPSPs, with amplitudes of 106.2 ± 4.7% (Fig. 2H, left) (n = 5, p < 0.05) and 102.9 ± 8.0% (n = 5, p < 0.01) for the second and fifth simEPSPs, respectively. TTX also reduced the integrated area under the train of simEPSPs from 469.5 ± 12.0 to 260.9 ± 42.0 mV · ms (Fig. 2H, right) (n = 5, p < 0.01). Together, these results strongly suggest that INaP plays an active role in temporal summation as well as amplification of excitatory synaptic events.
Both subthreshold simEPSPs and ON-evoked suprathreshold firing responses exhibit voltage-dependent amplification
Because the EPSP integral is thought to be a more important determinant of whether an EPSP initiates an action potential than its peak amplitude (Jack et al., 1983), the voltage-dependent amplification of both duration and peak amplitude of subthreshold simEPSPs suggests that INaP boosts not only subthreshold but also suprathreshold synaptic responses of ET cells to ON inputs. To test this, we recorded suprathreshold firing responses to ON stimulation followed by simEPSP experiments in each cell with the membrane potential held at nine different levels from −53 to −69 mV (2 mV/step). In the suprathreshold response experiments, the spike number per response to each ON stimulus was used to assess the response strength. As shown in Figure 3, A and C, spike number per response significantly increased when the membrane holding voltage increased from −57 to −53 mV, indicating a voltage-dependent amplification of suprathreshold firing responses.
Figure 3.

Voltage dependence of suprathreshold and subthreshold postsynaptic responses. A, B, Current-clamp recordings showing the effect of holding membrane potential (from −53 to −69 mV; 2 mV increments) on suprathreshold postsynaptic firing responses to ON stimulation (A) and subthreshold simEPSPs evoked by an inverted EPSC (20 pA) injection in the absence (black) or presence (red) of 1 μm TTX (B). C, Plot of six cells showing that ON-evoked spikes per response significantly increase with membrane depolarization in the range from −59 to −53 mV. D, E, Plots showing that both amplitude (D) and half-width (E) of simEPSPs from five cells are significantly reduced by TTX (1 μm) in the voltage range from −57 to −53 mV. *p < 0.05; **p < 0.01. Error bars indicate SEM.
In the simEPSP experiments, the amplitude of the inverted EPSC command was reduced to 20 pA to minimize the probability of simEPSP-evoked action potentials at holding potential of −53 mV. As shown in Figure 3, B, D, and E, when the membrane holding potential was increased from −69 to −53 mV (especially from −57 to −53 mV), simEPSPs exhibited an incremental change in both amplitude and duration. TTX (5 min) significantly reduced both amplitude and half-width of simEPSPs at voltages from −57 to −53 mV. The voltage range of this effect and its TTX sensitivity further implicates INaP as a key contributor to the amplification of excitatory synaptic responses.
The voltage range of simEPSP amplification overlaps with that of suprathreshold response amplification, indicating an active role of INaP in amplifying ET cell suprathreshold responses to ON inputs.
The low-voltage-activated Ca2+ conductance voltage-dependently amplifies subthreshold synaptic responses
ET cells have a prominent Ca2+ conductance that is activated at approximately −50 mV (without correction of junction potential) and has biophysical and pharmacological features of both T- and L-type Ca2+ currents (Liu and Shipley, 2008). To activate LVA Ca2+ conductance, relatively large EPSP amplitude is required, but once engaged, this current may boost EPSP amplitude sufficient to reach the threshold for action potentials. Thus, experiments were performed in the presence of 10 nm TTX, which shifted the threshold of action potentials to a relatively positive level but did not abolished synaptic transmission. In this condition, with membrane potential held at −65 mV, ON stimulation was adjusted to evoke EPSPs (Fig. 4A) with amplitudes of 13.9 ± 0.9 mV (n = 5) (Fig. 4B, left) and half-widths of 14.8 ± 0.8 ms (n = 5) (Fig. 4B, right). After 5 min of treatment with Bay K8644, an L-type Ca2+ channel activator that enhances LVA Ca2+ currents in ET cells (Liu and Shipley, 2008), both the amplitude and half-width of EPSPs increased significantly (Fig. 4B), suggesting that LVA Ca2+ conductance can amplify excitatory synaptic transmission in ET cells. Replacement of Bay K8644 with a T-type Ca2+ channel blocker, NNC55-0396, and an L-type Ca2+ channel blocker, nimodipine, in the same cells significantly decreased both the amplitude (Fig. 4B, left) and half-width (Fig. 4B, right) of EPSPs to 9.4 ± 0.3 mV (n = 5, p < 0.01 compared with control) and 12.5 ± 0.7 ms (n = 5, p < 0.001 compared with control), respectively, further suggesting that an LVA Ca2+ conductance is actively engaged and contributes to synaptic responses evoked by ON-input.
Figure 4.

Low-voltage-activated Ca2+ conductance voltage-dependently boosts subthreshold synaptic responses. A, Current-clamp recording showing that ON-evoked EPSP is boosted by Bay K8644 (Bay K; 5 μm, 5 min) in the presence of 10 nm TTX. Replacing Bay K8644 with both NNC55-0396 (NNC; 50 μm) and nimodipine (20 μm) for 10 min not only reverses the amplificatory effect but further attenuates EPSP compared with control. B, Plots showing that amplitude (left) and half-width (right) of ON-evoked EPSPs from five cells are significantly and reversibly enhanced by Bay K8644 (5 μm, 5 min) with holding potential at −65 mV. Note both amplitude and half-width of EPSPs after the replacement of Bay K8644 with both NNC55-0396 (50 μm) and nimodipine (20 μm) for 10 min are significantly smaller than in control. C, Current-clamp recordings showing that large but not small simEPSPs are enhanced by Bay K8644 (5 μm, 5 min). D, Pooled data from five cells showing that both the amplitude (left) and half-width (right) of large (top) but not small (bottom) simEPSPs are enhanced by Bay K8644 (5 μm, 5 min) with the holding potential at −55 mV. E, Current-clamp recording showing that large but not small simEPSPs are attenuated by nimodipine (20 μm, 10 min). F, Graphs showing that both the amplitude (left) and half-width (right) of large (top) but not small (bottom) simEPSPs from five cells are reduced by NNC55-0396 (50 μm, 10 min) with the holding potential at −55 mV. G, Recording showing that large but not small simEPSPs are attenuated by NNC55-0396 (50 μm, 10 min). Nim., Nimodipine. H, Plots showing that both amplitude (left) and half-width (right) of large (top) but not small (bottom) simEPSPs from five cells are reduced by nimodipine (20 μm, 10 min) with the holding potential at −55 mV. Error bars indicate SEM. **p < 0.01; ***p < 0.001.
Because the minimal membrane potential of ET cells is about −55 mV rather than −65 mV, and because EPSPs evoked by ON inputs (especially weak inputs) may not always reach 10 mV, we assessed the role of LVA Ca2+ conductance in shaping synaptic responses at a holding potential of −55 mV with simulated EPSPs whose amplitude could be varied by changing the amplitude of the injected EPSC. Two sets of inverted EPSCs with amplitudes of 50 and 100 pA were applied to each ET cell before and after drug treatment in the presence of NBQX and APV, but without TTX. In the first set of experiments, Bay K8644 (5 min) increased both the amplitude and half-width of the simEPSPs (Fig. 4C,D, top) evoked by the larger EPSC command, but had no effect on simEPSPs evoked by the smaller EPSC command (Fig. 4C,D, bottom). Either NNC55-0396, a selective T-type channel blocker (Fig. 4E,F), or nimodipine, a selective L-type channel blocker (Fig. 4G,H) significantly reduced both the amplitude and half-width of the larger simEPSP (Fig. 4F,H, top), with little effect on the smaller sEPSPs (Fig. 4F,H, bottom), indicating that T- and/or L-type channel-mediated LVA Ca2+ conductance boosts only large EPSPs, which depolarizes cell membrane to at least −50 mV (without correction of junction potential) and is consistent with our previous study (Liu and Shipley, 2008). Together, these results demonstrate that L- and/or T-type channel-mediated LVA Ca2+ currents actively amplify large excitatory synaptic responses in ET cells to ON input that depolarize the membrane to the activation voltage for these conductances.
The LVA Ca2+ conductance boosts suprathreshold synaptic responses
We next examined the role of LVA Ca2+ conductance in suprathreshold synaptic responses. Exposure to NNC55-0396, a selective T-type Ca2+ channel blocker, for 10 min reduced ON-evoked spike bursts to single spikes in all ET cells tested (Fig. 5A,B) (n = 5), indicating that T-type Ca2+ conductance amplifies suprathreshold synaptic responses to ON input. This was not attributable to presynaptic effects because the paired-pulse ratio (PPR), the amplitude ratio of the second to the first EPSCs (EPSC2/EPSC1) evoked by two consecutive ON stimuli with an interval at 50 ms, was unaffected by NNC55-0396 (Fig. 6H, top) (0.69 ± 0.03 in control vs 0.68 ± 0.06 after NNC, n = 5).
Figure 5.

T-type Ca2+ conductance amplifies suprathreshold synaptic responses. A, Current-clamp recording of suprathreshold postsynaptic responses to ON-stimulation before (left) and after (right) NNC55-0396 (50 μm, 10 min) treatment with holding potential at −60 mV. B, Pooled data from five cells showing that NNC55-0396 turns burst-firing (right) into single-spike-firing (left) suprathreshold responses. C, Plot showing the effect of sequentially bath-applied gabazine (10 μm), CGP55845 (10 μm), and l-741626 (5 μm) on the spikes per suprathreshold response. D, Pooled data from five cells showing that bath application of mixed blockers (10 μm gabazine, 10 μm CGP55845, and 5 μm l-741626) for 10 min significantly increases spikes per suprathreshold synaptic response; addition of NNC55-0396 (NNC; 50 μm, 10 min) in the presence of the blocker mixture still reduces spikes per suprathreshold synaptic response in the same cells. Bar graphs in D represent average value. ***p < 0.001.
Figure 6.

Blocking L-type Ca2+ conductance attenuates suprathreshold synaptic responses. A–C, Current-clamp recordings showing postsynaptic responses to suprathreshold ON-stimulation in the same cell before (A), after nimodipine (20 μm) treatment for 10 min (B), and after replacing nimodipine with Bay K8644 (5 μm) for 10 min (C). D–F, Pooled data from five cells showing the spikes per suprathreshold response before (D), after nimodipine treatment for 10 min (E), and after washout of nimodipine with addition of Bay K8644 for 10 min (F). G, Plot showing the increment of spikes per response in five cells by mixture of synaptic blockers (10 μm gabazine, 10 μm CGP55845, and 5 μm l-741626) for 10 min. Nimodipine (nim.; 20 μm, 10 min) significantly decreases spikes per suprathreshold response in the presence of blocker mixture in the same cells. Bar graphs represent the average values. H, Recording of paired-pulse EPSCs with interpulse interval at 50 ms before (left) and after NNC55-0396 (NNC; 50 μm; top right) or nimodipine (20 μm; bottom right) treatment for 10 min (holding potential, −60 mV). ***p < 0.001.
ET cells provide monosynaptic, glutamatergic input to (Hayar et al., 2004a; Liu and Shipley, 2008) and receive feedback synaptic inputs from local, intraglomerular GABA/dopaminergic PG cells (Hayar et al., 2005). Blocking GABAA receptors modulates spontaneous bursting activity in ET cells (Hayar and Ennis, 2007). The GABAA receptor blocker gabazine significantly increased the number of spikes in response to ON stimulation, but adding the GABAB receptor blocker CGP55845 and then dopamine (DA) D2 receptor blocker l-741626 did not further change the suprathreshold synaptic responses (Fig. 5C). However, the effects of blocking T-type Ca2+ conductance with NNC55-0396 on suprathreshold synaptic responses was not caused by an effect on local circuits because, if NNC55-0396 reduced GABA release from PG cells, the number of spikes per response should increase rather than decrease. This conclusion was further supported by experiments in which slices were exposed to a mixture of the three blockers, gabazine, CGP55845, and l-741626 for 10 min before NNC55-0396 treatment. As expected, the spikes per response to ON stimulation increased (n = 5, p < 0.001) (Fig. 5D) in the mixture. However, NNC55-0396 (10 min) still significantly reduced spikes per response (Fig. 5D). These results indicate that intrinsic T-type Ca2+ conductance actively boosts suprathreshold synaptic responses in ET cells.
Given that LVA Ca2+ conductance is at least partially mediated by L-type Ca2+ channels (Liu and Shipley, 2008), we next examined the effect of blocking L-type Ca2+ channels on ON-evoked suprathreshold synaptic response in ET cells. After 10 min of exposure to the L-type blocker nimodipine, ON stimuli triggered only single spikes in all five cells tested (Fig. 6B,E). This was completely reversed by 10 min of washout of nimodipine and the addition of Bay K8644 (Fig. 6C,F). The PPR of ON-evoked EPSCs was not affected by nimodipine (Fig. 6H, bottom) (0.66 ± 0.1 in control vs 0.67 ± 0.1 after 15 min nimodipine, n = 5), indicating that the effect of nimodipine on suprathreshold synaptic response is not caused by presynaptic action. Nimodipine still significantly reduced the spikes/response (Fig. 6G) in the mixture of synaptic blockers (gabazine, CGP55845, and l-741626). This argues against a network origin of nimodipine effect on the suprathreshold synaptic responses and indicates that an intrinsic L-type Ca2+ conductance amplifies suprathreshold synaptic spiking responses to ON input in ET cells.
Consistent with a role of L-type Ca2+ conductance, Bay K8644 (10 min) dramatically increased the number of spikes in each response to ON stimuli (Fig. 7, compare A,D to B,E). After replacing Bay K8644 with nimodipine for 15 min, spike number per response decreased (Fig. 7C,F). The similar boosting effect of Bay K8644 on suprathreshold synaptic responses in the presence of GABAA, GABAB, and D2 blockers excludes a network contribution (Fig. 7G).
Figure 7.
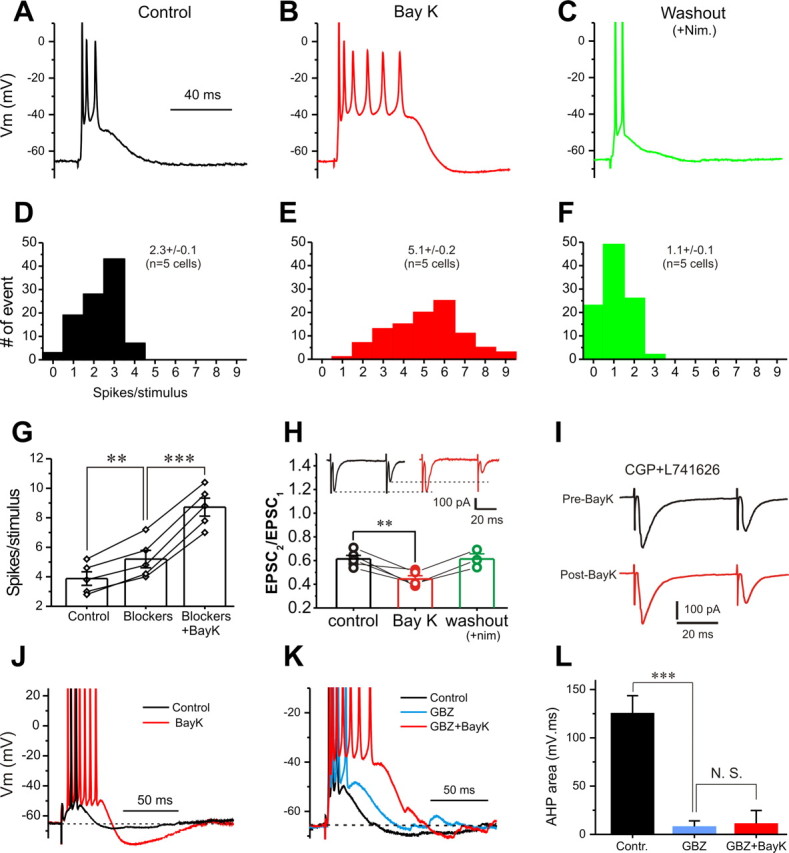
Activation of L-type Ca2+ conductance boosts suprathreshold postsynaptic responses. A–C, Current-clamp recordings of suprathreshold synaptic response before (A, black), after Bay K8644 (Bay K; 5 μm) for 5 min (B, red), and after replacement of Bay K8644 by nimodipine (Nim; 20 μm) for 15 min (C, green). D–F, Pooled data from five cells showing spikes per suprathreshold response before (D, black), after Bay K8644 treatment for 10 min (E, red), and after replacing Bay K8644 with nimodipine for 15 min (F, green). G, Graph showing that exposure to the mixture of synaptic blockers (10 μm gabazine, 10 μm CGP55845, and 5 μm l-741626 for 10 min) increases spikes per suprathreshold response; addition of Bay K8644 (5 μm, 5 min) further increases spikes per response. H, Plot showing that Bay K8644 significantly reduces the PPR in all five cells tested; reduced PPR is reversed by washout of Bay K8644 with the addition of nimodipine for 15 min in all three cells tested. Inset, Voltage-clamp recordings of paired EPSCs (interpulse interval 50 ms) before (black) and after (red) Bay K8644 (5 μm, 5 min). Note that the amplitude of the first EPSC from the paired-pulse EPSCs in the inset is not affected by Bay K8644 (5 μm, 10 min). I, Paired-pulse EPSCs before (black) and after (red) Bay K8644 (5 μm, 10 min) in the presence of both CGP55845 (10 μm) and l-741626 (5 μm). J, Comparison of the AHP after suprathreshold responses before (black) and after (red) Bay K8644 (5 μm, 5 min) in the absence of gabazine. K, Comparison of the AHP after suprathreshold responses in the same cell before (black), after gabazine (5 μm, 10 min; cyan), and after Bay K8644 (5 μm, 5 min; red) in the presence of gabazine. L, Graphs showing that the average AHP area integral is significantly reduced by gabazine but not affected by Bay K8644 in the presence of gabazine (n = 5 cells). **p < 0.01; ***p < 0.001. N.S., Not significant.
In attempt to test the effect of Bay K8644 on presynaptic transmitter release, we found that Bay K8644 (5 min) significantly reduced ON-evoked EPSC PPR in ET cells (Fig. 7H, insets). After replacing Bay K8644 with nimodipine for 15 min, PPR recovered. The PPR reduction by Bay K8644 may be attributable to increased glutamate release from ON terminals or to increased GABA and/or dopamine released from PG cells in the glomerular local circuits, or both. As shown in the insets in Figure 7H, Bay K8644 had little effect on the amplitude of the first EPSC in response to paired-pulse stimulation (204.4 ± 21.4 pA in control vs 206.3 ± 26.3 pA after Bay K8644, n = 5), suggesting no direct effect on glutamate release from ON terminals. Therefore, we tested the second possibility by treating slices with the GABAB blocker CGP55845 and the D2 blocker l-741626 for 10 min before Bay K8644. In this condition, Bay K8644 (5 min) did not affect the PPR (Fig. 7I) (0.74 ± 0.10 in control vs 0.73 ± 0.10 after Bay K8644, n = 5, p > 0.05), This suggests that Bay K8644 enhances ON-evoked PPR in ET cells by increasing GABA and/or dopamine release from PG cells in local circuits. In this scenario, the first stimulation causes PG cells to release more GABA and/or dopamine, which inhibits subsequent glutamate release from the ON terminals in responses to the second stimulation, thus reducing the amplitude of the second EPSC.
The possibility that Bay K8644 enhances GABA release from PG cells is also suggested by its effect on the afterhyperpolarization (AHP) in ET cells after burst-firing responses to ON stimulation. As shown in Figure 7J, Bay K8644 significantly boosted AHP from 137.9 ± 15.5 to 452.3 ± 31.4 mV · ms (n = 5, p < 0.001). The GABAA receptor blocker gabazine reduced this AHP (Fig. 7K,L), and in this condition the remaining AHP was unaffected by Bay K8644 (5 min) (Fig. 7K,L). This suggests that activation of GABAA receptors underlies majority of the AHP after each burst-firing response to ON stimulation, and that its increase after Bay K8644 is attributable to increased GABA release onto ET cells. Together, all of these results support the idea that L- and/or T-type-mediated intrinsic LVA conductance in ET cells can amplify both subthreshold and suprathreshold synaptic responses to ON inputs.
Ih plays a role in integrating excitatory synaptic responses to ON input
TTX reduced both the amplitude and decay time constant of simEPSPs at −55 mV. However, the decay time constant (18.8 ± 2.7 ms, n = 5) of simEPSPs at −55 mV in the presence of TTX is still longer than that (9.6 ± 1.2 ms, n = 5, p < 0.05) at −75 mV (Fig. 8A, inset). This suggests that in addition to INaP, other voltage-dependent conductances active near resting membrane potential contribute to synaptic integration. One candidate is the hyperpolarization-activated nonselective cation current, Ih. ET cells have a prominent Ih current, which plays an important role in setting their minimal membrane potential and input resistance and in the generation of spontaneous bursting (Liu and Shipley, 2008). Because this current is active at membrane potentials below −45 mV, we tested its potential role in shaping synaptic responses. As shown in Figure 8B, blocking Ih with ZD7288 increased both the amplitude and decay time constant of ON-evoked EPSP, suggesting that Ih contributes to the voltage-dependent difference in decay time constant of simEPSPs between −55 and −75 mV. To further investigate this current, we again used simEPSPs so that its role in shaping EPSP could be tested in the presence of TTX with the holding potential at −55 mV, the average minimal membrane potential for most ET cells. In this condition, blocking Ih with ZD7288 significantly increased the integrated area under simEPSP (Fig. 8C,D right) with little effect on simEPSP amplitude (Fig. 8D, left).
Figure 8.
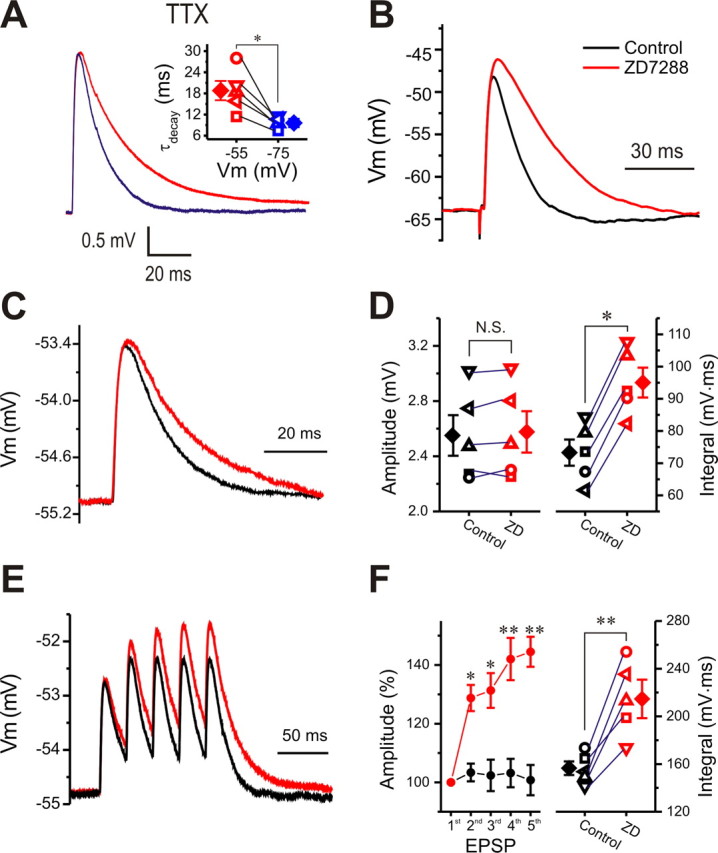
Blocking Ih prolongs EPSPs and produces temporal summation. A, Comparison of simEPSPs from the same cell with holding potential at −55 or −75 mV in the presence of TTX (1 μm). Inset, The decay time constant of simEPSPs from five cells at −55 mV is significantly higher than that from the same cells at −75 mV. B, Comparison of ON-evoked EPSPs from the same cell before (black) and after ZD7288 (10 μm, 10 min; red). C, Comparison of simEPSPs from the same cell held at −55 mV before (black) and after ZD7288 (10 μm, 10 min; red). D, Graphs showing that the area integral (right) but not the amplitude (left) of single simEPSPs from five cells is increased by ZD7288 (ZD). E, Trains of five simEPSPs (40 Hz) from the same cell at −55 mV before (black) and after (red) ZD7288 (10 μm, 10 min). F, Graphs showing both the relative amplitudes (normalized to the first, left) of and the integrated area (right) under multiple simEPSPs from five cells are significantly increased by ZD7288. *p < 0.05; **p < 0.01. N.S., Not significant.
Because its main impact is on the decay of EPSPs, Ih may play a role in EPSP summation. Next, we determined the effect of blocking Ih on multiple simEPSPs in response to a train of five EPSCs at 40 Hz. As shown in Figure 8, E and F, in the presence of TTX, the relative amplitude of the second and fifth simEPSPs were 103.4 ± 3.0% and 100.8 ± 5.2%, respectively, indicating no summation. However, treatment with ZD7288 (10 min) increased the relative amplitude of the second and the fifth simEPSPs to 128.8 ± 4.5% (n = 5, p < 0.05) and 144.5 ± 5.1% (n = 5, p < 0.01) (Fig. 8F, left), respectively. The integrated area under multiple simEPSPs was also dramatically increased (Fig. 8F, right). These results indicate that Ih reduces temporal summation of subthreshold excitatory synaptic responses.
What is the role of Ih in suprathreshold synaptic responses to ON stimulation? As shown in Figure 9, ZD7288 (10 min) hyperpolarized cells and significantly reduced spikes per burst in response to each ON stimulation (Fig. 9A–C), confirming the active role of Ih in setting membrane potential (Liu and Shipley, 2008). Because blocking Ih increased EPSP duration (Fig. 8), the broadened EPSPs, if they have any influence on the suprathreshold firing responses, should increase rather than decrease the spikes/response. Thus, the reduction in spikes/response in the presence of ZD7288 might simply be attributable to membrane hyperpolarization, which attenuates suprathreshold synaptic firing responses by terminating INaP (Fig. 3). Consistent with this idea, when the membrane potential was restored to its original level by injected current in the presence of ZD7288, the number of spike per response significantly increased (Fig. 9B,C).
Figure 9.

Effects of blocking Ih on suprathreshold synaptic responses. A, B, Current-clamp recordings of suprathreshold postsynaptic response to ON stimulation before (A, black), after ZD7288 (ZD; 10 μm, 10 min; B, red), and after ZD7288 plus maintained 30 pA depolarizing current injection (B, green). C, Graph showing that the spike number per suprathreshold response from five cells is significantly reduced by ZD7288 alone, but is increased by ZD7288 with compensation of membrane potential change. D, Paired-pulse EPSCs from the same cell holding at −60 mV before (black) and after ZD7288 (10 μm, 10 min; red). Error bars indicate SEM. *p < 0.05; ***p < 0.001.
Because Ih plays a role in transmitter release at some synapses (Southan et al., 2000; Mellor et al., 2002), it was necessary to determine whether presynaptic effects contributed to the observed changes in suprathreshold postsynaptic responses. To assess this, we examined PPR of ON-evoked EPSCs before and after ZD7288. Blocking Ih had no effect on the ON-evoked PPR (Fig. 9D) (0.68 ± 0.06 control vs 0.68 ± 0.06 after ZD7288, n = 5), indicating that Ih does not engage in presynaptic transmitter release from ON terminals and confirming its active role in shaping suprathreshold postsynaptic responses.
Ih can transform inhibitory inputs to postsynaptic excitation in ET cells
Thus far, we have considered excitatory synaptic input because the membrane depolarization produced by EPSPs engages the inward currents mediated by INaP, IT/L, and Ih. In contrast to INaP and IT/L, however, Ih is an inward current that is activated by membrane hyperpolarization such as occurs in response to inhibitory inputs. ET cells receive inhibitory input from GABAergic PG cells in the glomerular local circuit (Hayar et al., 2004a, 2005; Hayar and Ennis, 2007). As shown in Figure 10, in voltage clamp, both ON-evoked and spontaneous IPSCs were revealed by holding the cells at −20 (Fig. 10A) or 0 mV (Fig. 10A,B). The complete elimination of these IPSCs by gabazine (Fig. 10A,B) shows that they are mediated by GABAA receptors.
Figure 10.

Ih transforms inhibitory inputs to postsynaptic excitation. A, Voltage-clamp recordings of ON-evoked EPSCs from the same cell with holding potential at either −20 or 0 mV before (black) and after (red) gabazine (10 μm, 10 min) showing GABAA receptor-mediated IPSCs. Note that the IPSC onset latency is longer than that of the EPSC. B, Voltage-clamp recordings of spontaneous synaptic currents from the same cell with holding potential at −55, 0, or 0 mV in the presence of gabazine (10 μm, 10 min) showing inward spontaneous EPSCs and GABAA receptor-mediated outward spontaneous IPSCs (sIPSCs). C, Top, Superimposed sIPSCs from B with holding potential at 0 mV; the red trace represents the average IPSC. Bottom, An inverted average IPSC from the top (left) and two inverted IPSCs paired at 50 Hz (right) are used as simIPSCs to evoke voltage responses. D, Current-clamp recordings showing that a rebound burst response (black) evoked by injection of the two simIPSCs in C to an ET cell at −55 mV is eliminated by ZD7288 (10 μm; red). Note that restoring the hyperpolarized membrane potential caused by ZD7288 to control level does not restore the rebound burst-firing response (green). E, Comparison of responses to two simIPSCs injected in the same cell held at −65 mV before (black) and after (red) ZD7288 (10 μm, 10 min) showing that the rebound depolarization is completely abolished by ZD7288. Inset, Graph showing that the rebound depolarization evoked by a train of two simIPSCs from five cells is completely eliminated by ZD7288. Error bars indicate SEM. ***p < 0.001.
Do hyperpolarizing IPSPs activate Ih in ET cells, and if so, what are the functional consequences? To address this question, we used the simulated IPSP (simIPSP) approach. An average IPSC was taken from the spontaneous IPSCs in a typical ET cell held at 0 mV (Fig. 10B,C). This average IPSC has an amplitude of 32 pA, a peak rise time (20 to 80%) of 0.3 ms, and is well fitted by a single exponential function with a decay time constant of 4.3 ms. This IPSC (Fig. 10C) was then inverted and applied as a current-clamp command to evoke simulated voltage responses (simIPSPs) in ET cells. Injection of a single inverted IPSC triggered rebound burst-firing responses in 1 of 10 cells tested (data not shown). However, injection of two inverted IPSCs paired at 50 Hz evoked burst-firing responses in 7 of 10 cells (Fig. 10D). A 50 Hz train of five inverted IPSCs evoked burst-firing responses in 10 of 10 ET cells (data not shown). These findings indicate that inhibitory inputs can elicit a rebound excitatory response in ET cells.
We then investigated whether Ih was required for this excitatory effect of IPSPs. Exposure to ZD7288 (5 min) abolished the burst-firing responses after simIPSPs (Fig. 10D). This was not attributable to the membrane hyperpolarization caused by ZD7288 treatment because restoring the membrane potential to its control level by current injection did not restore simIPSP-evoked bursting (Fig. 10D). The role of Ih in mediating this effect was further analyzed by examining the rebound depolarization after simIPSPs. To observe rebound depolarization without action potentials, and to determine the effect of blocking Ih, cells were held at −65 mV by current injection. In this condition, injection of two inverted IPSCs (50 Hz) evoked a rebound depolarization (2.2 ± 0.1 mV, n = 5), which was completely eliminated by ZD7288 (10 min) treatment (0.04 ± 0.06 mV, n = 5, p < 0.001) (Fig. 10E, inset), indicating that Ih can convert inhibitory input into excitatory response in ET cells by mediating rebound depolarization.
Discussion
ET cells play a pivotal role in the glomerular network. They receive monosynaptic ON input and provide the major excitatory drive on intraglomerular and interglomerular inhibitory circuits. We recently demonstrated that several voltage-dependent conductances function cooperatively to regulate autonomous, rhythmical spontaneous bursting in external tufted cells (Liu and Shipley, 2008). Because these same conductances are active at or near resting membrane potential, they might play roles in the integration of synaptic inputs. Indeed, the present study demonstrates that three subthreshold inward conductances significantly shape postsynaptic responses in ET cells to subthreshold and suprathreshold ON and inhibitory inputs: (1) INaP amplifies and strongly enhances temporal summation of EPSPs. (2) ILVA mediated by L- and/or T-type Ca2+ channels boosts both subthreshold and suprathreshold excitatory postsynaptic responses. (3) Ih attenuates excitatory postsynaptic response and reduces temporal summation. (4) In addition, Ih transforms hyperpolarizing inhibitory inputs to postsynaptic rebound excitation. Thus, in addition to their essential roles in regulating spontaneous bursting behavior, intrinsic conductances significantly determine the impact of sensory and inhibitory synaptic inputs to ET cells.
INaP amplifies and strongly enhances temporal summation of postsynaptic responses to sensory input
The present study revealed a marked voltage-dependent amplification of subthreshold EPSPs in ET cells. The amplifying mechanism has rapid onset but slower decay kinetics. Using simEPSPs, we were able to demonstrate that Na+ conductance accounts for most of the amplification of both amplitude and duration of subthreshold EPSPs. ET cells have a prominent persistent sodium current, INaP (Hayar et al., 2004b; Liu and Shipley, 2008). The lack of specific blockers to INaP and INaT makes it impossible to differentiate their separate roles in subthreshold postsynaptic responses but, two points strongly argue that INaP is the key amplificatory current: (1) INaP has the relatively negative activation voltage required to amplify subthreshold EPSPs; (2) its fast activation and slow inactivation correspond well to the observed amplification of both amplitude and duration of subthreshold EPSPs. The ability of INaP to significantly prolong EPSP duration suggested that it might enhance temporal summation of multiple subthreshold EPSPs. Indeed, we confirmed that there is pronounced, TTX-sensitive temporal summation of EPSPs in ET cells. These results are consistent with previous studies indicating that INaP can amplify EPSPs and enhance temporal summation in other neurons (Stuart and Sakmann, 1995; Lipowsky et al., 1996; Andreasen and Lambert, 1999; González-Burgos and Barrionuevo, 2001).
By amplifying subthreshold EPSPs, INaP should increase the reliability of suprathreshold responses. Indeed, the voltage-dependent amplification of both subthreshold and suprathreshold postsynaptic responses correlated very well in the voltage range in which INaP is active. Thus, INaP not only boosts subthreshold EPSPs, but also amplifies suprathreshold postsynaptic firing responses to ON inputs.
ILVA contributes to both subthreshold and suprathreshold excitatory postsynaptic responses
ET cells have a prominent low-voltage-activated conductance that is mediated by T- and/or L-type channels (Liu and Shipley, 2008). In contrast to INaP, the ability of this LVA Ca2+ conductance to amplify subthreshold EPSPs depends on EPSP amplitude and resting membrane potential (Deisz et al., 1991; Markram and Sakmann, 1994; Stuart and Sakmann, 1995; Gillessen and Alzheimer, 1997; Urban et al., 1998; Andreasen and Lambert, 1999). Consistently with this, we found that ILVA does not amplify small EPSPs. However, when the amplitude of either simEPSPs or ON-evoked EPSPs drives the membrane potential >50 mV, the synaptic response is strongly boosted by ILVA. The simplest explanation for this is that the depolarization caused by small simEPSPs is insufficient to reach the threshold of ILVA because our previous study showed that LVA Ca2+ spikes activate at approximately −50 mV. Thus, ILVA amplifies large EPSPs and enhances summated EPSPs when the membrane potential is sufficiently depolarized.
ILVA also amplifies suprathreshold postsynaptic responses. Pharmacological block of ILVA significantly decreased the number of action potentials produced by ON synaptic input. Enhancing ILVA with Bay K8644 significantly increased the number of action potentials produced by each synaptic input. These effects were not attributable to presynaptic mechanisms or network contributions. Thus, direct activation of ILVA in ET cells enhances their suprathreshold as well as subthreshold responses.
Role of Ih in integrating excitatory postsynaptic responses
In contrast to the amplificatory roles of INaP and ILVA, Ih attenuates subthreshold EPSPs, mainly by increasing their rate of decay, an effect consistent with studies in other mammalian CNS neurons (Schwindt and Crill, 1997; Magee, 1998; Williams and Stuart, 2000; Angelo et al., 2007). Activation of Ih reduces ET cell input resistance (Liu and Shipley, 2008) so that the same amount of synaptic current produces briefer membrane depolarization. However, this does not explain why blocking Ih increased the amplitude of ON-evoked EPSPs, but not small simEPSPs. EPSPs reach their peak very quickly (<10 ms), during which time, because of its slow activation and deactivation kinetics, Ih is not appreciably recruited. Thus, the amplitudes of smaller simEPSPs may be less influenced by Ih than those of larger ON-evoked EPSPs. Consistent with its main effect on EPSP decay, Ih, as expected, reduced temporal summation of multiple simEPSPs.
Ih transforms inhibitory inputs to postsynaptic excitatory responses
A novel function of Ih in ET cells is to transform inhibitory inputs into excitatory rebound burst firing. ON stimulation elicits monosynaptic EPSCs followed by a burst of GABAA receptor-mediated IPSCs in ET cells. This feedback inhibition from PG cells may play a role in terminating the spike burst (Hayar et al. 2005). Thus, when ET cells are at relatively depolarized membrane potentials, feedback from PG cells provides classic inhibition. However, near the minimal membrane potential of ET cells, where Ih is active, a short barrage of IPSPs produces a rebound burst-firing response that is abolished by the Ih blocker ZD7288. Therefore, Ih may play an important role in transforming inhibitory inputs to postsynaptic excitation by mediating rebound depolarization. ET cells express high levels of α3 subunits of the GABAA receptor (Panzanelli et al., 2005), which mediate slow IPSCs (Bosman et al., 2002) and thus could expand the frequency spectrum of IPSP summation. IPSP summation further facilitates the inhibition-to-excitation transformation by Ih. Thus, inhibitory inputs can result in either classic inhibition or excitation in the form of rebound bursting, depending on the membrane potential of the ET cell when the IPSPs occur.
Functional implications
ET cells receive monosynaptic ON input and provide excitatory burst input to ∼70% of the GABAergic PG cells in a glomerulus triggering GABA release (Aungst et al., 2003; Hayar et al., 2004a, 2005; Shao et al., 2008). This rhythmic, spontaneous ET cell drive on PG cells generates a level of extracellular GABA, which presynaptically inhibits sensory inputs via GABAB receptors on ON terminals (Aroniadou-Anderjaska et al., 2000; Wachowiak et al., 2005). This presynaptic inhibition appears to have a significant tonic component because of the steady pool of released GABA and the relatively slow-onset, slow-offset GABAB receptors on the ON terminals (Aungst et al., 2003; Pírez and Wachowiak, 2008) (Z. Shao, A. C. Puche, E. Kiyokage, G. Szabo, M. T. Shipley, unpublished observations). In contrast, GABA released from PG cells by spontaneous and ON-evoked ET bursts activates GABAA receptors on the apical dendrites of MT cells. Because GABAA receptors are rapid, postsynaptic inhibition may be more strongly influenced by phasic sensory inputs than presynaptic inhibition. The ET cell is the key player in both presynaptic and postsynaptic inhibition, providing both spontaneous and sensory driven input to the majority of the glomerular inhibitory interneurons. Its remarkable capacity to amplify and potently summate sensory inputs makes this cell the gatekeeper to glomerular network operations.
The glomerular network appears to be optimally organized to impose brief temporal windows within which MT cells can be excited by rhythmic ON inputs. Odors are sampled by sniffing. During a sniff, activity in olfactory nerve terminals increases during a temporal window with a rise time of ∼100 ms (Spors et al., 2006; Verhagen et al., 2007). ET cells receive asynchronous monosynaptic ON inputs throughout this window. The amplification of EPSP duration mediated by ET cell intrinsic conductances is ideally suited to provide strong temporal summation of asynchronous sensory synaptic inputs during sniffing. Asynchronous sensory inputs are thus converted to all-or-none ET cell bursts, which in turn drive PG cells to generate presynaptic and postsynaptic intraglomerular inhibition. Higher-frequency sniffing attenuates presynaptic ON inputs more than does lower-frequency sniffing (Verhagen et al., 2007; Pírez and Wachowiak, 2008). Interestingly, temporal summation of ON inputs by ET cells would amplify higher-frequency inputs more than lower-frequency inputs. Postsynaptic summation might thus compensate for presynaptic attenuation in an inverse frequency-dependent manner; this might optimize postsynaptic inhibition across a range of sniff frequencies. Thus, the same intrinsic conductances that regulate spontaneous rhythmical bursting of ET cells actively shape their responses to excitatory and inhibitory inputs in ways that significantly impact intraglomerular inhibitory circuits that regulate MT cell output to olfactory cortex.
Feedback inhibitory synaptic inputs to ET cells can reduce the strength of the excitatory burst. However, the present results showed that inhibitory inputs arriving between bursts are transformed to excitatory rebound responses. This would reset burst frequency and could function to synchronize bursting among ET cells. The transformation of inhibitory inputs to postsynaptic excitation may also impact interglomerular network operations. Recent findings indicate that some DAergic/GABAergic PG cells have dendrites that extend to multiple distant glomeruli (Kiyokage et al., 2007; Kosaka and Kosaka, 2007; Pírez and Wachowiak, 2008). If these interglomerular inhibitory connections target ET cells, they could trigger Ih-dependent, rebound excitation. Rebound excitation of ET cells would cause glutamatergic excitation of PG cells resulting in the release of GABA, which inhibits MT cells. Consistent with this intriguing possibility, interglomerular inhibition of MT cells is eliminated by GABAA antagonists, as expected, and also by blockers of glutamatergic transmission (Aungst et al., 2003). Thus, IPSP-triggered, Ih-dependent rebound excitation of ET cells could amplify interglomerular inhibition of MT cells.
Footnotes
This work was supported by National Institutes of Health Grant DC005676. We thank Dr. Scott M. Thompson and Dr. Fu-Sun Lo for critical comments on this manuscript.
References
- Andreasen M, Lambert JD. Somatic amplification of distally generated subthreshold EPSPs in rat hippocampal pyramidal neurones. J Physiol. 1999;519:85–100. doi: 10.1111/j.1469-7793.1999.0085o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo K, London M, Christensen SR, Häusser M. Local and global effects of I(h) distribution in dendrites of mammalian neurons. J Neurosci. 2007;27:8643–8653. doi: 10.1523/JNEUROSCI.5284-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Zhou FM, Priest CA, Ennis M, Shipley MT. Tonic and synaptically evoked presynaptic inhibition of sensory input to the rat olfactory bulb via GABA(B) heteroreceptors. J Neurophysiol. 2000;84:1194–1203. doi: 10.1152/jn.2000.84.3.1194. [DOI] [PubMed] [Google Scholar]
- Aungst JL, Heyward PM, Puche AC, Karnup SV, Hayar A, Szabo G, Shipley MT. Centre-surround inhibition among olfactory bulb glomeruli. Nature. 2003;426:623–629. doi: 10.1038/nature02185. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Rosahl TW, Brussaard AB. Neonatal development of the rat visual cortex: synaptic function of GABAA receptor alpha subunits. J Physiol. 2002;545:169–181. doi: 10.1113/jphysiol.2002.026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisz RA, Fortin G, Zieglgänsberger W. Voltage dependence of excitatory postsynaptic potentials of rat neocortical neurons. J Neurophysiol. 1991;65:371–382. doi: 10.1152/jn.1991.65.2.371. [DOI] [PubMed] [Google Scholar]
- Ennis M, Zhou FM, Ciombor KJ, Aroniadou-Anderjaska V, Hayar A, Borrelli E, Zimmer LA, Margolis F, Shipley MT. Dopamine D2 receptor-mediated presynaptic inhibition of olfactory nerve terminals. J Neurophysiol. 2001;86:2986–2997. doi: 10.1152/jn.2001.86.6.2986. [DOI] [PubMed] [Google Scholar]
- Feinstein P, Mombaerts P. A contextual model for axonal sorting into glomeruli in the mouse olfactory system. Cell. 2004;117:817–831. doi: 10.1016/j.cell.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Gillessen T, Alzheimer C. Amplification of EPSPs by low Ni(2+)- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Neurophysiol. 1997;77:1639–1643. doi: 10.1152/jn.1997.77.3.1639. [DOI] [PubMed] [Google Scholar]
- González-Burgos G, Barrionuevo G. Voltage-gated sodium channels shape subthreshold EPSPs in layer 5 pyramidal neurons from rat prefrontal cortex. J Neurophysiol. 2001;86:1671–1684. doi: 10.1152/jn.2001.86.4.1671. [DOI] [PubMed] [Google Scholar]
- Hayar A, Ennis M. Endogenous GABA and glutamate finely tune the bursting of olfactory bulb external tufted cells. J Neurophysiol. 2007;98:1052–1056. doi: 10.1152/jn.01214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M, Shipley MT. External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci. 2004a;24:6676–6685. doi: 10.1523/JNEUROSCI.1367-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Shipley MT, Ennis M. Olfactory bulb glomeruli: external tufted cells intrinsically burst at theta frequency and are entrained by patterned olfactory input. J Neurosci. 2004b;24:1190–1199. doi: 10.1523/JNEUROSCI.4714-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Shipley MT, Ennis M. Olfactory bulb external tufted cells are synchronized by multiple intraglomerular mechanisms. J Neurosci. 2005;25:8197–8208. doi: 10.1523/JNEUROSCI.2374-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack JB, Doble D, Tsien RW. Electrical current flow in excitable cells. Oxford: Oxford UP; 1983. [Google Scholar]
- Karnup S, Stelzer A. Temporal overlap of excitatory and inhibitory afferent input in guinea-pig CA1 pyramidal cells. J Physiol. 1999;516:485–504. doi: 10.1111/j.1469-7793.1999.0485v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokage E, Pan Y, Shao Z, Szabo G, Kobayashi K, Puche AC, Shipley MT. Dopaminergic periglomerular cells form novel multiglomerular circuits. Soc Neurosci Abs. 2007;37:277–15. [Google Scholar]
- Kosaka T, Kosaka K. Tyrosine hydroxylase-positive GABAergic juxtaglomerular neurons are the main source of the interglomerular connections in the mouse main olfactory bulb. Neurosci Res. 2007;60:349–354. doi: 10.1016/j.neures.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C. Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol. 1996;76:2181–2191. doi: 10.1152/jn.1996.76.4.2181. [DOI] [PubMed] [Google Scholar]
- Liu S, Shipley MT. Multiple conductances cooperatively regulate spontaneous bursting in mouse olfactory bulb external tufted cells. J Neurosci. 2008;28:1625–1639. doi: 10.1523/JNEUROSCI.3906-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Sakmann B. Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proc Natl Acad Sci U S A. 1994;91:5207–5211. doi: 10.1073/pnas.91.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science. 2002;295:143–147. doi: 10.1126/science.1064285. [DOI] [PubMed] [Google Scholar]
- Mombaerts P. Axonal wiring in the mouse olfactory system. Annu Rev Cell Dev Biol. 2006;22:713–737. doi: 10.1146/annurev.cellbio.21.012804.093915. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Darcy DP, Isaacson JS. Intraglomerular inhibition: signaling mechanisms of an olfactory microcircuit. Nat Neurosci. 2005;8:354–364. doi: 10.1038/nn1403. [DOI] [PubMed] [Google Scholar]
- Panzanelli P, Perazzini AZ, Fritschy JM, Sassoè-Pognetto M. Heterogeneity of gamma-aminobutyric acid type A receptors in mitral and tufted cells of the rat main olfactory bulb. J Comp Neurol. 2005;484:121–131. doi: 10.1002/cne.20440. [DOI] [PubMed] [Google Scholar]
- Pírez N, Wachowiak M. In vivo modulation of sensory input to the olfactory bulb by tonic and activity-dependent presynaptic inhibition of receptor neurons. J Neurosci. 2008a;28:6360–6371. doi: 10.1523/JNEUROSCI.0793-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Modification of current transmitted from apical dendrite to soma by blockade of voltage- and Ca2+-dependent conductances in rat neocortical pyramidal neurons. J Neurophysiol. 1997;78:187–198. doi: 10.1152/jn.1997.78.1.187. [DOI] [PubMed] [Google Scholar]
- Southan AP, Morris NP, Stephens GJ, Robertson B. Hyperpolarization-activated currents in presynaptic terminals of mouse cerebellar basket cells. J Physiol. 2000;526:91–97. doi: 10.1111/j.1469-7793.2000.t01-1-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spors H, Wachowiak M, Cohen LB, Friedrich RW. Temporal dynamics and latency patterns of receptor neuron input to the olfactory bulb. J Neurosci. 2006;26:1247–1259. doi: 10.1523/JNEUROSCI.3100-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Urban NN, Henze DA, Barrionuevo G. Amplification of perforant-path EPSPs in CA3 pyramidal cells by LVA calcium and sodium channels. J Neurophysiol. 1998;80:1558–1561. doi: 10.1152/jn.1998.80.3.1558. [DOI] [PubMed] [Google Scholar]
- Verhagen JV, Wesson DW, Netoff TI, White JA, Wachowiak M. Sniffing controls an adaptive filter of sensory input to the olfactory bulb. Nat Neurosci. 2007;10:631–639. doi: 10.1038/nn1892. [DOI] [PubMed] [Google Scholar]
- Wachowiak M, Shipley MT. Coding and synaptic processing of sensory information in the glomerular layer of the olfactory bulb. Semin Cell Dev Biol. 2006;17:411–423. doi: 10.1016/j.semcdb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Wachowiak M, McGann JP, Heyward PM, Shao Z, Puche AC, Shipley MT. Inhibition [corrected] of olfactory receptor neuron input to olfactory bulb glomeruli mediated by suppression of presynaptic calcium influx. J Neurophysiol. 2005;94:2700–2712. doi: 10.1152/jn.00286.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic I(h) in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]