

Abstract
The speed, accuracy, efficiency, and cost-effectiveness of DNA sequencing have been improving continuously since the initial derivation of the technique in the mid-1970s. With the advent of massively parallel sequencing technologies, DNA sequencing costs have been dramatically reduced. No longer is it unthinkable to sequence hundreds or even thousands of genes in a single individual with a suspected genetic disease or complex disease predisposition. Along with the benefits offered by these technologies come a number of challenges that must be addressed before wide-scale sequencing becomes accepted medical practice. Molecular diagnosticians will need to become comfortable with, and gain confidence in, these new platforms, which are based on radically different technologies compared to the standard DNA sequencers in routine use today. Experience will determine whether these instruments are best applied to sequencing versus resequencing. Perhaps most importantly, along with increasing read lengths inevitably comes increased ascertainment of novel sequence variants of uncertain clinical significance, the postanalytical aspects of which could bog down the entire field. But despite these obstacles, and as a direct result of the promises these sequencing advances present, it will likely not be long before next-generation sequencing begins to make an impact in molecular medicine. In this review, technical issues are discussed, in addition to the practical considerations that will need to be addressed as advances push toward personal genome sequencing.
The speed, accuracy, efficiency, and cost-effectiveness of DNA sequencing have been improving continuously since the initial derivation of the technique by Maxam and Gilbert1 and Sanger et al.2 Cumbersome chemical methods gave way to enzymatic procedures, and manual techniques were replaced by even-faster automated instruments using capillary electrophoresis or high-density microarrays.3 More recently, the advent of massively parallel sequencing has made the $100,000 genome a reality, and further advances of the type discussed here4,5,6 demonstrate that the $1000 genome is not far away.7
The impact that these “next-generation” sequencing innovations will have in clinical genetics will certainly be substantial. The low-scale, targeted gene/mutation analysis that currently dominates the clinical genetics field will ultimately be replaced by large-scale sequencing of entire disease gene pathways and networks, especially for the so-called complex disorders. Eventually, the perceived clinical benefit of whole-genome sequencing will outweigh the cost of the procedure, allowing for these tests to be performed on a routine basis for diagnostic purposes, or perhaps in the form of a screening program that could be used to guide personalized medical treatments throughout the lifetime of the individual.
Indeed, technical advances in sequencing have been compounding at such a pace that keeping up may be difficult even for those well-versed in molecular biology, let alone those who are the more clinically based end-users of the technology. This review is intended as a current snapshot of the state of the art, with emphasis on which of the available “next-generation” technologies are most amenable and appropriate for clinical diagnostic use. Technical issues are discussed, in addition to such practical considerations as the ethical challenges that will need to be addressed as technological advances push toward personal genome sequencing.
“Next-Generation” Sequencing Platforms
Due to space constraints, this section provides only a cursory explanation of the technical aspects of the commercially available, next-generation sequencing platforms. For more detailed discussions of these topics, interested readers are referred elsewhere.8,9,10,11,12
The 454 Genome Sequencer 20 (454 Life Sciences, Roche Applied Sciences, Indianapolis, IN) was the first commercially available, next-generation sequencing instrument, with the current 454 FLX system representing the upgraded version. The 454 instrument carries out pyrosequencing13,14 reactions in parallel by partitioning hundreds of thousands of beads coated with homogeneous DNA fragments into individual wells of a PicoTiterPlate.15 Beads are generated by the immobilization of fragmented DNA at a concentration that, on average, results in the addition of one fragment per bead. The fragments are then independently amplified in an oil emulsion mixture that creates separate microreactors for each bead. As currently configured, the 454 FLX system can sequence 100 Mb DNA in 8 hours at an average read length of 250 bp (Table 1). Soon-to-be-released reagent and software upgrades are expected to increase sequencing output at least fivefold and read length almost twofold. The raw accuracy per nucleotide sequenced on the FLX system (99.5%), like all massively parallel sequencers, is low in relation to Sanger sequencing. However, extremely high confidence base calls can be achieved via oversampling and the resulting redundancy of the output data.
Table 1.
Commercially Available Next-Generation Sequency Platforms
| Sequencing systema | Estimated system cost | Consumable cost per single-end run (paired-end run) | Read length per single-end run (paired-end) | Gigabases sequenced per single-end run (paired-end) | Run time per single-end run (paired-end) | Raw accuracy |
|---|---|---|---|---|---|---|
| 454 Genome Sequencer FLX | $500,000b | n/ac | 250–300 bp (2 × 110 p)d | 0.1 Gbe (0.1 Gb) | 7.5 hours (7.5 hours) | 99.5% |
| Illumina Genome Analyzer | ∼$400,000 | $3000 (n/a)f | 36 bpg (2 × 36 bp) | 1.5 Gb (3.0 Gb) | 2.5 days (5 days) | >98.5% |
| ABI SOLiD™ System | $525,000 | $3390h ($4390) | 35 bp (2 × 25 bp)i | 3 Gbj (4 Gb) | 5–7 daysk (10 days) | 99.94% |
| Helicos Heliscope | n/a | n/a | 25–35 bpl | 7.5–10 Gb | 3–7 days | >99% |
All prices and specifications obtained via communication with company contacts except Helicos. Helicos specifications were obtained from Helicos (Cambridge, MA).
Price includes a server to run the instrument. The server stores up to 50 instrument runs, including the raw image files.
Pricing unavailable due to pricing variability from country to country.
110-bp tags separated by 3-kb genomic spacing.
In late 2008, 454 is expected to launch reagent and software upgrades that will extend read lengths to 400–500 bp and result in a minimum of 500 Mb sequenced per run.
Pricing for paired-end sequencing not yet released.
36-bp reads standard; system enables up to 50-bp reads.
US list prices per full slide run. The SOLiD™ System can accommodate two full slides per run.
Mate pair insert size is variable from 0.6 kb to 10 kb.
Mappable data per two slide run. This is not the raw data, but the useable data defined by the amount of data that uniquely maps to a reference sequence. If the sequence is not unique or does not map, regardless of the quality of the read, it is not, by definition, mappable.
Run time for two full slides.
Read length with the optimal throughput. Longer read lengths are possible, but reduce overall throughput.
The Illumina Genome Analyzer (Illumina Inc., San Diego, CA) can sequence 600 Mb DNA per day, although the tradeoff for this increased sequencing depth is that the reads are quite short, approximately 36 bp in length (Table 1). The Illumina sequencer achieves parallelization by the in situ amplification of DNA fragments immobilized onto the flow cell of the instrument at a concentration that promotes a dense array of non-overlapping fragment colonies. Each fragment colony is then sequenced one base at a time by the cyclical addition of fluorescently labeled nucleotides that are conjugated with a reversible terminator.
The Applied Biosystems SOLiD System (Foster City, CA) sequences by multiple cycles of hybridization and ligation.16 The sequencing reaction is initiated via the ligation of a universal anchoring primer to an 8-mer sequence derived from a population of fluorescently labeled 8-mers. After ligation of the first 8-mer, the emitted fluorescence is read followed by cleavage of the three downstream universal bases and another cycle of ligation. The ability of DNA ligase to discriminate between populations of fluorescently labeled oligonucleotides facilitates the sequencing reactions that generate approximately 500 Mb of sequence per day and read lengths up to 35 bp in length (Table 1). The main advantage of the SOLiD technology is that each base is interrogated twice, resulting in very accurate raw reads (>99.9%) that require a lower amount of oversampling to reach a threshold value of confidence for base calling.
Like the Illumina platform, the Helicos Heliscope (Cambridge, MA) immobilizes fragments on a flow cell at dilute concentrations. However, the unamplified fragments are then directly sequenced by monitoring the incorporation of nucleotides by a single polymerase.17 Labeled nucleotides are independently added to the reaction and, after the removal of unincorporated bases, the emitted fluorescence (or lack thereof) is determined for each individual fragment. The fluorescent group is then cleaved off the fragment and the other three nucleotides are added iteratively, assaying for incorporation after each step. The process can produce reads up to 55 bp in length, although optimal coverage is obtained with shorter reads (Table 1). The Heliscope is reportedly able to sequence up to 2000 Mb per day, making it the instrument with the highest throughput on the market.
Genome Enrichment
Although technological innovations in DNA sequencing may soon allow for whole-genome sequencing to become standard medical practice, resequencing selected portions of the genome remains the most prudent way forward at this time in terms of both cost and clinical utility. Targeted sequencing requires substantially less throughput per sample than genomic sequencing, but due to the large “appetite” for templates of massively parallel sequencers, unprecedented demands are placed on the upfront methods of sample preparation.
PCR amplification using a high-fidelity thermostable polymerase has proven to be a reliable method for isolating genomic areas of interest in preparation for Sanger sequencing. However, given its limited multiplex capability, traditional PCR is an impractical method of genomic enrichment for next-generation sequencers. One method developed to achieve further PCR multiplexing is to prime with oligonucleotides immobilized onto a solid surface (Figure 1A), thus reducing the aberrant reactions from undesired primer combinations that normally plague multiplex PCR.18 Furthermore, by including common sequences on the 5′ ends of the immobilized primers, subsequent rounds of amplification can be performed in solution using these universal primer sequences to amplify all products. This so-called megaplex PCR is capable of greatly increasing the multiplex capacity of PCR; however, it should be noted that currently only 80% of targets are captured per reaction.
Figure 1.
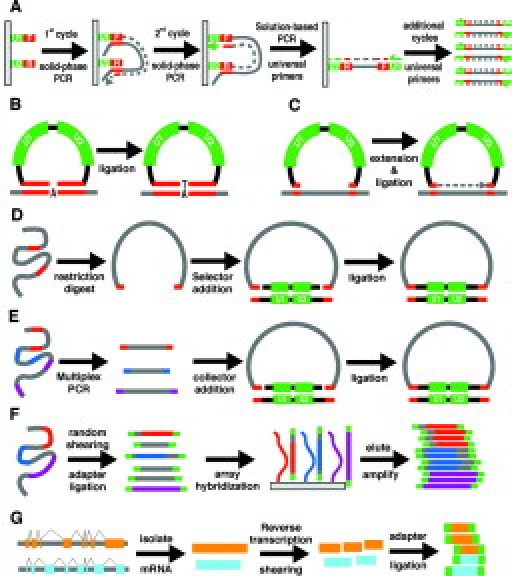
Genome Enrichment Strategies. A: Megaplex PCR. Surface bound primers are extended in the first and second PCR cycle. Additional cycles are performed using universal primers in solution. U1 and U2 (green) are universal primer sequences. F and R (red) are unique primer sequences. B: Molecular inversion probe circularization. The 5′ and 3′ ends (red) of the MIP probe (black) hybridize to complementary genomic DNA, leaving a 1-bp gap that is filled in by DNA ligase. U1 and U2 (green) are universal primer sequences within the MIP probe. C: Connector inversion probe circularization. The 5′ and 3′ ends (red) of the CIPer probe (black) hybridize to complementary genomic DNA (gray), leaving a sizeable gap that is extended by DNA polymerase and ligated to the 3′ arm with DNA ligase. U1 and U2 (green) are universal primer sequences within the CIPer probe. D: Selector probe circularization. Genomic DNA (gray) is fragmented with a restriction endonuclease. Selector probes (black) with single-stranded overhangs (red) are ligated to the genomic restriction fragment ends (red) complementary to the selector probe overhangs. U1 and U2 (green) are universal primer sequences within the selector probe. E: Gene-collector circularization. A multiplex PCR is performed generating amplicons with unique sequence ends. Gene-collector probes (black) with single-stranded overhangs (red) are ligated to single-stranded overhangs (red) complementary to ends generated from the PCR reaction. F: Microarray pull-down assay. Genomic DNA (gray, with regions of interest in red, blue, and purple) is randomly sheared then ligated to universal adapter sequences (green). Adapter-ligated DNA is then hybridized to the array, eluted, and amplified with universal primers. G: cDNA sequencing. mRNA is isolated, converted to cDNA, sheared, ligated with adapters (green), and sequenced.
Several other methods have sought to increase the multiplex capability of PCR by selecting for productive reactions following circularization.19,20,21,22,23,24 These methods are modified versions of the padlock and the molecular inversion probe (MIP) methods designed to capture up to tens of thousands of single nucleotide polymorphisms (SNPs) in a single reaction.25,26,27,28 In the MIP protocol, the termini of the probes duplex with complementary genomic sequences, thus forming a circle with a 1-bp gap that corresponds to the polymorphic base (Figure 1B). DNA ligase and mononucleotides (dATP, dCTP, dGTP, or dTTP) are then added to each of the four tubes containing the reaction mixture. For each probe, circle formation is accomplished in the tube containing the free nucleotide that is complementary to the genomic base that spans the gap. Circularized probes are then enriched by treatment with exonucleases that specifically digest linear DNA, followed by an amplification reaction. The strategy developed by Akhras et al,19 which utilizes a so-called connector inversion probe (CIPer), is a simple modification of the MIP protocol whereby all dNTPs are added to one tube and circles are formed after 3′ extension to the 5′ end of the probe approximately 100 bp downstream (Figure 1C).
Another method of circle formation is initiated by digesting genomic DNA with an appropriate restriction enzyme mixture. “Selector” probes, which hybridize to both the 5′ and 3′ ends of specific digestion products, facilitate the circularization of genomic fragments of interest. Selector probe binding brings together the ends of the targeted fragment, allowing DNA ligase to generate a circularized product via the formation of a covalent bond between the fragment ends (Figure 1D).20,21,24
The gene-collector procedure is yet another method that utilizes circularization to isolate regions of interest in multiplex.22 Following a high-complexity multiplex reaction, the desired amplicons are enriched from the mixture using a gene-collector probe complementary to specific PCR primer ends, allowing for the circularization of targeted fragments (Figure 1E). The main benefit of the gene-collector method is the high uniformity obtained among the targeted products, requiring less overall sequencing depth to obtain acceptable sequencing coverage for all targets.
The scalability of the gene-collector and selector technique has been demonstrated on a somewhat limited scale, while the scalability of the CIPer technique has been more thoroughly tested. Fredriksson et al22 isolated 90% of the 167 exons that they were attempting to capture using the gene-collector method, while Dahl et al21 detected 93% of target sequences from 177 exons using 503 selector probes, 83 of which were redesigned after the original probe yielded no product. Porreca et al23 used a population of 55,000 70-mer oligonucleotides to capture more than 10,000 exons in the human genome using a CIPer-like procedure. Although the number of exons captured by Porreca and colleagues23 is quite impressive, the fact that less than 20% of the desired regions of interest were captured illustrates that, although promising, these circularization techniques will require a fair amount of optimization to function on a genome-wide scale.
For each of the circularization enrichment techniques, long oligonucleotides are needed to capture each region of interest, making the cost per reaction potentially prohibitive as the number of targets becomes substantial. However, due to the multiplicative nature of these capture techniques, a population-based oligo synthesis method would represent a substantial cost reduction from conventional oligo production that requires independent synthesis and pooling. The synthesis of thousands of oligos in situ on a microarray, followed by cleavage and release of the probes from the slide,23 is one such method. Provided that each probe is synthesized with universal primer ends that can be removed following amplification, a renewable source of probes can be created in this manner.
An alternative to circularization enrichment techniques are nucleic acid pull-down assays in which targeted regions of the genome are selected by direct hybridization to oligonucleotide microarray probes.29,30,31 The genome is first randomly fragmented and universal adapter sequences are ligated to the ends of the fragments (Figure 1F). The resulting mixture is then hybridized to a microarray containing long oligonucleotide probes that correspond to sequences from the regions of interest. The array is then denatured and free nucleic acids are enriched by PCR using adapter sequences. Oligonucleotide microarray pull-down assays appear to be amenable to large-scale capture of regions of interest. Okou et al31 reproducibly obtained a 99% base-calling rate using resequencing arrays that sampled 50 kb of the 304-kb region targeted for pull-down. Albert et al29 were able to enrich the sequence from 6726 exons, corresponding to 5 Mb DNA, approximately 400-fold. On average, 80% of sequence reads that aligned to the human genome mapped to targeted exons and more than 90% of targeted nucleotides were contained in at least one read. Hodges et al30 scaled up the pull-down assay even further, targeting 43 Mb of sequence that corresponded to the 200,000 protein-coding exons in the Refseq database.32 Oligonucleotide capture probes were divided among six arrays, each tiling 5 to 6 Mb of exon sequence. An average genomic enrichment of 323-fold was achieved with 55% to 85% of aligned sequence reads that corresponded to selected targets, numbers similar to those determined by Albert et al29 In addition, Hodges and colleagues30 demonstrated that genomic DNA fragmented to an average length of 500 bp can be enriched almost three times as effectively as fragments 100 to 200 bp in length. However, the smaller fragments resulted in better sequence coverage as a result of increased sequencing efficiency, presumably due to a size bias inherent to the Illumina instrument.
A more customary method of enriching for functional portions of the genome is to directly sequence cDNA transcripts (Figure 1G). High-throughput sequencing of cDNAs can be a viable alternative to expression arrays since the difference in sequence read counts between one gene and another should reflect the relative difference in transcript levels present in the cell before extraction. This type of analysis will prove most useful in diseases such as cancer in which mutation scanning and gene expression profiling are extremely powerful predictors of disease progression33 and, via this technology, can be consolidated on a single platform. However, even with the highest throughput sequencers, it may take multiple runs to gain an accurate representation of the gene expression landscape. Alternatively, adequate transcript sampling can be efficiently obtained by sacrificing sequence content for increased transcript read density using serial analysis of gene expression (SAGE) or a related technique (i.e., LongSAGE, DeepSAGE).34,35,36 SAGE sequence alignment is also greatly simplified relative to cDNA sequencing that does not align well to genomic DNA and is not well-served by transcript sequence databases that are incomplete.
While most of the aforementioned enrichment procedures are still being optimized, each is not without its limitations. Selectors rely on restriction sites flanking targeted regions, while CIPers have difficulty capturing regions of high GC content.23 Also, the gene-collector method might be problematic on a very large scale since competition among primers can decrease the yield of some desired amplification products. Finally, the microarray enrichment method is less specific to non-unique portions of the genome than the other techniques that are capable of targeting the distinct sequences flanking these regions. Depending on the structure of the assay being performed, one might prefer one method to another. For example, the array pull-down assay would likely be the preferable method when sequencing thousands of targets, especially considering that the microarrays used for these procedures can be used twice without a discernable loss in quality of enriched sample.31 However, if one would like to isolate a small number of genes from many individuals, a circularization technique or megaplex PCR may be cheaper and faster. It is also worth considering that the genomic pull-down method could be more applicable on a moderate to small scale if a solution-based method were developed.
Tagging Methods
While next-generation sequencing platforms can easily sequence thousands of targets isolated from one sample, these instruments are unable to differentiate matching targets isolated from multiple samples. However, there are a number of work-around methods that address this predicament to different degrees. One solution is to mix separate tests with one another. For example, if genes A, B, and C are sequenced in patient #1, genes D, E, and F in patient #2, and genes G, H, and I in patient #3, the patient samples may all be combined because the target genes to be sequenced are unique and known for each patient. Another alternative is to rely on physical divisions built into the sequencers themselves that provide sequence data independent of each other. Illumina's Genome Analyzer has eight such independent channels, while the ABI SOLiD system has eight channels per slide and can run two slides per run. The Heliscope Sequencer also runs two slides per run, but with each flow cell containing 25 independent channels. The 454 Genome Sequencer FLX PicoTiterPlate plate can be partitioned into two, four, or 16 regions, but doing so reduces overall coverage. However, the long sequencing reads produced by the 454 instrument allows for efficient addition of “DNA barcode tags,” unique nucleotide signatures that allow one to mark and track individual samples. Barcodes can be added to the ends of fragments by either tagging during PCR37 or following the isolation of targeted sequences.38
Although barcodes can be used in a similar fashion with the higher throughput sequencers, the short read lengths produced by these instruments require that the tags be kept short or else the resulting genomic sequence may be difficult to align. Of course, this is considerably less of a problem if the reference sequence is confined to only a small portion of the genome or if paired-end sequencing is used to, in effect, increase the sequencing read length. Paired-end sequencing is a strategy whereby both ends of immobilized fragments are sequenced, effectively doubling the read length.39 An added benefit of paired-end sequencing is that it can allow for the identification of translocations in fragments containing breakpoints.40 Mate-pair sequencing is a modification of the pair-end strategy whereby mate pairs are generated by ligating the ends of size-selected genomic fragments to a common linker. Each fragment end is then cleaved at a known distance from the linker and paired-end sequenced. Since the expected distance between paired ends is known, deviations from this value allows for the identification of sequence copy number variations in the region.41
Additional Technical Considerations
Because the cost of a massively parallel sequencing system can be quite substantial, the benefits and limitations of each instrument should be carefully considered to determine which platform is best suited for a particular laboratory's needs. Table 1 lists the current price for each instrument along with reagent costs per full sequencing run and key sequencing specifications. It should be kept in mind that the values in Table 1 will change rapidly as market forces act and as systems are upgraded to improve performance.
Additional factors to consider with these instruments are data management and storage. The 454 FLX sequencer produces a modest amount of data per run (∼18 gigabytes of data, including all raw images) relative to the higher throughput sequencing instruments. In contrast, a lab running a higher throughput instrument 2 to 3 times per week could be expected to spend in excess of $100,000 per year in hardware alone to store, access, and back up the 100+ terabytes of total data generated. In such circumstances, it is much more cost-effective to save the DNA samples, which can be resequenced if the patient results are ever questioned, rather than the processed image files that take up the bulk of the data storage space. It is not known whether this approach will conflict with CLIA ‘8842 and certain state and professional regulations that specify test results should be archived for at least 5 years, with some proposing that genetic test results be archived for 10 or 20 years.43
Although these sequencing systems are phenomenal in what they can do, it is also important to understand their limitations. With the exception of the 454 FLX instrument that produces reads averaging 250 bp in length, each of the currently available next-generation sequencing platforms produce sequence reads of very short length (Table 1). These short reads can be very difficult, if not impossible, to align if the read is repeated elsewhere in the genome or if it harbors even relatively modest variations from the reference sequence. In part, these issues will be easier to address as better alignment programs are developed [such as Maq (Mapping and Assembly with Quality); http://maq.sourceforge.net/].44 However, increasing the read length, by either paired-end sequencing runs or future improvements to the process, still represents the most ideal solution to the alignment problem.
Another issue to consider, particularly when scanning for mutations in recessively inherited disorders, is that the determination of whether intragenic mutations are in cis or trans can be extremely difficult. Traditional Sanger sequencing can be used to obtain longer sequencing reads, but this technique will only work if the variants in question are very close together. Targeted sequencing in parents could help resolve genotypic phasing, but parents are not always available and may sometimes be unwilling to undergo genetic testing. Alternatively, for some extensively studied genes (e.g., CFTR), the haplotypes of the major mutations are generally known, which can help to rule out cis-inheritance. Unfortunately, this information will not usually be known for less studied genes and rare diseases, which paradoxically have the most to benefit from these next-generation methods.
Clinical Potential and Utility
At time of this writing, next-generation sequencing approaches are primarily being used in the research setting for either rapid whole-genome sequencing or specific region resequencing to aid gene mapping and population genetics studies. Indeed, there has been little call for whole-gene sequencing, much less for whole-genome sequencing, in the clinical setting thus far. However, as sequencing costs continue to fall and as the underlying basis of polygenic disorders becomes clear, large-scale sequencing will become a more attractive option. One can envision, for example, the brute-force sequencing of hundreds of genes involved in drug metabolism as a means toward “next-generation” pharmacogenetic testing, beyond the individual CYP450 genes and handful of SNPs that are considered today. Also, we can speculate that someday we will be called on to sequence hundreds of genes (once we know them) involved in complex/multifactorial diseases, such as hypertension or atherosclerosis.
For the present, however, the ability to interpret the data presents a much more imposing obstacle to the clinical utility of next-generation sequencing than does the ability to obtain the three billion bases of human genome sequence.45 Certain large genes with great heterogeneity in their mutations across the affected patient population have been examined extensively at the level of whole-gene sequencing, the most prominent to date being BRCA1/BRCA2 and CFTR. This level of scrutiny is justified in the case of the BRCA genes because hereditary breast/ovarian cancer is dominantly inherited, and failing to detect even a single rare mutation in a woman at risk could exclude her from potentially lifesaving interventions, such as heightened surveillance and prophylactic surgery. The indications are somewhat less clear in the case of CFTR since cystic fibrosis is recessive and a much more limited panel of relatively common mutations, detected by allele-specific targeting rather than sequencing, is the accepted standard for population carrier screening.46 However, full-gene sequencing may be useful for molecular confirmation of atypical cases that are not fully elucidated by the screening panel, or for determining both parental mutations in a timely fashion to allow for prenatal diagnosis in a subsequent pregnancy.47 The data accrued for CFTR and the BRCA genes thus far is both informative and humbling. In all three genes, there seems to be an almost limitless variety of mutations, many of them so rare that they are essentially “private,” that is, found in only a single family. Determining the penetrance, expression, and severity of such rare variants can be quite difficult or even impossible in the face of scant clinical or molecular data available on the patients in question. Sometimes, a missense change that was initially designated as a pathological mutation turns out, on further study, to be a benign variant.48 Indeed, given the nature of the genetic code, the physicochemical similarity of classes of amino acids, and negative evolutionary selection for changes that affect reproductive fitness, the number of benign variants (here designated as polymorphisms) is likely to far exceed the number of disease-causing mutations in most genes. Even for genes as extensively studied as BRCA1 and BRCA2, undoubtedly the most thoroughly sequenced genes in the human genome, previously unseen variants continue to be detected frequently. At one reference laboratory alone (Myriad Genetics, Salt Lake City, UT), these two genes have been sequenced completely in over 150,000 people. In the process, upwards of 10,000 deleterious mutations and missense variants of negligible or uncertain clinical significance have been identified and recorded in a database49 (B. Ward, personal communication). Yet every week, 1% to 2% of patients currently being tested demonstrate missense variants not seen before (B. Ward, personal communication), and each of these must be carefully analyzed to attempt to assess its likely clinical effect before reporting out the result. While there are a number of deductive and informatics methods for making these assessments,50 in many cases it is simply impossible to draw any conclusion without extensive clinical follow-up of those individuals carrying the variants (Myriad maintains extensive tracking and correlation data, and will sometimes revise the clinical classification of a missense variant years after its first detection).
Since there is no reason to assume, a priori, that the BRCA genes are any more mutable or unstable than most other genes in the genome, these findings should give us pause when contemplating high-throughput clinical sequencing of many genes at once, or even the full genome. If such a service were offered on a large scale, we can expect thousands, perhaps millions, of novel variants to be unveiled in both affected patients and healthy individuals, potentially creating uncertainty and anxiety with no obvious clinical benefit or intervention to be offered. In fact, a term has been coined to define this constellation of spurious findings—the “incidentalome”51—and it has the very real potential to drown out genuine diagnostic findings in the genome itself.
In an effort to expand our ability to decipher interindividual variation in the human genome, several research endeavors have been launched with the intent to sequence the genomes of more than a thousand individuals.52,53,54 Although these projects will aid in preemptively identifying benign variants, further efforts beyond discovery will continue to be essential for determining which variants are predictive of patient health.
Challenges for Test Reporting
Reporting of complex molecular genetic tests has been a subject of much attention in view of the life-changing nature of the results, the basing of irreversible clinical decisions on residual risks rather than absolutes, and the lack of sophistication in this field among both patients and providers. This is true even when testing for just a single, common mutation such as factor V Leiden.55 These challenges are exacerbated when reporting results of full-gene sequencing, and we can thus expect them to be orders of magnitude more problematic when reporting partial or full genome sequencing using the next-generation instruments. The challenges of dealing with such huge masses of sequence data, as discussed above, are difficult enough in a research laboratory, but will be absolutely unprecedented when applied to real patients in a clinical setting. How much of the sequence data should be reported to the ordering physician as opposed to stored in the clinical laboratory or hospital information system? How can such large data sets even be handled without overloading the system? How can raw sequence information be made understandable to ordering physicians and patients? The American College of Medical Genetics has established guidelines for the testing and reporting of ultra-rare genetic disorders56,57; since those tests are almost uniformly performed by gene sequencing, many of the recommendations would also pertain to next-generation sequencing, especially considering that most of the variants detected by these methods will be “rare” or novel. Special considerations include citation in the report of the “normal” reference sequence used, deduced effect of any detected change at both the DNA and protein (and RNA, if involving a splice site) levels, and use of standard mutation nomenclature to document results. Missense variants of uncertain significance should be assessed as thoroughly as possible and a conclusion made as to whether they are more likely to be benign, deleterious, or indeterminate. At least in the early stages of testing a gene or genomic region, it is recommended that even apparently benign variants be reported for the record and to assist in genetic epidemiology studies. Later, after extensive study of a locus has established certain common, recurring polymorphisms that are proven to be of no clinical significance, they need no longer be mentioned specifically in the test report.58 However, as noted above, at least for the foreseeable future, any next-generation sequencing program is likely to reveal far more novel variants of uncertain significance than clear-cut mutations, making the test reporting immensely complicated.
Ethical Considerations
The science of genetics has always been fraught with the potential for abuse and discrimination, both real and imagined. Eugenics movements have arisen at one time or another in almost every country of the world, their implementation ranging from controlled marriages to mandatory sterilizations to outright genocide. With the advent of molecular genetics, the potential for new and more insidious eugenics practices has raised widespread concerns, even influencing both state and federal legislation in the form of genetic nondiscrimination laws designed to protect individuals found to carry deleterious genetic markers and to hopefully lessen a major barrier to genetic testing among the population. These fears to date have largely centered on predictive testing for defined single-gene disorders such as Huntington disease and familial cancers. Now that next-generation sequencing technology is upon us, opening the way for whole-genome analysis on individual patients, the perceived risks and potential for harm are amplified many-fold over those genetic tests that examine just one gene at a time. For a patient confronting a Huntington disease test who consents to the analysis of just that single gene, it is highly unlikely that any unrelated findings will emerge from the targeted mutation testing technology (in that case, measurement of the length of the CAG repeat). With next-generation sequencing, on the other hand, thousands of genes or even the whole genome are addressed simultaneously. A patient requesting testing because of concern about one particular disease, or class of diseases, is quite likely to discover some other potential health threat relevant to an entirely different organ system—not to mention inevitable sequence variants of uncertain significance that can cause unnecessary anxiety.
The potential for harm resulting from these “incidental” findings can apply to any genome-scanning technique, and has already been considered, for example, in the context of high-density oligonucleotide microarrays.59 While many of the issues will be the same, there is one important difference: with a microarray, one can pick and choose which sequence variants to target or address, and any uncertain or undesired ones can be excluded from the array entirely. This is not the case with sequencing where there is no masking or ignoring each and every variant that is present within the range of the sequencing reactions. In the case of whole-genome sequencing, theoretically every deviation from “normal” (whatever that is) will be detected and presumably made known to the patient with all of its unattended risks and adversities. Of course, one could simply choose not to report certain findings, but this too is problematic, since a variant that appears unimportant today may, in a few years, be shown to be life-threatening, and failure to divulge it at a time when preventive measures could have been taken invites legal liability for the testing laboratory.
Perhaps the most prudent course to take at this time is that which we follow for all genetic testing: ample pre-test counseling and education, informed consent when applicable (especially for predictive genetic tests,60 a small category now, but one that almost all next-generation sequencing tests will encompass), maintenance of genetic privacy and confidentiality, and sensitivity to family, community, and ethnicity issues. At the same time, we must be wary of overly Draconian measures designed to protect patients but that end up having the effect of impeding patient access to the technology and in fact blocking its development before it reaches its full promise. As long as this balance is struck, there is no reason why next-generation sequencing cannot transition from the research setting to the clinical setting in the same way that so many other molecular biology innovations have, from Southern blot to PCR, despite the formidable technical and ethical hurdles.
References
- 1.Maxam A, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanger F, Nicklen S, Coulson A. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meldrum D. Automation for genomics, part two: sequencers, microarrays, and future trends. Genome Res. 2000;10:1288–1303. doi: 10.1101/gr.157400. [DOI] [PubMed] [Google Scholar]
- 4.Braslavsky I, Hebert B, Kartalov E, Quake S. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levene M, Korlach J, Turner S, Foquet M, Craighead H, Webb W. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 6.Winters-Hilt S, Vercoutere W, DeGuzman V, Deamer D, Akeson M, Haussler D. Highly accurate classification of Watson-Crick basepairs on termini of single DNA molecules. Biophys J. 2003;84:967–976. doi: 10.1016/S0006-3495(03)74913-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Service R. Gene sequencing. The race for the $1000 genome. Science. 2006;311:1544–1546. doi: 10.1126/science.311.5767.1544. [DOI] [PubMed] [Google Scholar]
- 8.Hudson M. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol Ecol Res. 2008;8:3–17. doi: 10.1111/j.1471-8286.2007.02019.x. [DOI] [PubMed] [Google Scholar]
- 9.Hutchison C. DNA sequencing: bench to bedside and beyond. Nucleic Acids Res. 2007;35:6227–6237. doi: 10.1093/nar/gkm688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Käller M, Lundeberg J, Ahmadian A. Arrayed identification of DNA signatures. Expert Rev Mol Diagn. 2007;7:65–76. doi: 10.1586/14737159.7.1.65. [DOI] [PubMed] [Google Scholar]
- 11.Mardis E. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Shendure J, Mitra R, Varma C, Church G. Advanced sequencing technologies: methods and goals. Nat Rev Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- 13.Gharizadeh B, Herman Z, Eason R, Jejelowo O, Pourmand N. Large-scale pyrosequencing of synthetic DNA: a comparison with results from Sanger dideoxy sequencing. Electrophoresis. 2006;27:3042–3047. doi: 10.1002/elps.200500834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11:3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- 15.Margulies M, Egholm M, Altman W, Attiya S, Bader J, Bemben L, Berka J, Braverman M, Chen Y, Chen Z, Dewell S, Du L, Fierro J, Gomes X, Godwin B, He W, Helgesen S, Ho C, Irzyk G, Jando S, Alenquer M, Jarvie T, Jirage K, Kim J, Knight J, Lanza J, Leamon J, Lefkowitz S, Lei M, Li J, Lohman K, Lu H, Makhijani V, McDade K, McKenna M, Myers E, Nickerson E, Nobile J, Plant R, Puc B, Ronan M, Roth G, Sarkis G, Simons J, Simpson J, Srinivasan M, Tartaro K, Tomasz A, Vogt K, Volkmer G, Wang S, Wang Y, Weiner M, Yu P, Begley R, Rothberg J. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shendure J, Porreca G, Reppas N, Lin X, McCutcheon J, Rosenbaum A, Wang M, Zhang K, Mitra R, Church G. Accurate multiplex colony sequencing of an evolved bacterial genome. Science. 2005;309:1728–1732. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 17.Harris T, Buzby P, Babcock H, Beer E, Bowers J, Braslavsky I, Causey M, Colonell J, DiMeo J, Efcavitch J, Giladi E, Gill J, Healy J, Jarosz M, Lapen D, Moulton K, Quake S, Steinmann K, Thayer E, Tyurina A, Ward R, Weiss H, Xie Z. Single-molecule DNA sequencing of a viral genome. Science. 2008;320:106–109. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- 18.Meuzelaar L, Lancaster O, Pasche J, Kopal G, Brookes A. MegaPlex PCR: a strategy for multiplex amplification. Nat Methods. 2007;4:835–837. doi: 10.1038/nmeth1091. [DOI] [PubMed] [Google Scholar]
- 19.Akhras M, Unemo M, Thiyagarajan S, Nyrén P, Davis R, Fire A, Pourmand N. Connector inversion probe technology: a powerful one-primer multiplex DNA amplification system for numerous scientific applications. PLoS ONE. 2007;2:e915. doi: 10.1371/journal.pone.0000915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahl F, Gullberg M, Stenberg J, Landegren U, Nilsson M. Multiplex amplification enabled by selective circularization of large sets of genomic DNA fragments. Nucleic Acids Res. 2005;33:e71. doi: 10.1093/nar/gni070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahl F, Stenberg J, Fredriksson S, Welch K, Zhang M, Nilsson M, Bicknell D, Bodmer W, Davis R, Ji H. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci USA. 2007;104:9387–9392. doi: 10.1073/pnas.0702165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredriksson S, Banér J, Dahl F, Chu A, Ji H, Welch K, Davis R. Multiplex amplification of all coding sequences within 10 cancer genes by Gene-Collector. Nucleic Acids Res. 2007;35:e47. doi: 10.1093/nar/gkm078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porreca G, Zhang K, Li J, Xie B, Austin D, Vassallo S, LeProust E, Peck B, Emig C, Dahl F, Gao Y, Church G, Shendure J. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 24.Stenberg J, Dahl F, Landegren U, Nilsson M. PieceMaker: selection of DNA fragments for selector-guided multiplex amplification. Nucleic Acids Res. 2005;33:e72. doi: 10.1093/nar/gni071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faruqi A, Hosono S, Driscoll M, Dean F, Alsmadi O, Bandaru R, Kumar G, Grimwade B, Zong Q, Sun Z, Du Y, Kingsmore S, Knott T, Lasken R. High-throughput genotyping of single nucleotide polymorphisms with rolling circle amplification. BMC Genomics. 2001;2:4. doi: 10.1186/1471-2164-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardenbol P, Banér J, Jain M, Nilsson M, Namsaraev E, Karlin-Neumann G, Fakhrai-Rad H, Ronaghi M, Willis T, Landegren U, Davis R. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nature Biotechnol. 2003;21:673–678. doi: 10.1038/nbt821. [DOI] [PubMed] [Google Scholar]
- 27.Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, Brundage T, Boudreau A, Chow S, Eberle J, Erbilgin A, Falkowski M, Fitzgerald R, Ghose S, Lartchouk O, Jain M, Karlin-Neumann G, Lu X, Miao X, Moore B, Moorhead M, Namsaraev E, Pasternak S, Prakash E, Tran K, Wang Z, Jones H, Davis R, Willis T, Gibbs R. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 2005;15:269–275. doi: 10.1101/gr.3185605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary B, Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2088. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- 29.Albert T, Molla M, Muzny D, Nazareth L, Wheeler D, Song X, Richmond T, Middle C, Rodesch M, Packard C, Weinstock G, Gibbs R. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 30.Hodges E, Xuan Z, Balija V, Kramer M, Molla M, Smith S, Middle C, Rodesch M, Albert T, Hannon G, McCombie R. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 31.Okou D, Steinberg K, Middle C, Cutler D, Albert T, Zwick M. Microarray-based genomic selection for high-throughput resequencing. Nat Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- 32.Pruitt K, Tatusova T, Maglott D. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sotiriou C, Piccart M. Taking gene-expression profiling to the clinic: When will molecular signatures become relevant to patient care? Nat Rev Cancer. 2007;7:545–553. doi: 10.1038/nrc2173. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen K, Høgh A, Emmersen J. DeepSAGE–digital transcriptomics with high sensitivity, simple experimental protocol and multiplexing of samples. Nucleic Acids Res. 2006;34:e133. doi: 10.1093/nar/gkl714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saha S, Sparks A, Rago C, Akmaev V, Wang C, Vogelstein B, Kinzler K, Velculescu V. Using the transcriptome to annotate the genome. Nature Biotechnol. 2002;20:508–512. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- 36.Velculescu V, Zhang L, Vogelstein B, Kinzler K. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 37.Binladen J, Gilbert M, Bollback J, Panitz F, Bendixen C, Nielsen R, Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer M, Stenzel U, Myles S, Prüfer K, Hofreiter M. Targeted high-throughput sequencing of tagged nucleic acid samples. Nucleic Acids Res. 2007;35:e97. doi: 10.1093/nar/gkm566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunn J, McCorkle S, Everett L, Anderson C. Paired-end genomic signature tags: a method for the functional analysis of genomes and epigenomes. Genet Eng (NY) 2007;28:159–173. doi: 10.1007/978-0-387-34504-8_9. [DOI] [PubMed] [Google Scholar]
- 40.Campbell P, Stephens P, Pleasance E, O'Meara S, Li H, Santarius T, Stebbings L, Leroy C, Edkins S, Hardy C, Teague J, Menzies A, Goodhead I, Turner D, Clee C, Quail M, Cox A, Brown C, Durbin R, Hurles M, Edwards P, Bignell G, Stratton M, Futreal P. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaughlin S, Peckham H, Zhang Z, Malek J, Sorenson J. Whole-genome resequencing with short reads: accurate mutation discovery with mate pairs and quality values. ASHG Annual Meeting 2007, 2620/F.
- 42.Centers for Disease Control Regulations for implementing Clinical Laboratory Improvement Amendments of 1988: a summary. MMWR. 1992;41(RR-2):1–17. [PubMed] [Google Scholar]
- 43.Schwartz M. Genetic testing and the clinical laboratory improvement amendments of 1988: present and future. Clin Chem. 1999;45:739–745. [PubMed] [Google Scholar]
- 44.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 45.Olson M. Human genetics: Dr. Watson's base pairs. Nature. 2008;452:819–820. doi: 10.1038/452819a. [DOI] [PubMed] [Google Scholar]
- 46.Grody W, Cutting G, Klinger K, Richards C, Watson M, Desnick R. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 2001;3:149–154. doi: 10.1097/00125817-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 47.McGinniss M, Chen C, Redman J, Buller A, Quan F, Peng M, Giusti R, Hantash F, Huang D, Sun W, Strom C. Extensive sequencing of the CFTR gene: lessons learned from the first 157 patient samples. Hum Genet. 2005;118:331–338. doi: 10.1007/s00439-005-0065-1. [DOI] [PubMed] [Google Scholar]
- 48.Claustres M, Altiéri J, Guittard C, Templin C, Chevalier-Porst F, Des Georges M. Are p.l148Tp.R74W and p.D1270N cystic fibrosis causing mutations? BMC Med Genet. 2004;5:19. doi: 10.1186/1471-2350-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Easton D, Deffenbaugh A, Pruss D, Frye C, Wenstrup R, Allen-Brady K, Tavtigian S, Monteiro A, Iversen E, Couch F, Goldgar D. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kazazina H, Boehm C, Seltzer W. Recommendations for standards for interpretation of sequence variations. Genet Med. 2000;2:302–303. [Google Scholar]
- 51.Kohane I, Masys D, Altman R. The incidentalome: a threat to genomic medicine. JAMA. 2006;296:212–215. doi: 10.1001/jama.296.2.212. [DOI] [PubMed] [Google Scholar]
- 52.Church G. Genomics: being well informed. Nature. 2007;449:627. [Google Scholar]
- 53.Hayden E. International genome project launched. Nature. 2008;451:378–379. doi: 10.1038/451378b. [DOI] [PubMed] [Google Scholar]
- 54.Qiu J, Hayden E. Genomics sizes up. Nature. 2008;451:234. doi: 10.1038/451234a. [DOI] [PubMed] [Google Scholar]
- 55.Grody W, Griffin J, Taylor A, Korf B, Heit J. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med. 2001;3:139–148. doi: 10.1097/00125817-200103000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grody W, Richards C. New Quality assurance standards for rare disease testing. Genet Med. 2008;10:320–324. doi: 10.1097/GIM.0b013e31817283ba. [DOI] [PubMed] [Google Scholar]
- 57.Maddalena A, Bale S, Das S, Grody W, Richards S. Technical standards and guidelines: molecular genetic testing for ultra-rare disorders. Genet Med. 2005;7:571–583. doi: 10.1097/01.gim.0000182738.95726.ca. [DOI] [PubMed] [Google Scholar]
- 58.Richards C, Bale S, Bellissimo D, Das S, Grody W, Hegde M, Lyon E, Ward B. Recommendations for standards for interpretation of sequence variations: revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 59.Grody W. Ethical issues raised by genetic testing with oligonucleotide microarrays. Mol Biotechnol. 2003;23:127–138. doi: 10.1385/MB:23:2:127. [DOI] [PubMed] [Google Scholar]
- 60.Holtzman N, Murphy P, Watson M, Barr P. Predictive genetic testing: from basic research to clinical practice. Science. 1997;278:602–605. [PubMed] [Google Scholar]