

Abstract
Previously, the smart amplification process version 2 (SMAP-2) was developed to detect mutations from tissue and in crude cell lysates and has been used for rapid diagnosis of specific somatic mutations with single-nucleotide precision. The purpose of this study was to develop a rapid and practical method to detect cancer and metastasis in specimens using the SMAP-2 assay. We developed modified SMAP-2 assays that enabled detection of any change in a single codon using a single assay. Rapid SMAP-2 screening assays are suitable for routine clinical identification of critical amino acid substitutions such as codon 12 mutations in KRAS. Primers bracketing the first two nucleotides of KRAS codon 12 were designed so that all possible alleles would be amplified by the SMAP-2 assay. In combination with the peptide nucleic acid (PNA) with exact homology to the wild-type allele, our assay amplified all mutant alleles except for the wild-type sequence. With this new assay design (termed PNA-clamp SMAP-2), we could detect KRAS mutations within 60 minutes, including sample preparation. We compared results from PNA-clamp SMAP-2 assay, polymerase chain reaction-restriction fragment length polymorphism, and direct sequencing of clinical samples from pancreatic cancer patients and demonstrated perfect concordance. The PNA-clamp SMAP-2 method is a rapid, simple, and highly sensitive detection assay for cancer mutations.
Previously we developed a rapid single nucleotide polymorphism detection system named the smart amplification process version 2 (SMAP-2).1 The SMAP-2 can rapidly detect mutations from tissue, and has been used for rapid diagnosis of specific somatic mutations.2 However, instead of using several specific point mutation assays in parallel to analyze clinical specimens, it was more desirable to create a SMAP-2 assay that would identify many possible mutations in a single tube. This would enable screening of genes for amino acid substitutions at specific codons in a way that saves significantly on cost and time and perhaps provides enough information to make more effective clinical decisions. To test this principle, we focused on KRAS point mutations at codon 12 since they are well characterized and observed frequently in human pancreatic cancer.3,4,5
Many methods such as polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP), PCR-SSCP, PCR sequencing, and MASA, have been used to detection KRAS mutations in clinical samples, but all of these methods require purification of DNA, involve many steps and require skilled technicians to perform.6,7,8,9 These assays are very complicated and time-consuming. A KRAS mutation detection system using the SMAP-2 may be useful for routine intraoperative clinical screening such as the rapid detection of micrometastasis, which has not been possible until now.
To suppress any misamplification of highly related target species, SMAP-2 utilizes two fundamental technologies.1 The first is a unique asymmetric design of the primers flanking the target sequence. These primers are referred to as the folding primer and turn-back primer and are engaged in amplifying the target through a self-priming mechanism. The second approach to minimize background amplification uses Thermus aquaticus (Taq) MutS in combination with the isothermal amplification procedure.
Oligonucleotides of modified chemistry such as peptide nucleic acid (PNA)-based primers have also been shown to be effective for the enrichment of mutant alleles in PCR screening.10 PNA is a synthetic oligonucleotide where the ribose-phosphate backbone is completely replaced by (2-aminoethyl)-glycine units linked by amide bonds. PNA cannot serve as a primer for polymerization because it is not recognized by the polymerase as natural DNA, and it cannot be a substrate for exonuclease activities of Taq polymerase. In addition, the melting temperature of a perfectly matched PNA-DNA duplex is higher than that of DNA-DNA of the same length, but a single mismatch destabilizes the PNA-DNA hybrids, causing a melting temperature shift of 10–18°C.11 Because of these features, PNA has been used to specifically block primer annealing or chain elongation on undesirable templates without interfering with PCR amplification of target templates with which their sequence is not a perfect match.12,13,14 Here, we report the use of PNA in a modified SMAP-2 assay and demonstrate the complete suppression of background amplification, which enables the accurate and rapid detection of all KRAS codon 12 point mutations in a single reaction. This is the first report of PNA oligonucleotides effectively applied to enhance an isothermal amplification technique.
Materials and Methods
Cell Lines and Plasmid
To assist in the development of the SMAP-2 assay, positive control DNA was extracted from the colon cancer cell line SW480 (American Type Culture Collection, Manassas, VA), which harbors a homozygous mutation at the second base of codon 12 in the KRAS gene (GGT>GTT), and the pancreatic cancer cell line KP-1N (Health Science Research Resources Bank, Osaka, Japan), which harbors a heterozygous mutation at second base of codon 12 in the KRAS gene (GGT>GAT). DNA was extracted from cell lines by QIAamp DNA Mini Kit (Qiagen, Tokyo, Japan) and serially diluted to a concentration representing 10 genomic copies/μL. The dilution needed for specificity studies were performed by mixing the cell line DNA with wild-type human genomic DNA (Promega, Tokyo, Japan), which is homozygous wild-type for both KRAS alleles. All experiments using wild-type human genomic DNA in this study refer to the control DNA from Promega (Tokyo, Japan). The human genomic DNA and all DNA templates extracted from cell lines were stored at −20°C before use.
It has been reported that six nonsynonymous point mutations of codon 12 of the KRAS gene (GAT, GCT, GTT, AGT, CGT, TGT) were observed in human tumors.4 To assist in validating the SMAP-2 primer set, each of these six mutations were engineered into a clone of the wild-type KRAS gene by PCR-based site-directed mutagenesis. These mutants were cloned into the plasmid pGEM-T (Promega, Tokyo, Japan), for use as template DNA in SMAP-2 assay development, and the sequence of all of the synthesized mutant templates were verified by sequencing. The synthesized templates were suspended in TE buffer and stored at −20°C before use.
The SMAP-2 Assay for KRAS Point Mutation Detection
The PNA-clamp SMAP-2 assay was engineered in a way to amplify all six point mutations of KRAS gene that occur in the first two nucleotides of codon 12 (Figure 1). The PNA competition probe was designed with a full match for the wild-type sequence and spanned across the codon 12 region (Figure 2, A and B). Consequently, hybridization of the wild-type PNA competitive probe (CP) would interfere with inhibition of chain elongation from turn-back primer, resulting in suppressing amplification of the wild-type allele.
Figure 1.

Schematic of PNA-clamp SMAP-2 assay. For this assay, the SMAP-2 primer design principle is engineered to allow amplification of all possible sequences dictated by the first two nucleotides in codon 12 of KRAS. PNA-clamp competitive probe is designed for the wild-type allele sequence. The greater stability of the PNA probe in hybridization inhibiting to SMAP-2 amplification, and consequently the wild-type allele amplification is suppressed. This effect is theoretically manifest at the initial elongating event from the turn-back primer on wild-type template (case 1) or on a folding primer-synthesized strand (case 2) also early in the reaction.
Figure 2.

PNA-clamp SMAP-2 primer set sequences for the KRAS mutation codon 12 detection assay. A: Sequence alignment of the KRAS mutation at codon 12 and SMAP-2 primer positioning. Wild-type sequence of KRAS gene is illustrated. The arrowheads indicate the position of the mutations found in codon 12. DNA sequences used for primer design are marked. B: SMAP-2 primer sequences for the KRAS mutation at codon 12 detection assay. Unlike other SMAP-2 assays previously reported, none of the sequences are a discrimination probe, since they bracket the actual site of sequence variability.
SMAP-2 reactions were assembled on ice and incubated at 60°C for 60 minutes. We used the Mx3000P system (Stratagene, La Jolla, CA) for maintaining isothermal conditions and monitoring the transition in fluorescence intensity of intercalating SYBR Green I (Invitrogen, Tokyo, Japan) during the reaction. The SMAP-2 method for detecting KRAS point mutation was performed in a total reaction volume of 26 μl. Enzymatic components (Aac DNA polymerase) for the SMAP-2 assays were supplied by K.K. DNAFORM (Kanagawa, Japan). Each reaction contained 3.0 μmol/L each of folding primer and turn-back primer, 1.5 μmol/L BP, 0.4 μmol/L OP1 and OP2 (primers), 0.7 μmol/L PNAs, 1.4 mmol/L deoxynucleoside-5′-triphosphates, 5% dimethyl sulfoxide, 10 mmol/L (NH4)2SO4, 10 mmol/L KCl, 8 mmol/L MgSO4, 1/100,000 diluted original SYBR Green I, 20 mmol/L Tris-HCl (pH 8.8), 0.1% Tween 20, 9 units of Aac DNA polymerase, and 1 μl of prepared sample.
In data analysis and diagnostic judgment, we evaluated SMAP-2 results according to the principle of amplification versus nonamplification compared to threshold values. Generally, we considered amplification to be positive if the fluorescence (delta raw fluorescence: dR), a baseline-subtracted fluorescence reading, strength was higher than a value of 1000 and no amplification occurred if the signal was less than 1000.
Collection and Preparation of Clinical Specimens
Clinical samples from 25 consecutive patients who underwent operation for pancreatic ductal adenocarcinoma in the Yokohama City University Hospital from September 2004 to June 2007 were evaluated. All tumor tissues were diagnosed for pancreatic cancer by hematoxylin and eosin stain. The tissues of nerve plexus around superior mesenteric artery (SMA plexus) were also evaluated for cancer by hematoxylin and eosin stain and judged to be negative for metastasis. After surgical removal, all tumor samples were immediately frozen and stored at −80°C until analyzed. All patients gave informed consent in writing for use of their pancreatic cancer tissue and SMA plexus in research. Institutional approval for conducting research using human material was obtained from the Ethical Advisory Committee of Yokohama City University Graduate School of Medicine and the RIKEN Institute before initiating the study.
Sample Preparation
For the PNA-clamp SMAP-2 assay, approximately 5 mg of tumor sample and SMA plexus were minced and mixed with 1 ml of 30 mmol/L NaOH, stirred, and incubated at 90°C for a minute. After chilling on ice, the tumor lysis extract was diluted 1:10 in water, and 1 μl of the diluted solution was used as the template for SMAP assays performed in the same way as described in the previous sections. To verify the results of the SMAP-2, genomic DNA was extracted from tumor tissues and SMA plexus by using QIAamp DNA Mini Kit and subject to PCR-RFLP analysis and direct sequencing.
PCR-RFLP Analysis and Direct Sequencing of Clinical Samples
For confirmation of the result of SMAP-2, the mutation status at codon 12 of the KRAS gene was examined by PCR-RFLP and direct sequencing. The method of PCR-RFLP analysis was done as described previously.6 Genomic DNA extracted from tumor sample was amplified by PCR and digested with BstNI overnight at 60°C. The digested products were analyzed on a 2% agarose gel, which was stained with ethidium bromide. PCR products from wild-type allele should be digested by BstNI; however, PCR product from mutant type allele cannot be digested. DNA typing was based on the gel pattern of the restricted PCR fragments as previously described.6
For the direct sequencing, following primers were used to amplify the KRAS gene: forward primer, 5′-CCTAAACTCTTCATAATGCTTGCTC-3′; reverse primer, 5′-CCACAAAATGGATCCAGACA-3′. PCR products were cycle-sequenced using the Big Dye Terminator v1.1 cycle-sequencing kit (Applied Biosystems; Tokyo, Japan) according to the manufacturer's instructions. Sequence reactions were then subjected to electrophoresis on an ABI PRISM 3100 (Applied Biosystems) instrument.
Direct Sequencing of the Amplification Product of Our Assay
In our assay, we could not tell which mutation is present in given sample. So we developed the method to identify a particular mutation in each sample after SMAP-2 amplification screening. After 1000 times diluted SMAP-2 amplification products, we purified it by Wizard SV Gel and PCR Clean-Up System (Promega, Tokyo, Japan). Purified SMAP-2 products were cycle-sequenced in the same way as described in the previous sections.
Results
Development of a PNA-Clamp SMAP-2 Assay for the KRAS Point Mutation at Codon 12
Controlled tests on known genomic templates were performed with the PNA-clamp SMAP-2 assay to validate its performance before testing unknown clinical samples. Each genomic DNA purified from the SW480 and KP-1N cell lines was used as a template after dilution to a concentration representing 6000 genomic copies/μl.
SMAP-2 primer sets with PNA specific for detecting the KRAS point mutation were shown to rapidly amplify the SW480 (homozygous for mutation) and KP-1N (heterozygous for mutation) cell line DNA within 40 minutes, while the same primer set was not capable of amplifying wild-type genomic DNA even after 60 minutes. A “no-template” control reaction was also negative at 60 minutes (Figure 3).
Figure 3.
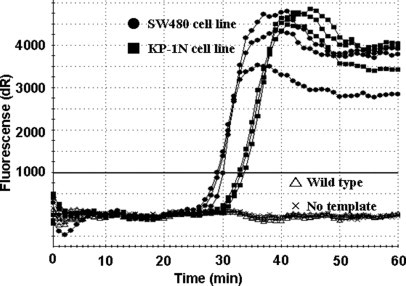
Assay validation for detection of the KRAS point mutations at codon 12. Amplification curves of PNA clamp SMAP-2 assay for KRAS mutation detection indicate a detection time of ∼25–35 minutes on homogeneous cell populations harboring KRAS mutations at codon 12. As expected, the heterozygous cell line (closed squares) (KP-1N; GAT, heterozygous) displays slightly slower amplification, likely because it has 50% less actual target sequences than the homozygous cell line (closed circles) (SW480; GTT, homozygous). No amplification is observed for wild-type DNA (open triangles) or the no template DNA (x) controls.
Sensitivity of Mutation Detection by PNA-Clamp SMAP-2 Assay in Mixed-Cell Populations
In the clinical setting, there are frequently situations where only very small amounts of tumor sample can be obtained, such as from cytology samples or biopsies. To be a useful as a diagnostic tool for mutation detection, assays need to display high sensitivity and be able to detect mutations in a background of wild-type genomic DNA. To explore these characteristics of the PNA-clamp SMAP-2 assay, we conducted serial dilution experiments to examine detection sensitivities. In the serial dilution experiments using purified SW480 cell line genomic DNA alone, the assay could detect 10 copies of the KRAS point mutation in a 26-μl amplification reaction within 50 minutes (Figure 4A).
Figure 4.
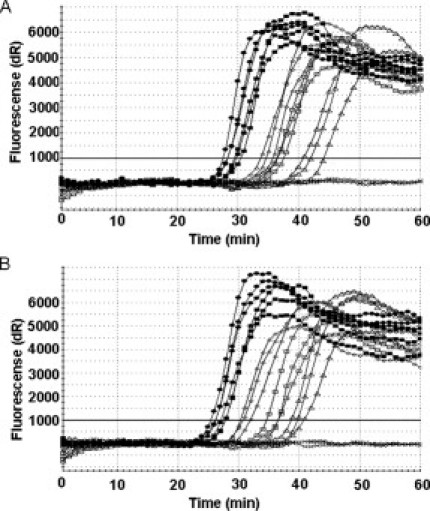
Assay sensitivity for detection of KRAS point mutations at codon 12 in homogeneous and mixed cell populations. A: Sensitivity of the PNA clamp SMAP-2 assay on a homogeneous target. PNA clamp SMAP-2 assay with serial dilutions of SW480 cell line genomic DNA. Shown are 1000 copies (closed circles), 500 copies (closed squares), 100 copies (open circles), 50 copies (open squares), 10 copies (open triangles), and 0 copy (x). Ten copies of mutant DNA are detected in a 26-μl reaction in ∼45 minutes. B: Sensitivity of the PNA clamp SMAP-2 assay on a heterogeneous target. PNA clamp SMAP-2 assay detects mutations in a mixture of wild-type genomic DNA. Shown are 50% (closed circles), 10% (closed squares), 1% (open circles), 0.5% (open squares), 0.1% (open triangles), and 0% (x) of SW480 cell line genomic DNA. Even 0.1% of mutant DNA can be detected in a 26-μl reaction in ∼50 minutes.
Furthermore, because tumor samples frequently consist of numerous subpopulations of cancer cells, we assessed the PNA-clamp SMAP-2 assay for detection of mutations in a heterogenous background. Mutant cell line DNA was mixed with increasing amounts of wild-type DNA, and SMAP-2 assay was performed to determine the minimal detection limits in a background of wild-type DNA. Using the SW480 cell line genomic DNA and the primers for detection of the KRAS point mutation, the mutant sequences could be detected within 50 minutes even when it presented at only 0.1% (Figure 4B). These serial dilution experiments showed good linearity (see Supplementary Figure 1, A and B, at http://jmd.amjpathol.org/).
Equivalent Detection of Six Mutant Alleles by the PNA-Clamp SMAP-2 Assay
The PNA-clamp SMAP-2 assay was engineered to detect all six known mutant alleles of KRAS codon 12. This design assumption was examined using the six mutant templates made by PCR-based site-directed mutagenesis. The assay results demonstrated approximately equal efficiencies for the amplification of all mutant templates in 40 minutes (Figure 5).
Figure 5.
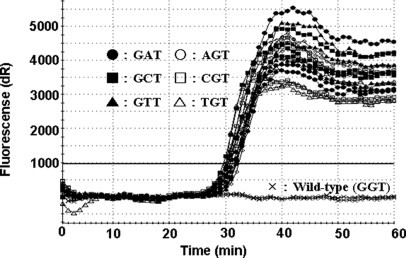
KRAS codon 12 mutant allele detection of six known variants. Amplification of six mutant sequences corresponding to known mutations of KRAS codon 12 is shown. Each assay was performed on 6000 copies of template. The assay showed little to no bias, validating its effectiveness as a general screening tool for the six mutations of interest.
PNA Is a Superior Competitive Probe for Suppression of Background Amplification in Detection of KRAS Mutation
Previously, we reported CP for suppression of background amplification.15 For suppression of background amplification, the mechanism of PNA is similar to CP and therefore we compared PNA with CP in their ability to suppress background amplification.
Control studies were first performed without the use of a competitive probe (Figure 6A). Amplifications of wild-type and mutant alleles occurred with nearly identical kinetics. The concentration of CP used in the assays was 30 μmol/L compared to 0.7 μmol/L for PNA. Furthermore, several different CPs were designed and tested. The data illustrating the best performing CP is reported (Figure 6B). Unlike that of CP, PNA was found to completely suppress the background amplification (Figure 6C).
Figure 6.
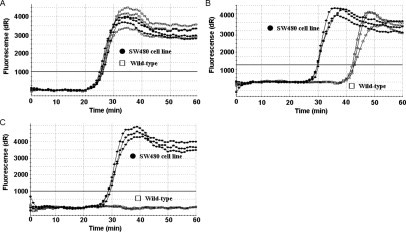
Comparison of PNA and conventional CP for suppression of background amplification in detection of KRAS mutations at codon 12. Shown are 40 ng of SW480 cell line DNA (GTT, homozygous) as a mutant template (closed circles) and 40 ng of wild-type human genomic DNA (GGT, homozygous) as a wild-type template (open squares). A: SMAP-2 amplification without the CP and PNA shows that amplifications of both wild-type and mutant allele were detected at the same time (∼25 minutes). B: SMAP-2 amplification with a conventional CP cannot suppress wild-type allele amplification completely. In this study, 28 different CP sequence designs were tested. Typical data are shown. C: SMAP-2 amplification with a PNA-clamp competitive probe could suppress wild-type allele background amplification completely.
The PNA-Clamp SMAP-2 Assay Detects KRAS Mutations in Clinical Samples
By using only a crude sodium hydroxide extraction procedure and SMAP-2 analysis, we were able to diagnose KRAS mutations from clinical samples within 60 minutes. In this study, the KRAS gene was evaluated from a series of 25 pancreatic cancer samples (23 primary pancreatic cancer specimens and two peritoneal dissemination specimens) and seven SMA plexus samples by both PNA clamp SMAP-2 assay and PCR-RFLP. The clinical samples represented a group of 11 males and 14 females with an age at diagnosis ranging from 50 to 81 (median 64.3) years. Among the 25 pancreatic cancer patients, six (24.0%) were diagnosed as well-differentiated adenocarcinomas, 16 (64.0%) as moderate differentiated adenocarcinomas, and three (12.0%) as poorly differentiated adenocarcinomas.
In 21 (19 primary pancreatic cancer specimens and two peritoneal dissemination specimens) of the 25 pancreatic cancer specimens, KRAS mutations were detected by the PNA-clamp SMAP-2 assay. Also, we could detect micrometastasis of KRAS mutations in one SMA plexus. These results that were proven to have mutations by PNA-clamp SMAP-2 assay were also verified independently by PCR-RFLP and direct sequence, and correlated completely. We also sequenced the amplification product of our assay. The types of mutation of SMAP-2 amplification product was in concordance with the result of direct sequence perfectly (Table 1). In summary, by using the PNA-clamp SMAP-2 assay, we could easily and rapidly detect KRAS mutations in primary cancer specimens, peritoneal dissemination, and micrometastasis in SMA plexus.
Table 1.
Detection of KRAS Codon 12 Mutations in Clinical Samples
| KRAS mutation | SMAP | PCR-RFLP | Direct sequence | SMAP-sequence | ||
|---|---|---|---|---|---|---|
| Primary pancreatic cancer (n = 23) | Mutant type; 19 | Mutant type; 19 | GAT; 15 | GTT; 4 | GAT; 15 | GTT; 4 |
| Wild type; 4 | Wild type; 4 | GGT; 4 | GGT; 4 | |||
| Peritoneal dissemination (n = 2) | Mutant type; 2 | Mutant type; 2 | GAT; 2 | GAT; 2 | ||
| SMA plexus (n = 7) | Mutant type; 1 | Mutant type; 1 | GAT; 1 | GAT; 1 | GTT; 4 | |
| Wild type; 6 | Wild type; 6 | GGT; 6 | GGT; 6 | |||
The PNA clamp SMAP-2 assay was used to analyze 32 clinical samples from patients with pancreatic cancer. The assay results were compared to PCR-RFLP data for each clinical sample and showed a perfect match.
Discussion
The SMAP method has several advantages for KRAS point mutation detection over conventional PCR-based technologies such as PCR-RFLP and other methods. The SMAP-2 technique is simple, requires no DNA purification, and is performed in a closed tube, which reduces the risk of contamination. Conventional PCR-based methods generally require careful DNA purification, because impurities of DNA interfere with the activity of Taq DNA polymerase and similar enzymes. The SMAP-2 assay uses a strand-displacing DNA polymerase and can amplify and detect point mutation directly from clinical samples requiring only a simple heat lysis and denaturation step. It is not known why some strand-displacing polymerases can effectively amplify DNA from such crude samples, but this feature is one of the crucial merits of the SMAP-2 assay.
The KRAS point mutations at codon 12 have been reported to be mainly six different alleles,3,4,5 which by conventional SMAP-2 design approaches using discrimination primers, would require six different assays. For routine clinical use, a new method was desired to detect all six possible mutations from only one primer set. To accomplish this task, we used a PNA-based competition probe in the SMAP-2 assay to suppress amplification of the wild-type allele. This made it possible to amplify all six mutant alleles equally with a single primer set.
In a previous report analyzing KRAS mutations by PNA-clamp PCR methodology, the mismatch amplification was reported to occur depending on the position of PNA and DNA primer.16 This may be due to the temperature cycling conditions of PCR, which have differential effects on the various primers and their actual hybridization affinity for the target. This is the first report for use of PNA competitive probes in an isothermal DNA amplification technique. It may be that PNA clamp approaches are better suited for isothermal amplification where conditions are constant and primers exist in a state of equilibrium with respect to hybridization kinetics. Under these conditions, temperature cycling variation effects are eliminated perhaps allowing for a more effective translation of the optimized parameters of the competitive probe and amplification primer design. The basic design of this KRAS assay is also unique with respect to previous SMAP design strategies. By deliberately bracketing two nucleotides of a key codon to allow amplification of all possible sequences, and suppressing wild-type allele amplification through use of a PNA probe, a general approach to identify all possible amino acid substitution mutations in a single assay was developed. This may prove to have wider appeal for detection of other important cancer mutation markers.
Previously we reported the detection of various single nucleotide polymorphisms, mutations, and deletions using SMAP-2, such as ALDH2 and CYP2C19*2,1 EGFR,2 and UGT1A1.15 As applications of this assay technology continue to expand, challenging targets are likely to be encountered. With new variations on SMAP-2 including CP usage15 or PNA clamping, the versatility of the system continues to expand, providing an opportunity to customize design approaches in a manner suitable for the objective (eg, mutation detection, single nucleotide polymorphism, deletions) and appropriate for the target sequence.
The benefit of SMAP-2 assay is the use of crude lysates of frozen tissue. However, SMAP-2 assay also can be applied to paraffin-embedded archival tumor tissues. After deparaffinization and DNA extraction by QIAamp DNA Micro Kit (Qiagen), we tested 25 paraffin-embedded tumor tissue sample from same patients of our study. The results of PNA-clamp SMAP-2 assay for frozen tissue and paraffin-embedded tissue demonstrated perfect concordance (mutant type, 21; wild type, 4).
The detection of micrometastasis during operations has not been feasible due to technical issues related to DNA extraction and PCR complexity and assay processing time. In general, several hours are necessary for the detection of mutations. The KRAS mutation detection at codon 12 assay reported here is an effective tool to rapidly diagnose cancer and metastasis. We have shown that the PNA-clamp SMAP-2 assay is capable of detecting the KRAS point mutation system within 60 minutes including sample preparation time. Amplification-based techniques are clearly more sensitive than normal pathological examinations.
In summary, our study is the first report of a PNA-clamp being used in an isothermal amplification process. Furthermore, the primer strategy used for detection of the mutants by the SMAP-2 method is unique, versatile, and powerful for codon mutation screening. This approach can be more widely applied for detection of cancer mutations where codon changes result in amino acid substitutions. Many examples exist where single amino acid changes to proto-oncogenes or tumor suppressor genes have been characterized and found to play a role in tumorigenesis. The PNA-clamp SMAP-2 technology makes it possible to screen for all possible mutations leading to an amino acid substitution in one assay.
Acknowledgements
We thank Kazuhito Nomura (RIKEN) for development of Aac polymerase.
Footnotes
Supported by a research grant for the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Y.H.) and the Yokohama City University Advanced Medical Research Center Project.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Supplementary data
References
- 1.Mitani Y, Lezhava A, Kawai Y, Kikuchi T, Oguchi-Katayama A, Kogo Y, Itoh M, Miyagi T, Takakura H, Hoshi K, Kato C, Arakawa T, Shibata K, Fukui K, Masui R, Kuramitsu S, Kiyotani K, Chalk A, Tsunekawa K, Murakami M, Kamataki T, Oka T, Shimada H, Cizdziel PE, Hayashizaki Y. Rapid SNP diagnostics using asymmetric isothermal amplification and a new mismatch-suppression technology. Nat Methods. 2007;4:257–262. doi: 10.1038/nmeth1007. [DOI] [PubMed] [Google Scholar]
- 2.Hoshi K, Takakura H, Mitani Y, Tatsumi K, Momiyama N, Ichikawa Y, Togo S, Miyagi T, Kawai Y, Kogo Y, Kikuchi T, Kato C, Arakawa T, Uno S, Cizdziel PE, Lezhava A, Ogawa N, Hayashizaki Y, Shimada H. Rapid detection of epidermal growth factor receptor mutations in lung cancer by the SMart-amplification process. Clin Cancer Res. 2007;13:4974–4983. doi: 10.1158/1078-0432.CCR-07-0509. [DOI] [PubMed] [Google Scholar]
- 3.Motojima K, Urano T, Nagata Y, Shiku H, Tsunoda T, Kanematsu T. Mutations in the Kirsten-ras oncogene are common but lack correlation with prognosis and tumor stage in human pancreatic carcinoma. Am J Gastroenterol. 1991;86:1784–1788. [PubMed] [Google Scholar]
- 4.Mu DQ, Peng YS, Xu QJ. Values of mutations of K-ras oncogene at codon 12 in detection of pancreatic cancer: 15-year experience. World J Gastroenterol. 2004;10:471–475. doi: 10.3748/wjg.v10.i4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 6.Hatzaki A, Razi E, Anagnostopoulou K, Iliadis K, Kodaxis A, Papaioannou D, Labropoulos S, Vasilaki M, Kosmidis P, Saetta A, Mihalatos M, Nasioulas G. A modified mutagenic PCR-RFLP method for K-ras codon 12 and 13 mutations detection in NSCLC patients. Mol Cell Probe. 2001;15:243–247. doi: 10.1006/mcpr.2001.0367. [DOI] [PubMed] [Google Scholar]
- 7.Mulcahy HE, Lyautey J, Lederrey C, qi Chen X, Anker P, Alstead EM, Ballinger A, Farthing MJ, Stroun M. A prospective study of K-ras mutations in the plasma of pancreatic cancer patients. Clin Cancer Res. 1998;4:271–275. [PubMed] [Google Scholar]
- 8.Yamashita K, Tatebayashi T, Shinoda H, Okayasu I. Simplified rapid non-radioactive PCR-SSCP method applied to K-ras mutation analysis. Pathol Int. 1996;46:801–804. doi: 10.1111/j.1440-1827.1996.tb03553.x. [DOI] [PubMed] [Google Scholar]
- 9.Takeda S, Ichii S, Nakamura Y. Detection of K-ras mutation in sputum by mutant-allele-specific amplification (MASA) Hum Mutat. 1993;2:112–117. doi: 10.1002/humu.1380020209. [DOI] [PubMed] [Google Scholar]
- 10.Demers DB, Curry ET, Egholm M, Sozer AC. Enhanced PCR amplification of VNTR locus D1S80 using peptide nucleic acid (PNA) Nucleic Acid Res. 1995;23:3050–3055. doi: 10.1093/nar/23.15.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyger EM, Krevolin MD, Powell MJ. Detection of the hereditary hemochromatosis gene mutation by real-time fluorescence polymerase chain reaction and peptide nucleic acid clamping. Anal Biochem. 1998;260:142–148. doi: 10.1006/abio.1998.2687. [DOI] [PubMed] [Google Scholar]
- 12.Sun X, Hung K, Wu L, Sidransky D, Guo B. Detection of tumor mutations in the presence of excess amounts of normal DNA. Nature Biotechnol. 2002;20:186–189. doi: 10.1038/nbt0202-186. [DOI] [PubMed] [Google Scholar]
- 13.Thiede C, Bayerdörffer E, Blasczyk R, Wittig B, Neubauer A. Simple and sensitive detection of mutations in the ras proto-oncogenes using PNA-mediated PCR clamping. Nucleic Acids Res. 1996;24:983–984. doi: 10.1093/nar/24.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taback B, Bilchik AJ, Saha S, Nakayama T, Wiese DA, Turner RR, Kuo CT, Hoon DS. Peptide nucleic acid clamp PCR: a novel K-ras mutation detection assay for colorectal cancer micrometastases in lymph nodes. Int J Cancer. 2004;111:409–414. doi: 10.1002/ijc.20268. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe J, Mitani Y, Kawai Y, Kikuchi T, Kogo Y, Oguchi-Katayama A, Kanamori H, Usui K, Itoh M, Cizdziel PE, Lezhava A, Tatsumi K, Ichikawa Y, Togo S, Shimada H, Hayashizaki Y. Use of a competitive probe in assay design for genotyping of the UGT1A1*28 microsatellite polymorphism by the smart amplification process. Biotechniques. 2007;43:479–484. doi: 10.2144/000112563. [DOI] [PubMed] [Google Scholar]
- 16.Luo JD, Chan EC, Shih CL, Chen TL, Liang Y, Hwang TL, Chiou CC. Detection of rare mutant K-ras DNA in a single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe. Nucleic Acids Res. 2006;34:e121–e127. doi: 10.1093/nar/gnj008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.