

Abstract
Resuscitation and prolonged ventilation using 100% oxygen after cardiac arrest is standard clinical practice despite evidence from animal models indicating that neurologic outcome is improved using normoxic compared with hyperoxic resuscitation. This study tested the hypothesis that normoxic ventilation during the first hour after cardiac arrest in dogs protects against prelethal oxidative stress to proteins, loss of the critical metabolic enzyme pyruvate dehydrogenase complex (PDHC), and minimizes subsequent neuronal death in the hippocampus. Anesthetized beagles underwent 10 mins ventricular fibrillation cardiac arrest, followed by defibrillation and ventilation with either 21% or 100% O2. At 1 h after resuscitation, the ventilator was adjusted to maintain normal blood gas levels in both groups. Brains were perfusion-fixed at 2 h reperfusion and used for immunohistochemical measurements of hippocampal nitrotyrosine, a product of protein oxidation, and the E1α subunit of PDHC. In hyperoxic dogs, PDHC immunostaining diminished by approximately 90% compared with sham-operated dogs, while staining in normoxic animals was not significantly different from nonischemic dogs. Protein nitration in the hippocampal neurons of hyperoxic animals was 2–3 times greater than either sham-operated or normoxic resuscitated animals at 2 h reperfusion. Stereologic quantification of neuronal death at 24 h reperfusion showed a 40% reduction using normoxic compared with hyperoxic resuscitation. These results indicate that postischemic hyperoxic ventilation promotes oxidative stress that exacerbates prelethal loss of pyruvate dehydrogenase and delayed hippocampal neuronal cell death. Moreover, these findings indicate the need for clinical trials comparing the effects of different ventilatory oxygen levels on neurologic outcome after cardiac arrest.
Keywords: brain oxidative metabolism, global cerebral ischemia, hyperoxia, immunohistochemistry, mitochondria, nitrotyrosine
Introduction
The most common clinical scenario that results in global ischemia/reperfusion brain injury is cardiac arrest and resuscitation. The typical clinical practice of using 100% ventilatory O2 (FiO2) for cardiac arrest victims during CPR and for prolonged periods after resuscitation has been questioned by results obtained with animal models showing improved neurologic outcome using postischemic FiO2 of as low as 21% O2 (Liu et al, 1998; Mickel et al, 1987; Zwemer et al, 1994).
Oxidative stress and impaired cerebral energy metabolism contribute to delayed neural cell death after global cerebral ischemia (Facchinetti et al, 1998; Starkov et al, 2004; Sugawara and Chan, 2003). While the specific molecular targets of reperfusion injury responsible for metabolic failure in different neural cell types are not identified, damage to the mitochondrial pyruvate dehydrogenase complex (PDHC) is implicated in metabolic dysfunction and reperfusion brain injury (Bogaert et al, 1994; Fukuchi et al, 1998; Martin et al, 2004; Schoder et al, 1998). For example, canine frontal cortex PDHC enzymatic activity is reduced by up to 70% within 30 mins of reperfusion after 10 mins of cardiac arrest (Bogaert et al, 1994). Reperfusion-dependent loss of enzyme activity and immunoreactivity is the greatest in selectively vulnerable brain regions in both canines and rats (Bogaert et al, 2000; Zaidan and Sims, 1997). This enzyme constitutes the sole bridge between anaerobic and aerobic cerebral energy metabolism, and is critical for generating reducing power nicotinamide adenine dinucleotide (reduced) (NADH) used by the electron transport chain to drive the process of oxidative phosphorylation. As the first 30 to 60 mins of reperfusion is accompanied by a net oxidized shift in the cerebral cortex NADH/nicotinamide adenine dinucleotide (NAD) (oxidized) redox state (Rosenthal et al, 1995), impairment of PDHC may limit postischemic oxidative phosphorylation. Oxidative stress is one likely mechanism by which PDHC activity is lost during ischemia/reperfusion. Brain protein oxidation, as measured by the presence of protein carbonyl groups, increases during reperfusion concomitant with loss of PDHC activity (Bogaert et al, 1994; Liu et al, 1993). Moreover, PDHC in vitro is highly sensitive to inactivation by the presence of reactive O2 and N2 species, including hydroxyl radical (Bogaert et al, 1994) and peroxynitrite (Martin et al, 2004).
Several lines of evidence indicate that reperfusion brain injury is exacerbated by hyperoxygenation. Supranormal tissue O2 levels during reoxygenation after exposure of rat cortical brain slices to severe hypoxia correlate with severity of neuronal damage (Halsey Jr et al, 1991). Gerbils treated with 100% O2 after 15 mins of bilateral carotid occlusion sustain increased white matter damage compared with those exposed to room air (Mickel et al, 1987, 1990). On a neurochemical level, hyperoxic reperfusion exacerbates brain lipid oxidation and tissue lactic acidosis (Liu et al, 1998; Mickel et al, 1987), and worsens the postischemic oxidized shift in tissue redox state (Feng et al, 1998).
In an attempt to understand further why postischemic neurologic impairment is reduced by normoxic compared with hyperoxic resuscitation, we tested the hypothesis that normoxic reperfusion protects against hippocampal tyrosine nitration and loss of PDHC activity. The results of this study performed with a clinically relevant large animal model of cardiac arrest and resuscitation indicate that normoxic resuscitation improves the neurologic outcome by minimizing oxidative stress and preserving cerebral aerobic energy metabolism.
Materials and methods
Animal Model of Cardiac Arrest and Resuscitation
All animal experiments were approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee. The animal model used for these studies is well described and has been used extensively by us and others to study global cerebral ischemia and reperfusion (Fiskum et al, 1992; Krajewska et al, 2004; Leonov et al, 1990; Liu et al, 1998; Rosenthal et al, 2003; Zwemer et al, 1994). Adult pure breed female beagles weighing 10 to 15 kg were anesthetized initially with an intravenous injection of veterinary thiopental (8 to 12 mg/kg). Prolonged anesthesia was then induced with an infusion of α-chloralose (75 mg/kg). After establishment of appropriate anesthesia, animals were intubated endotracheally and subjected to controlled ventilation on room air. Cut down catheters were placed in the left femoral artery and vein. The arterial catheter was attached to a transducer for continuous monitoring of arterial pressure. The venous catheter was advanced to the level of the inferior vena cava for the purpose of resuscitative drug delivery. An intravenous infusion of normal saline (3 ml/kg h) was started at this point and continued for the remainder of the experiment. A left lateral thoracotomy was performed through the fourth intercostal space. The pericardium was incised and reflected. Core temperature was monitored continuously and maintained greater than 37°C with a heating blanket and heat lamps.
After surgical preparation, cardiac arrest was induced with an electrical train of currents generated by a Grass stimulator, applied directly to the epicardium of the heart for 45 secs. The presence of cardiac arrest was verified by EKG rhythm, consistent with ventricular fibrillation (VF) in the presence of systolic arterial pressure (SAP) < 20 mm Hg. At the initiation of VF, artificial ventilation was discontinued. Ventricular fibrillation was allowed to continue without treatment for 10 mins. At the end of 10 mins of VF, ventilation was resumed (respiratory rate = 28) simultaneous with the beginning of manual open chest cardiopulmonary resuscitation (OCCPR) at a rate of 56/min. Open chest cardiopulmonary resuscitation was standardized such that SAP remained between 90 and 120 mm Hg throughout. Epinephrine (0.2 mL/kg of 1:10,000 solution) and sodium bicarbonate (1 mEq/kg) were injected at the beginning of resuscitation through the femoral central venous catheter. Open chest cardiopulmonary resuscitation was continued for 3 mins at which point internal defibrillation was performed at 5 J. After successful defibrillation, atrial fibrillation that occurred occasionally in the first 5 mins after restoration of spontaneous circulation (ROSC) was cardioverted internally at 5 J. Arterial blood gas (ABG) determinations were made immediately after resuscitation and at 10, 30, 60, and 90 mins, and every 60 mins thereafter.
Animals were randomized to one of two resuscitative protocols. Hyperoxic: Resuscitation was performed with 100% oxygen and dogs breathed 100% oxygen for 1 h. During the next 1 to 23 h, the ventilator settings were adjusted to maintain pO2 > 80 and < 120 mm Hg. Normoxic Dogs were resuscitated with room air, and then inspired oxygen was immediately adjusted to maintain pO2 > 80 and < 120 mm Hg. The pO2 values for sham-operated, nonischemic control animals were also within this range. pCO2 was maintained at > 25 and < 35 mm Hg in all animals. Exclusion criteria included baseline T < 37° or > 39°, SAP < 60 at any time after resuscitation or < 80 for 10 mins, or inability to maintain pO2 or pCO2 within the stated limits.
All animals received intensive care for up to 24 h; an experienced animal care technician was present in the animal laboratory at all times during the postresuscitative period. In animals receiving 24 h of intensive care, the chest was closed in four layers, a chest tube inserted, the chest evacuated, and the chest tube connected to a Heimlich chest valve beginning 1 h after resuscitation; analgesia was maintained with a morphine drip (0.1 mg/ kg h for 10 h followed by 0.05 mg/kg h, with additional boluses as needed). Pancuronium (0.1 mg/kg) was used intermittently, only as needed for muscle relaxation to facilitate ventilation, and only after verification that the animal was adequately sedated, and feeling no pain. All animals enrolled in this study survived.
Tissue Preparation
Animals were perfused with a fixative according to a protocol originally described for macaque monkeys (Hof and Nimchinsky, 1992; Hof and Morrison, 1995) and adapted by us for use with canines (Hof et al, 1996). Perfusion occurred at either 2 h or 24 h after cardiac arrest and at 2 h after anesthetization and simulated cardiac arrest for sham-operated control animals. The descending aorta was clamped and the animals were perfused transcardially with cold 1% paraformaldehyde in phosphate buffer for 1 min, followed by cold 4% paraformaldehyde for 10 mins. After removing the brain from the skull, it was postfixed for 7 days in 4% paraformaldehyde. Coronal blocks cut so that they contained the entire hippocampus were postfixed in 2.5% acrolein (Polysciences, EM grade, Warrington, PA, USA) solution in 4% paraformaldehyde, pH 6.8, for 2 h. After 24 h postfixation in 4% paraformaldehyde, pH 6.8, the blocks were transferred into 30% sucrose. Once the blocks sunk to the bottom of the containers, they were cut on a sliding microtome while frozen on crushed dry ice. The coronal sections were cut 25 μm thick and 12 series per animal were produced. These sections were kept in cryoprotectant antifreeze solution consisting of glycerol, ethylene glycol, distilled H2O, and phosphate-buffered saline (3:3:3:1 volume ratio) at −20°C until further processing was initiated (Watson Jr et al, 1986).
Immunohistochemistry
For single- and double-labeling immunohistochemistry, the free-floating sections were processed by ABC technique to visualize the different antigens (ABC Elite Kits, Vector Laboratories, Burlingame, CA, USA), as described previously (Hoffman et al, 1992). For single immunohistochemistry, we used ABC immunoperoxidase staining and a nickel enhanced DAB solution as the chromogen. Cell regions of the hippocampus were defined by using immunocytochemical localization of neuron nuclear antigen (NeuN), a neuron-specific antigen. A mouse monoclonal anti-NeuN was purchased from Chemicon (Temecula, CA, USA) and used at a concentration of 1:70,000. Using a mouse monoclonal anti-E1α subunit of PDHC (Molecular Probes, Eugene, OR, USA), staining was performed by conventional Nickel-intensified DAB solution (Berghorn et al, 1994). Briefly, the sections were rinsed free of cryoprotectant with 0.05 mol/L potassium phosphate-buffered saline (KPBS). The slides were incubated in 1% sodium borohydrate solution, then rinsed multiple times until bubbles were eliminated. Sections used for localization of neurons with NeuN were placed into primary antibody at a dilution of 1:70,000 in KPBS + 0.4% triton-X100 for 1 h at room temperature, followed by 48 h at 4°C. On the third day, after multiple rinses in KPBS, the samples were exposed to biotinylated horse anti-mouse antibody for 1 h at room temperature at a 1:600 dilution in KPBS + 0.4% triton-X100. After multiple rinses with KPBS, the sections were incubated with ABC solutions (45 μl each A and B in 10 mL of KPBS + 0.4% triton-X100, Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. The samples were then rinsed three times at 5 mins each in KPBS, and then rinsed three times at 5 mins each in 10 mL of 0.175 mol/L sodium acetate. The sections were then placed into a Ni-DAB H2O2 chromogen solution (250 mg Ni sulfate and 2 mg DAB and 8.3 μmL of 3% H2O2/10 mL of 0.175 mol/L sodium acetate solution). Staining was performed for 10 mins and then was terminated by transfer of the sections into sodium acetate solution.
The samples to be stained for anti-E1α were rinsed and treated with sodium borohydride as above. They then were placed in preheated 0.01 mol/L potassium citrate buffer (pH 6.0) and heated further in a microwave to improve antigen accessibility. After multiple rinses in KPBS, the samples were exposed to the anti-PDHC E1α subunit antibody at a 1:1000 dilution in KPBS + 0.4% triton-X100 for 1 h at room temperature, followed by 48 h at 4°C. The procedure described above was then followed. To test the specificity of the anti-E1α subunit antibody, we preincubated the diluted antibody solution with 200 μg/mL porcine PDHC (Sigma, St Louis, MO, USA) for 90 mins at room temperature. The preadsorbed solution was substituted for the primary antibody. This procedure was slightly modified compared with that described by others using other antibodies to pyruvate dehydrogenase that were not specific for the E1α subunit (Sheu et al, 1983, 1984, 1985; Milner et al, 1987). Staining was negligible when the preabsorbed solution was used (not shown).
For single immunohistochemistry using a rabbit anti-3-nitrotyrosine antiserum at a 1:2000 dilution (Upstate, Waltham, MA, USA), we used the identical staining protocol described for PDH, except that the secondary antibody was biotinylated goat anti-rabbit antibody. To test the specificity of the antinitrotyrosine, we preabsorbed the antibody at the same concentration used for staining with 10 mmol/L 3-nitro-L-tyrosine (Sigma, St Louis, MO, USA) for 90 mins at room temperature (Viera et al, 1999) and substituted this solution for the primary antibody. As with the anti-E1α antibody, staining with the preabsorbed anti-3-nitrotyrosine antibody was also negligible (not shown).
For double-fluorescence labeling, the procedure of Shindler and Roth (1996) was followed. This protocol consists of first detecting PDHC E1α with biotinylated tyramide amplification and immunofluorescence detection, and then using immunofluorescence localization of the second antigen with a direct-tagged secondary antibody. The first primary antigen, PDH < E1α subunit, was localized with a mouse monoclonal anti-E1α subunit of PDHC (Molecular Probes, Eugene, OR, USA) at a concentration of 1:500 for 48 h at 4°C. We purchased a Tyramide Signal Amplification Kit (Molecular Probes, Eugene, OR, USA) and followed the protocol described by Berghorn et al (1994) for tyramide amplification, employing the reagents from an ABC Elite kit (Vector Laboratories) after the biotin tyramide, and then used streptavidin Alexa Fluor 488 (Molecular Probes) detection of the deposited biotin. The anti-PDHC E1α concentration, while effective when tyramine-amplified fluorescence is used, is too low to enable any fluorescence detection without amplification, thereby providing the basis for successful double labeling with a second mouse monoclonal antibody (Shindler and Roth, 1996). Detection of the second primary antigen employed a monoclonal mouse anti-NeuN at a 1:7000 dilution incubated with the tissue for 48 h at 4°C. Neuron nuclear antigen immunoreactivity was visualized using an Alexa 546-tagged secondary antibody (at 1:500 dilution) incubated for 3 h at 37°C. The sections were rinsed in saline and then mounted onto glass slides, dried overnight and dehydrated briefly with ascending alcohols. Sections were cleared in xylene and cover-slipped with Krystalon (EM Science Harleco, Kansas City, MO, USA).
Quantification of Pyruvate Dehydrogenase Complex E1α Immunostaining
One series per animal was processed for E1α subunit of PDHC immunoreactivity using standard immunocytochemical techniques. Microwave antigen reveal treatment was used to improve detection. The staining was performed with Ni-DAB chromogen solution, which results in a black insoluble stain at the site of the antigen. Quantitative analyses were performed with computer-assisted image analysis consisting of a Nikon Eclipse 800 photomicroscope, a Retiga cooled CCD digital camera (Biovision Technologies), and a Macintosh G4 computer with IP Spectrum software (Scanalytics, Fairfax, VA, USA). Quantification was performed by an individual masked to the treatment protocol. Across the septotemporal axis, three slides per animal were selected for quantitative analyses: one at the rostral, one in the middle, and one in the caudal part of the ventral hippocampus (Figure 1), carefully matching the rostrocaudal levels of these sections with the aid of a stereotaxic atlas of the dog brain. After defining the cornus ammonis (CA1), CA3, and dentate gyrus (DG) under low magnification (×2), under high magnification (×60, immersion oil lens, numerical aperture (NA) 1.4) three fields in each subfield were captured (Figure 2) at 0.5-μm intervals throughout the entire section and the stack of images was deconvolved, enabling precise resolution of the reactive mitochondria throughout the thickness of the section, while at the same time eliminating any stained structures that were out of focus at each plane. The image stack was then collapsed into one plane and PDHC E1α-stained structures were then counted by the program. Results are expressed as mean ± s.d. values for n = 3 animals per group. Statistical significance was calculated using one-way analysis of variance (ANOVA) with Tukey–Kramer multiple-comparison test post hoc analyses.
Figure 1.

Macroscopic anatomy of the adult beagle hippocampus. The hippocampus is ‘C’-shaped, lying caudally on the thalamus. Coronal sections used for quantification of PDHC and 3-nitrotyrosine immunoreactivity are shown using NeuN staining for neurons in three macroscopic regions: (A) Rostral −2700 to 4200 μm. (B) Middle − 7200 to 8100 μm. (C) Caudal − 10,800 to 11,700 μm. Scale bar = 500 μm.
Figure 2.

Distribution of high magnification fields used for quantification of PDHC E1α immunostaining. Within hippocampal regions CA1, CA3, and DG, three high-magnification fields of equal area were used for quantification. Scale bar = 500 μm.
Quantification of Nitrotyrosine Immunostaining
We used one hemisphere of each dog for the analyses. In all, 1 in 12 series per animal was processed for nitrotyrosine immunoreactivity using standard immunocytochemical techniques. The staining was performed with Ni-DAB chromogen solution, which results in a black insoluble stain at the site of the antigen. Quantitative analyses were performed with the same computer-assisted image analysis described above. Quantification was performed by an individual masked to the treatment protocol. Two sections per animal were selected for quantitative analyses. As the staining was not equally uniform after the intense microwave treatment, with some sections exhibiting even staining, others that were only partially stained, and a third group that failed to stain, we selected the two most evenly stained sections from each animal. After defining the CA1, CA3 and DG under low magnification (×2), three fields per subregion were used to measure the quantity of the staining under higher magnification (×20). The stage of the microscope was adjusted so that the cell layer was centered in the field and was oriented horizontally in the captured image. The amount of staining was expressed as the average area occupied by the black reaction product representing 3-nitrotyrosine immunoreactivity within the microscopic field. Results are expressed as mean ± s.d. values for n = 3 animals per group. Statistical significance was calculated using one-way ANOVA with Fisher’s least-square difference (LSD) multiple-comparison test post hoc analyses.
Cresyl Violet Staining for Histopathology
Free floating sections were rinsed free of cryoprotectant with KPBS, mounted on adhesion superfrost glass slides, and were stained after overnight drying. Slides were dipped in 75%, 95%, and 100% alcohol, and then immersed for 10 mins in each of the following: xylene (twice), 100% alcohol (twice), 95% alcohol, and 75% alcohol. The slides were then immersed in distilled water for 2 mins and stained with 0.5% cresyl violet for 15 mins. Slides were then dipped in water 4 to 5 times, and dehydrated through 75% alcohol for 2 mins. Then, they were dipped in 95% alcohol plus 10 to 16 drops of glacial acetic acid, then in 100% alcohol for 5 mins (twice), then in xylene for 2 mins (twice). Coverslips were subsequently applied.
Stereologic Quantifications
Quantitative analysis utilized one coronal block containing the entire hippocampus from one hemisphere per animal. Using a random starting point, every twelfth 25-μm-thick section was stained with cresyl violet and used for estimating the total neuronal numbers of normal and dying apoptotic or necrotic appearing neurons in the different CA regions of the hippocampus in the 24-h hyperoxic or normoxic ventilated dogs after 10 mins of cardiac arrest compared with nonischemic sham-operated animals.
Quantitative analysis was performed on a computer-assisted image analysis system consisting of a Nikon Eclipse 800 photomicroscope equipped with a Ludi I/F computer-controlled motorized stage, an Optronics video camera, a Dell microcomputer, and StereoInvestigator program (MicroBrightField, Wiliston, VT, USA), a custom-designed morphology and stereology software. Tracings were made from the rostral through the caudal extent of the hippocampus. In the tracings, a distinction was made among the CA1 and CA2/3 fields and the DG. Six or seven tracings were made per hippocampus, per hemisphere for each animal. The traced sections were evenly spaced 3300 μm apart. After outlining the boundaries of CA1, CA2/3, and DG fields at low magnification (×4) on the computer graphic display in each section separately, the software placed within each subfield boundary a set of optical disector frames (CA1, CA2/3: 55 ×55 μm, DG: 20 ×20 μm), in a systematic–random fashion. Normal neurons and dying neurons were then counted in optical disectors 3.5 μm in depth, according to stereologic principles (West et al, 1991; Schmitz and Hof, 2005). All analyses were performed using a 1.4 NA ×100 Plan Apo, oil objective with a 1.4 NA auxiliary condenser lens, and Koehler illumination to achieve optimal optical sectioning during disector analysis. Normal neurons were defined as large cells with identifiable nuclei and discrete nucleoli (Figure 3). Dying neurons were defined as those exhibiting either clear accumulation of dense, globular materials in the cytoplasm with evidence of nuclear fragmentation or cells showing shrunken perikarya and darkly stained nuclei of reduced size (Figure 3). In each region, at least 500 neurons were sampled to ensure the robustness of the data (Schmitz and Hof, 2000). For the sterologic cell counts on n = 3 animals per group, the statistical significance was calculated using a Mann–Whitney rank sum test.
Figure 3.
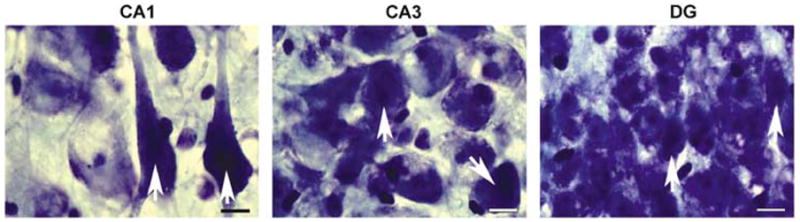
Canine hippocampal histopathology after cardiac arrest and resuscitation. The hippocampus from an animal that underwent 10 mins cardiac arrest and hyperoxic resuscitation was stained with cresyl violet. Under high magnification (×100, immersion oil), both normal and dying neurons (arrows) are evident throughout the CA1, CA3, and DG subfields. Scale bar = 10 μm.
Results
Physiological Variables
Physiological variables for the six animals in the 2-plus 24-h normoxic resuscitation groups were compared with the six animals in the hyperoxic resuscitation groups. No significant differences in prearrest values for arterial pO2, pCO2 and pH, heart rate, rectal T, and SAP were observed. At 30 mins after resuscitation, that is, midway through the 60-min period of different FiO2 for these groups, the pO2 for hyperoxic animals was significantly greater than that of the normoxic animals (384±68 s.e. versus 76±2 mm Hg; P = 0.001), as expected. In addition, the SAP for the hyperoxic groups was significantly greater than that of the normoxic animals (141±9 versus 105±2 mm Hg; P < 0.01). This result is not surprising as exposure of healthy humans to 100% O2 for 1 h can increase systemic vascular resistance and elevate mean arterial blood pressure (Waring et al, 2003). No differences among hyperoxic and normoxic groups were found for any other physiologic parameter. In addition, all animals enrolled in this study were successfully resuscitated with one countershock after open chest CPR, indicating no effect of normoxia on CPR effectiveness in this model.
Hippocampal Pyruvate Dehydrogenase Complex E1α Immunoreactivity
Previously, we reported the distribution of PDHC immunoreactivity in the canine frontal cortex and the neuronal subtype-selective reduction in immunostaining that occurs after 10 mins cardiac arrest and 2 h reperfusion (Bogaert et al, 2000). In this study, we focused on neuronal subregions within the hippocampus, another area highly sensitive to delayed neuronal death during global cerebral ischemia and reperfusion. As in vitro evidence suggests that oxidative stress is responsible for loss of PDHC enzyme activity during reperfusion, this study tested the hypothesis that loss of hippocampal PDHC immunoreactivity is minimized by reducing oxidative stress via lowering the resuscitative inspired [O2].
Pyruvate dehydrogenase complex E1α immunostaining in nonischemic animals was particularly intense in hippocampal neuronal cell bodies, as identified by NeuN immunostaining, and exhibited the punctuate appearance typical of mitochondrial antigens (Figure 4). The primary neuronal localization of PDHC immunoreactivity is further verified by the single and double immunostaining for E1 α and NeuN in the CA3 region of the canine hippocampus seen in Figure 5. A comparison between immunostaining in the hippocampus from a nonischemic control animal and one from an animal that underwent 10 mins cardiac arrest and 2 h reperfusion using the hyperoxic resuscitation protocol is also shown. While PDHC immunoreactivity is reduced dramatically in the hyperoxic resuscitated dog, NeuN immunostaining remains relatively unchanged. Loss of PDHC is therefore not simply due to loss of neurons, as expected since neuronal death occurs over many hours to days in this model (Rosenthal et al, 2003).
Figure 4.

Confocal image of PDHC E1 α and NeuN in the hippocampal CA1 region. Neuron nuclear antigen immunostaining (red) is present throughout the neuronal cell body, including the nucleus. Pyruvate dehydrogenase complex E1 α immunostaining (green) is localized to punctate structures indicative of mitochondria. Scale bar = 10 μ m.
Figure 5.

Confocal images of PDHC E1 α and NeuN immunoreactivity in the hippocampal CA3 region of a sham-operated (S) compared with a hyperoxic resuscitated animal (H). Robust PDHC immunostaining apparent in the cell bodies of pyramidal neurons in the nonischemic animal is lost in the hyperoxic resuscitated animal. Loss of PDHC immunoreactivity is not due to loss of neurons as NeuN immunostaining remains relatively unchanged. Scale bar = 50 μ m.
Quantitative analysis of PDHC E1 α immunostaining was restricted to the stratum pyramidale in the CA1 and CA3, and the stratum granulosum of the DG. The PDHC E1 α staining in sham-operated animals (Figures 6A to 8) was not different across the rostrocaudal levels of hippocampus in CA1 or DG. In the CA3 region, the middle part of the hippocampus exhibited significantly more intensive staining than the rostral and caudal parts of the hippocampus. In the hippocampus of the hyperoxic ischemia/reperfusion dogs, PDHC E1 α immunostaining in CA1, CA3, and DG was dramatically reduced (84% to 91%) compared with that observed in the nonischemic, sham-operated animals (P < 0.05) (Figures 6B to 8). In addition, the degree of reduction was similar throughout the entire rostrocaudal extent of the hippocampus. In the normoxic resuscitated animals, PDHC E1 α staining was not significantly different from the sham-operated dogs (Figures 6C to 8). There were, however, some minor differences in the distribution of immunostaining across the rostrocaudal extent of the hippocampus. Specifically, in the CA3 and in the DG, immunoreactivity was significantly lower in the rostral compared with the middle and caudal regions.
Figure 6.
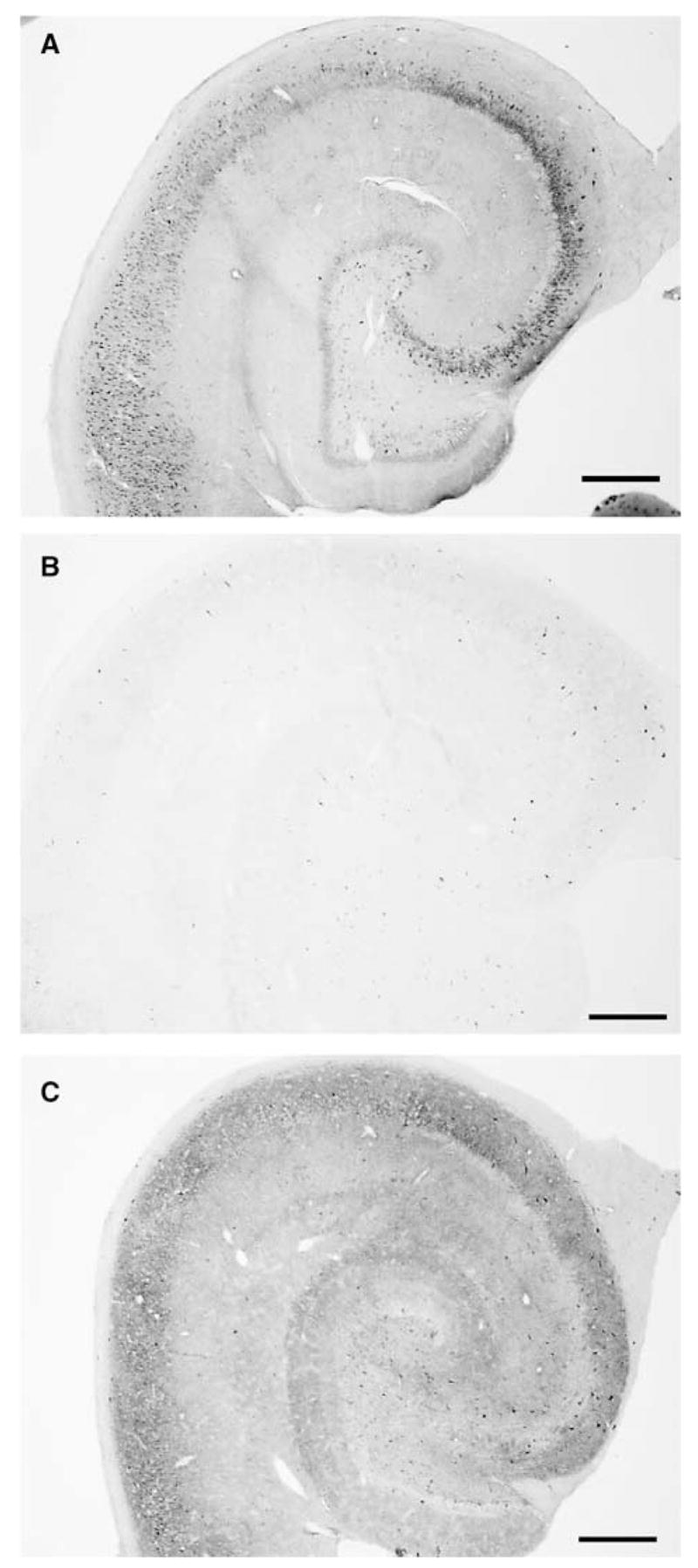
Comparison of hippocampal PDHC E1 α immunostaining between nonischemic and hyperoxic and normoxic resuscitated animals. Low-magnification photomicrographs were taken of PDHC E1 α immunostaining in the middle region of hippocampi from (A) sham-operated, (B) hyperoxic resuscitated, and (C) normoxic resuscitated animals. Scale bar = 500 μ m.
Figure 8.

Quantitative comparison of hippocampal PDHC E1α immunostaining in nonischemic and postischemic animals. Staining is expressed as number of particles in the different subfields of hippocampus: (A) CA1, (B) CA3, and (C) DG. In hyperoxic ventilated animals, the staining diminished significantly in all subfields of the hippocampus through the entire rostrocaudal extent, whereas in the normoxic ventilated animals the staining statistically was not significantly different from the sham-operated animals. Results are expressed as mean ± s.d. values; n = 3 per animal group. Statistical significance was calculated using one-way ANOVA with Tukey–Kramer multiple-comparison test post hoc analyses, P < 0.05. a*: Significantly different compared with sham-operated and normoxic animals in the same rostrocaudal level. b*: Significantly different in rostrocaudal distribution within the same animal group.
3-Nitrotyrosine Immunoreactivity
Immunocytochemical staining for 3-nitrotyrosine was used as a marker for oxidative protein alterations (Lorch et al, 2004). In sham-operated animals, we observed some vascular periluminal 3-nitrotyrosine immunostaining, but very weak immunoreactivity elsewhere (Figure 9A). After 2 h of reperfusion using the hyperoxic ventilation protocol, 3-nitrotyrosine immunostaining increased dramatically (Figure 9B). In contrast to the immunoreactivity apparent in the nonischemic samples, hyperoxic reperfused brains exhibited intense staining within neurons. 3-Nitrotyrosine immunoreactivity present within the hippocampi after 2 h of reperfusion using the normoxic ventilation protocol was weak compared with that observed in the hyperoxic animal samples, and much more similar to that observed within samples from sham-operated, nonischemic animals (Figure 9C).
Figure 9.

Comparison of hippocampal nitrotyrosine immunostaining between nonischemic and hyperoxic and normoxic resuscitated animals. Low-magnification photomicrographs were taken of 3-nitrotyrosine immunostaining in the middle region of hippocampi from (A) sham-operated, (B) hyperoxic resuscitated, and (C) normoxic resuscitated animals. Scale bar = 500 μm.
Quantitative analysis of 3-nitrotyrosine immunoreactivity throughout the hippocampal CA1, CA3, and DG subfields reveal an approximately 300% increase in hyperoxic resuscitated animals compared with nonischemic controls for all regions (Figures 10 and 11). In contrast, there was no significant difference between the 3-nitrotyrosine staining of the normoxic and sham-operated animals in any of the hippocampal subfields. Thus, normoxic resuscitation both protected against the increase in tyrosine nitration and the loss of PDHC immunoreactivity that occurs throughout the hippocampus in dogs after normothermic cardiac arrest, and 2 h reperfusion using the standard clinical practice of hyperoxic ventilation.
Figure 10.
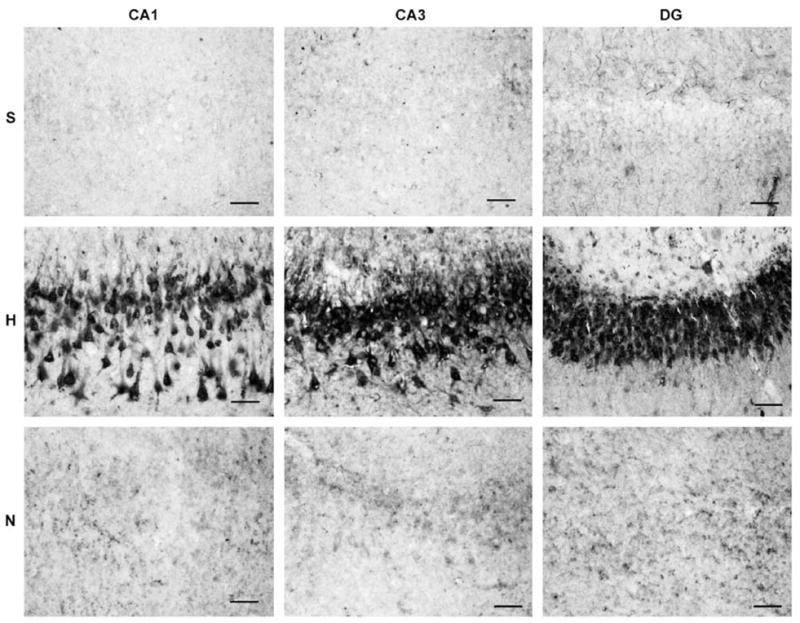
Photomicrographs illustrating hippocampal 3-nitrotyrosine immunostaining in microscopic fields used for quantification. In the hyperoxic resuscitated animal (H), the 3-nitrotyrosine immunostaining is robust and localized primarily in neurons. In contrast, staining intensity is considerably reduced in the hippocampus of a normoxic resuscitated animal (N) and a sham-operated control animal (S). Scale bar = 50 μm.
Figure 11.

Quantitative comparison of hippocampal 3-nitrotyrosine immunostaining in nonischemic and postischemic animals. Immunostaining is expressed as the area occupied by the black precipitate of Ni-DAB staining within the defined fields as shown in the figure 3-Nitrotyrosine staining was significantly 2 to 3 times greater in each hippocampal subregion of the hyperoxic resuscitated animals compared with either the normoxic reperfused animals or the sham-operated controls (P < 0.05). Results are expressed as mean ± s.d. values; n = 3 per animal group. Statistical significance was calculated using one-way ANOVA with Fisher’s LSD multiple-comparison test post hoc analyses.
While the differences in hippocampal immunostaining for PDHC E1 α and 3-nitrotyrosine between hyperoxic and normoxic resuscitated animals is consistent with the differences in short-term neurologic outcome we reported previously with this model of cardiac arrest (Liu et al, 1998), the relationship with hippocampal pathology was unknown as neuronal cell injury was not previously analyzed. We therefore engaged in a rigorous stereologic analysis of neuronal cell injury present at 24 h reperfusion, the time at which neurologic outcome had previously been documented. As expected, the percentage of injured hippocampal neurons was negligible in the nonischemic, sham-operated animals (Figure 12). The percentage of injured neurons present in the different subfields of the hippocampi from hyperoxic dogs ranging from 40% to 48% was significantly different from the percentage observed in the nonischemic samples (P < 0.001). Most importantly, the results shown in Figure 12 show a significant reduction in the percentage of dying hippocampal neurons present in normoxic (ranging from 20% to 33%) compared with hyperoxic animals in all three subfields. While the percentage of injured neurons in the normoxic animals was significantly different from that of the nonischemic animals in the CA2/3 subfield, the apparent differences in the CA1 region and the DG for these two groups were not significantly different.
Figure 12.

Quantification of hippocampal neuronal cell death after cardiac arrest and resuscitation using hyperoxic or normoxic protocols. The percentage of dying neurons was estimated using a stereologic approach, as described in Materials and methods and shown in Figure 3. The percentage of dying neurons at 24 h reperfusion was significantly lower in the normoxic resuscitated animals than in the hyperoxic animals in all hippocampal subfields. Results are expressed as mean ± s.d. values; n = 3 per animal group. The statistical significance was calculated using Mann–Whitney rank sum test.* P < 0.01, **P < 0.05.
Discussion
The primary conclusion reached from this study is that within the canine hippocampus, reperfusion-dependent loss of PDHC immunoreactivity is associated with oxidative stress, as observed by comparing PDHC and 3-nitrotyrosine immunoreactivities between nonischemic animals and those subjected to cardiac arrest and resuscitation using either hyperoxic or normoxic ventilation protocols. This study is the first to show the relationship between oxidative stress and loss of PDHC immunoreactivity using an animal or cell model of neuropathology, although this relationship was postulated based on animal models of cerebral ischemia/reperfusion and thiamine (vitamin B1) deficiency (Bubber et al, 2004; Martin et al, 2004). Enzyme activity measurements performed with purified PDHC indicate that this enzyme is very sensitive to inactivation by both hydroxyl radical and peroxynitrite (Bogaert et al, 1994; Martin et al, 2004). We also showed previously a loss of cortical PDHC immunoreactivity and enzyme activity at 2 h of reperfusion compared with nonischemic dogs, but did not use reperfusion paradigms that varied the levels of oxidative stress (Bogaert et al, 2000). As PDHC is a critical enzyme in oxidative cerebral energy metabolism, our new results strongly supporting oxidative stress as the mechanism responsible for loss of PDHC in vivo represent an important step in understanding the relationship between oxidative molecular modifications and metabolic failure in the context of reperfusion brain injury.
The use of immunocytochemistry for detection of changes in PDHC, while indicating a dramatic loss in animals resuscitated with 100% O2, must be interpreted cautiously. Immunocytochemical detection of an antigen requires that the antigen be present and accessible to the antibody. The techniques we used are extraordinarily sensitive and thus loss of immunoreactivity might not be linear with respect to enzyme content. Earlier studies suggest that changes in staining intensity of the enzyme tyrosine hydroxylase are not apparent in neurons until more than 70% of the enzyme is lost (Pearson et al, 1979). While our experience with tyrosine hydroxylase expression in hypothalamus suggests a much closer relationship between staining intensity and enzyme mRNA expression (Berghorn et al, 2001), whether linearity is achieved has not been determined. Loss of immunoreactivity might also not be strictly due to protein loss. For example, changes in staining intensity could reflect masking of the epitope recognized by the antibodies. This possibility is well illustrated by the observation that antibodies generated against the estrogen receptor alpha can be quite sensitive to circulating hormone levels due to the conformational change evoked by receptor ligand binding (Henry Jr et al, 1991). Considering the extreme size and complexity of the PDHC, conformational changes that could mask an epitope in one of the many subunits, for example, the E1 α antigen used in our study, are very possible. It is likely, however, that the change in hippocampal PDHC E1 α immunoreactivity is associated with a loss in PDHC enzyme activity as this relationship was observed previously in the frontal cortex of reperfused animals using a polyclonal antibody that detects E1 α and several other enzyme subunits (Bogaert et al, 2000). Reperfusion-dependent reduction in immunoreactivity and enzyme activity of glutamine synthetase, another large multi-subunit metabolic enzyme, have also both been associated with oxidative protein alterations (Oliver et al, 1990). Other studies suggest that these alterations promote the proteolytic degradation of this enzyme complex (Starke-Reed and Oliver, 1989). If loss of immunoreactivity is due to proteolysis, the mitochondrial matrix localization of PDHC suggests the involvement of the Lon protease, the enzyme responsible for the degradation of oxidatively modified mitochondrial aconitase (Bota and Davies, 2002).
Another important conclusion reached from comparisons of 3-nitrotyrosine immunoreactivity is that normoxic resuscitation reduces oxidative alterations to brain proteins during reperfusion after global cerebral ischemia. While there is evidence for reduction in brain lipid oxidation by normoxic compared with hyperoxic resuscitation (Liu et al, 1998), this is the first study to document the effects of resuscitative FiO2 on protein oxidation and specifically on tyrosine nitration. Elevated nitrotyrosine, measured using either antibodies or chemical analysis, is associated with neuronal death and poor neurologic outcome in animal models of both global and focal cerebral ischemia and traumatic brain injury (Eliasson et al, 1999; Hall et al, 2004; Karabiyikoglu et al, 2003; Martin et al, 2000; Ste-Marie et al, 2001; Takizawa et al, 1999; Tan et al, 2001; Tanaka et al, 1997). Moreover, elevated 3-nitrotyrosine immunostaining is apparent in the infarct area after ischemic stroke in humans (Forster et al, 1999). Postischemic tyrosine nitration is caused primarily by increased production of peroxynitrite, as a consequence of elevated generation of superoxide, nitric oxide, or both molecules. Elevated intracellular and intramitochondrial Ca2 + likely contribute to the increased production of these species during reperfusion (Haynes et al, 2004; Starkov et al, 2004). Brain tissue [O2] concentration rises above baseline during the first 30 mins or more of reperfusion (Halsey Jr et al, 1991), due to a transient postischemic hyperemia and decreased O2 utilization. While brain oxidative energy metabolism is saturated by O2 at levels approximately 50% less than normal tissue pO2, other activities including those that produce reactive O2 species have much lower affinities for O2 (Erecinska and Silver, 2001). In particular, superoxide production by respiring mitochondria rises with increasing [O2] up to 100% O2 saturation (Kudin et al, 2004; Turrens et al, 1982). The substantial elevation of hippocampal 3-nitrotyrosine immunostaining in the hyperoxic reperfused animals could therefore be due to increased 3-nitrotyrosine generation caused by elevated superoxide production by mitochondria and other sources, for example, cyclooxygenases and oxidases, among others. Increased production of nitric oxide by one or more O2 utilizing nitric oxide synthases may also contribute to elevated postischemic 3-nitrotyrosine formation (Griffiths et al, 2002). The influence of tissue oxygenation on the relative contributions of these sources of reactive oxygen and nitrogen species to oxidative protein modifications is important for understanding the pathophysiology of reperfusion brain injury and damage to other tissues, for example, the lung, caused by exposure to abnormally high levels of O2 (Lorch et al, 2004).
While the association between 3-nitrotyrosine immunoreactivity and loss of PDHC immunostaining supports the role of oxidative stress in reperfusion-dependent alterations to this important enzyme, the molecular basis for loss of PDHC E1α immunoreactivity is not yet known. Pyruvate dehydrogenase complex enzyme activity can be inhibited by exposure to peroxynitrite (Bubber et al, 2004), but it is similarly impaired in the presence of hydroxyl radical (Bogaert et al, 1994; Tabatabaie et al, 1996). In addition to determining the molecular mechanism(s) responsible for PDHC inactivation during ischemia/reperfusion, the impact that depressed PDHC activity has on cerebral energy metabolism is perhaps an even more important goal. Our previous finding that hyperoxic resuscitation exacerbates postischemic brain tissue lactic acidosis suggests that oxidative impairment of PDHC activity may limit postischemic cerebral aerobic energy metabolism (Liu et al, 1998). Although ischemia/ reperfusion can also inhibit the flow of electrons through respiratory chain complexes (Fiskum et al, 1999), the observation that there is an oxidized shift in the redox state of brain pyridine nucleotides (NAD(H), NADP(H)) during reperfusion indicates that the rate-limiting step in postischemic aerobic metabolism is proximal to the electron transport chain (Rosenthal et al, 1995). The additional finding that hyperoxic reperfusion exacerbates the oxidized shift in redox state indicates that the limiting step is sensitive to oxidative stress (Feng et al, 1998). Pyruvate dehydrogenase complex, as the only bridge between anaerobic and aerobic metabolism, is an attractive candidate as the limiting step; however, inhibition of other enzymes, for example, aconitase and 2-oxoglutarate dehydrogenase, could also explain postischemic metabolic dysfunction. The observations of ischemic neuroprotection afforded by administration of ketone bodies or acetyl-L-carnitine that can potentially be metabolized to acetylCoA, the product of the PDHC reaction and precursor to downstream citric acid cycle intermediates, is further evidence, albeit indirect, that damage to PDHC has important metabolic and neurologic consequences (Dardzinski et al, 2000; Gueldry and Bralet, 1994; Rosenthal et al, 1992; Sims and Heward, 1994). Metabolic studies using 13C-NMR spectroscopy in our clinically relevant canine model of global ischemic brain injury are underway to provide further insight into the relationship between hyperoxic resuscitation, oxidative stress, and metabolic dysfunction.
The results of this study using sterologic quantification of hippocampal neuronal injury at 24 h reperfusion also support our previous findings that short-term neurologic outcome is improved with normoxic compared with hyperoxic resuscitation. As described previously, this canine cardiac arrest/ resuscitation model produces widespread neuronal injury throughout the hippocampus as well as some neuronal layers of the superior frontal cortex (Rosenthal et al, 2003). The present observation that normoxic resuscitation reduces hippocampal injury in all hippocampal areas measured is further evidence that this intervention has a profound impact on the neuropathology associated with global cerebral ischemia and reperfusion. This conclusion should not, however, be extrapolated to other forms of acute brain injury, for example, stroke and trauma, as evidence from some models of these disorders suggest that hyperoxia can under some circumstances be beneficial (Flynn and Auer, 2002; Kim et al, 2005; Liu et al, 2004; Menzel et al, 1999; Singhal et al, 2005).
The time during which the brain is exposed to high O2 can also determine whether hyperoxia is helpful or detrimental. For instance, we found that when dogs were exposed to hyperbaric O2 1 to 2 h after cardiac arrest and resuscitation, both short-term neurologic and histologic outcome were improved (Rosenthal et al, 2003). Our current working hypothesis is that abnormally high brain O2 levels are toxic primarily during the first 30 mins after global cerebral ischemia, when abnormal intracellular Ca2 +, pH, and redox state all promote the production of reactive O2 and N2 species and impair their detoxification. At later times when these factors normalize, high tissue O2 can promote recovery, particularly if tissue oxygenation is the limiting factor for aerobic cerebral energy metabolism.
There are several limitations to the conclusions reached by this study based on the fact that only young, healthy female dogs were used and only short-term, that is, 24-h, histologic outcome was measured. Further studies comparing hyperoxic and normoxic reperfusion in males and in aged animals are therefore needed. Studies are in progress to determine if normoxic reperfusion reduces long-term neuronal death and neurologic impairment. Nevertheless, our present results together with our earlier studies and those of several other laboratories question the appropriateness of American Heart Association cardiac resuscitation guidelines that include the indiscriminate use of 100% ventilatory O2 immediately after cardiac arrest.
Figure 7.

Photomicrographs illustrating hippocampal PDHC E1α immunostaining in microscopic fields used for quantification. In the hyperoxic resuscitated animal (H), the PDHC E1α immunostaining is considerably reduced compared with the nonischemic sham-operated control animal (S) or to the normoxic resuscitated animal (N) in the CA1, CA3, and DG regions of the middle hippocampus. Scale bar = 20 μm.
Acknowledgments
The authors would like to thank Ms Kinny Jones and Dr Cynthia Cotta-Cumba for expert technical assistance and Dr Christos Chinopoulos, and Ms Takisha Schulterbrandt for helpful advice.
This work was supported by NIH grants NS34152 and NS49425 to G Fiskum and AHA grant 0215331U to E Martin.
References
- Berghorn KA, Bonnett JH, Hoffman GE. cFos immunoreactivity is enhanced with biotin amplification. J Histochem Cytochem. 1994;42:1635–42. doi: 10.1177/42.12.7983364. [DOI] [PubMed] [Google Scholar]
- Berghorn KA, Le WW, Sherman TG, Hoffman GE. Suckling stimulus suppresses messenger RNA for tyrosine hydroxylase in arcuate neurons during lactation. J Comp Neurol. 2001;438:423–32. doi: 10.1002/cne.1325. [DOI] [PubMed] [Google Scholar]
- Bogaert YE, Rosenthal RE, Fiskum G. Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic Biol Med. 1994;16:811–20. doi: 10.1016/0891-5849(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Bogaert YE, Sheu KF, Hof PR, Brown AM, Blass JP, Rosenthal RE, et al. Neuronal subclass-selective loss of pyruvate dehydrogenase immunoreactivity following canine cardiac arrest and resuscitation. Exp Neurol. 2000;161:115–26. doi: 10.1006/exnr.1999.7250. [DOI] [PubMed] [Google Scholar]
- Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–80. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- Bubber P, Ke ZJ, Gibson GE. Tricarboxylic acid cycle enzymes following thiamine deficiency. Neurochem Int. 2004;45:1021–8. doi: 10.1016/j.neuint.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Dardzinski BJ, Smith SL, Towfighi J, Williams GD, Vannucci RC, Smith MB. Increased plasma beta-hydroxybutyrate, preserved cerebral energy metabolism, and amelioration of brain damage during neonatal hypoxia ischemia with dexamethasone pre-treatment. Pediatr Res. 2000;48:248–55. doi: 10.1203/00006450-200008000-00021. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–8. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–76. doi: 10.1016/s0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- Facchinetti F, Dawson VL, Dawson TM. Free radicals as mediators of neuronal injury. Cell Mol Neurobiol. 1998;18:667–82. doi: 10.1023/a:1020685903186. [DOI] [PubMed] [Google Scholar]
- Feng ZC, Sick TJ, Rosenthal M. Oxygen sensitivity of mitochondrial redox status and evoked potential recovery early during reperfusion in post-ischemic rat brain. Resuscitation. 1998;37:33–41. doi: 10.1016/s0300-9572(98)00031-8. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Liu Y, Bogaert YE, Rosenthal RE. Acetyl-L-carnitine stimulates cerebral oxidative metabolism and inhibits protein oxidation following cardiac arrest in dogs. In: Krieglstein J, Oberpichler-Schwenk H, editors. Pharmacology of cerebral ischemia 1992. Stuttgart: Wissenschaftliche Verlagsgesellschaft mbH; 1992. pp. 487–91. [Google Scholar]
- Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neuro-degenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–69. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52:566–72. doi: 10.1002/ana.10322. [DOI] [PubMed] [Google Scholar]
- Forster C, Clark HB, Ross ME, Iadecola C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol (Berl) 1999;97:215–20. doi: 10.1007/s004010050977. [DOI] [PubMed] [Google Scholar]
- Fukuchi T, Katayama Y, Kamiya T, McKee A, Kashiwagi F, Terashi A. The effect of duration of cerebral ischemia on brain pyruvate dehydrogenase activity, energy metabolites, and blood flow during reperfusion in gerbil brain. Brain Res. 1998;792:59–65. doi: 10.1016/s0006-8993(98)00121-8. [DOI] [PubMed] [Google Scholar]
- Griffiths C, Garthwaite G, Goodwin DA, Garthwaite J. Dynamics of nitric oxide during simulated ischaemia–reperfusion in rat striatal slices measured using an intrinsic biosensor, soluble guanylyl cyclase. Eur J Neurosci. 2002;15:962–8. doi: 10.1046/j.1460-9568.2002.01930.x. [DOI] [PubMed] [Google Scholar]
- Gueldry S, Bralet J. Effect of 1,3-butanediol on cerebral energy metabolism. Comparison with beta-hydroxybutyrate. Metab Brain Dis. 1994;9:171–81. doi: 10.1007/BF01999770. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Halsey JH, Jr, Conger KA, Garcia JH, Sarvary E. The contribution of reoxygenation to ischemic brain damage. J Cereb Blood Flow Metab. 1991;11:994–1000. doi: 10.1038/jcbfm.1991.166. [DOI] [PubMed] [Google Scholar]
- Haynes V, Elfering S, Traaseth N, Giulivi C. Mitochondrial nitric-oxide synthase: enzyme expression, characterization, and regulation. J Bioenerg Biomembr. 2004;36:341–6. doi: 10.1023/B:JOBB.0000041765.27145.08. [DOI] [PubMed] [Google Scholar]
- Henry WW, Jr, Medlock KL, Sheehan DM, Scallet AC. Detection of estrogen receptor (ER) in the rat brain using rat anti-ER monoclonal IgG with the unlabeled antibody method. Histochemistry. 1991;96:157–62. doi: 10.1007/BF00315987. [DOI] [PubMed] [Google Scholar]
- Hof PR, Morrison JH. Neurofilament protein defines regional patterns of cortical organization in the macaque monkey visual system: a quantitative immunohistochemical analysis. J Comp Neurol. 1995;352:161–86. doi: 10.1002/cne.903520202. [DOI] [PubMed] [Google Scholar]
- Hof PR, Nimchinsky EA. Regional distribution of neurofilament and calcium-binding proteins in the cingulate cortex of the macaque monkey. Cereb Cortex. 1992;2:456–67. doi: 10.1093/cercor/2.6.456. [DOI] [PubMed] [Google Scholar]
- Hof PR, Rosenthal RE, Fiskum G. Distribution of neurofilament protein and calcium-binding proteins parvalbumin, calbindin, and calretinin in the canine hippocampus. J Chem Neuroanat. 1996;11:1–12. doi: 10.1016/0891-0618(96)00117-2. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Smith MS, Fitzsimmons MD. Detecting steroidal effects on immediate early gene expression in the hypothalamus. Neuroprotocols. 1992;1:52–66. [Google Scholar]
- Karabiyikoglu M, Han HS, Yenari MA, Steinberg GK. Attenuation of nitric oxide synthase isoform expression by mild hypothermia after focal cerebral ischemia: variations depending on timing of cooling. J Neurosurg. 2003;98:1271–6. doi: 10.3171/jns.2003.98.6.1271. [DOI] [PubMed] [Google Scholar]
- Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neurol. 2005;57:571–5. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Rosenthal RE, Mikolajczyk J, Stennicke HR, Wiesenthal T, Mai J, et al. Early processing of Bid and caspase-6, -8, -10, -14 in the canine brain during cardiac arrest and resuscitation. Exp Neurol. 2004;189:261–79. doi: 10.1016/j.expneurol.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–35. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- Leonov Y, Sterz F, Safar P, Radovsky A, Oku K, Tisherman S, et al. Mild cerebral hypothermia during and after cardiac arrest improves neurologic outcome in dogs. J Cereb Blood Flow Metab. 1990;10:57–70. doi: 10.1038/jcbfm.1990.8. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:343–9. doi: 10.1097/01.WCB.0000110047.43905.01. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. 1998;29:1679–86. doi: 10.1161/01.str.29.8.1679. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rosenthal RE, Starke-Reed P, Fiskum G. Inhibition of postcardiac arrest brain protein oxidation by acetyl-L-carnitine. Free Radic Biol Med. 1993;15:667–70. doi: 10.1016/0891-5849(93)90171-p. [DOI] [PubMed] [Google Scholar]
- Lorch SA, Munson D, Lightfoot RT, Ischiropoulos H. Oxygen tension and inhaled nitric oxide modulate pulmonary levels of S-nitrosocysteine and 3-nitrotyrosine in rats. Pediatr Res. 2004;56:345–52. doi: 10.1203/01.PDR.0000134256.30519.9B. [DOI] [PubMed] [Google Scholar]
- Martin E, Rosenthal RE, Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2004;79:240–7. doi: 10.1002/jnr.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Sieber FE, Traystman RJ. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with differential alterations in metabotropic glutamate receptor signaling pathways. J Cereb Blood Flow Metab. 2000;20:153–67. doi: 10.1097/00004647-200001000-00020. [DOI] [PubMed] [Google Scholar]
- Menzel M, Doppenberg EM, Zauner A, Soukup J, Reinert MM, Bullock R. Increased inspired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactate levels after severe human head injury. J Neurosurg. 1999;91:1–10. doi: 10.3171/jns.1999.91.1.0001. [DOI] [PubMed] [Google Scholar]
- Mickel HS, Kempski O, Feuerstein G, Parisi JE, Webster HD. Prominent white matter lesions develop in Mongolian gerbils treated with 100% normobaric oxygen after global brain ischemia. Acta Neuropathol (Berl) 1990;79:465–72. doi: 10.1007/BF00296104. [DOI] [PubMed] [Google Scholar]
- Mickel HS, Vaishnav YN, Kempski O, von Lubitz D, Weiss JF, Feuerstein G. Breathing 100% oxygen after global brain ischemia in Mongolian Gerbils results in increased lipid peroxidation and increased mortality. Stroke. 1987;18:426–30. doi: 10.1161/01.str.18.2.426. [DOI] [PubMed] [Google Scholar]
- Milner TA, Aoki C, Sheu KF, Blass JP, Pickel VM. Light microscopic immunocytochemical localization of pyruvate dehydrogenase complex in rat brain: topographical distribution and relation to cholinergic and catecholaminergic nuclei. J Neurosci. 1987;7:3171–90. doi: 10.1523/JNEUROSCI.07-10-03171.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci USA. 1990;87:5144–7. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson J, Brandeis L, Goldstein M. Tyrosine hydroxylase immunoreactivity in familial dysautonomia. Science. 1979;206:71–2. doi: 10.1126/science.39339. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Feng ZC, Raffin CN, Harrison M, Sick TJ. Mitochondrial hyperoxidation signals residual intracellular dysfunction after global ischemia in rat neocortex. J Cereb Blood Flow Metab. 1995;15:655–65. doi: 10.1038/jcbfm.1995.81. [DOI] [PubMed] [Google Scholar]
- Rosenthal RE, Silbergleit R, Hof PR, Haywood Y, Fiskum G. Hyperbaric oxygen reduces neuronal death and improves neurological outcome after canine cardiac arrest. Stroke. 2003;34:1311–6. doi: 10.1161/01.STR.0000066868.95807.91. [DOI] [PubMed] [Google Scholar]
- Rosenthal RE, Williams R, Bogaert YE, Getson PR, Fiskum G. Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-L-carnitine. Stroke. 1992;23:1312–7. doi: 10.1161/01.str.23.9.1312. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Hof PR. Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat. 2000;20:93–114. doi: 10.1016/s0891-0618(00)00066-1. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–31. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- Schoder H, Knight RJ, Kofoed KF, Schelbert HR, Buxton DB. Regulation of pyruvate dehydrogenase activity and glucose metabolism in post-ischaemic myocardium. Biochim Biophys Acta. 1998;1406:62–72. doi: 10.1016/s0925-4439(97)00088-4. [DOI] [PubMed] [Google Scholar]
- Sheu KF, Lai JC, Blass JP. Pyruvate dehydrogenase phosphate (PDHb) phosphatase in brain: activity, properties, and subcellular localization. J Neurochem. 1983;40:1366–72. doi: 10.1111/j.1471-4159.1983.tb13578.x. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Roth KA. Double immunofluorescent staining using two unconjugated primary antisera raised in the same species. J Histochem Cytochem. 1996;44:1331–5. doi: 10.1177/44.11.8918908. [DOI] [PubMed] [Google Scholar]
- Sims NR, Heward SL. Delayed treatment with 1,3-butanediol reduces loss of CA1 neurons in the hippocampus of rats following brief forebrain ischemia. Brain Res. 1994;662:216–22. doi: 10.1016/0006-8993(94)90815-x. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, et al. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36:797–802. doi: 10.1161/01.STR.0000158914.66827.2e. [DOI] [PubMed] [Google Scholar]
- Starke-Reed PE, Oliver CN. Protein oxidation and proteolysis during aging and oxidative stress. Arch Biochem Biophys. 1989;275:559–67. doi: 10.1016/0003-9861(89)90402-5. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–64. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Ste-Marie L, Hazell AS, Bemeur C, Butterworth R, Montgomery J. Immunohistochemical detection of inducible nitric oxide synthase, nitrotyrosine and manganese superoxide dismutase following hyperglycemic focal cerebral ischemia. Brain Res. 2001;918:10–9. doi: 10.1016/s0006-8993(01)02903-1. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- Tabatabaie T, Potts JD, Floyd RA. Reactive oxygen species-mediated inactivation of pyruvate dehydrogenase. Arch Biochem Biophys. 1996;336:290–6. doi: 10.1006/abbi.1996.0560. [DOI] [PubMed] [Google Scholar]
- Takizawa S, Fukuyama N, Hirabayashi H, Nakazawa H, Shinohara Y. Dynamics of nitrotyrosine formation and decay in rat brain during focal ischemia–reperfusion. J Cereb Blood Flow Metab. 1999;19:667–72. doi: 10.1097/00004647-199906000-00010. [DOI] [PubMed] [Google Scholar]
- Tan S, Bose R, Derrick M. Hypoxia–ischemia in fetal rabbit brain increases reactive nitrogen species production: quantitative estimation of nitrotyrosine. Free Radic Biol Med. 2001;30:1045–51. doi: 10.1016/s0891-5849(01)00499-3. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Shirai T, Nagata E, Dembo T, Fukuuchi Y. Immunohistochemical detection of nitrotyrosine in postischemic cerebral cortex in gerbil. Neurosci Lett. 1997;235:85–8. doi: 10.1016/s0304-3940(97)00725-8. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Freeman BA, Crapo JD. Hyperoxia increases H2O2 release by lung mitochondria and microsomes. Arch Biochem Biophys. 1982;217:411–21. doi: 10.1016/0003-9861(82)90519-7. [DOI] [PubMed] [Google Scholar]
- Viera L, Ye YZ, Estevez AG, Beckman JS. Immunohistochemical methods to detect nitrotyrosine. Methods Enzymol. 1999;301:373–81. doi: 10.1016/s0076-6879(99)01101-5. [DOI] [PubMed] [Google Scholar]
- Waring WS, Thomson AJ, Adwani SH, Rosseel AJ, Potter JF, Webb DJ, et al. Cardiovascular effects of acute oxygen administration in healthy adults. J Cardiovasc Pharmacol. 2003;42:245–50. doi: 10.1097/00005344-200308000-00014. [DOI] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–9. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–97. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Zaidan E, Sims NR. Reduced activity of the pyruvate dehydrogenase complex but not cytochrome c oxidase is associated with neuronal loss in the striatum following short-term forebrain ischemia. Brain Res. 1997;772:23–8. doi: 10.1016/s0006-8993(97)00833-0. [DOI] [PubMed] [Google Scholar]
- Zwemer CF, Whitesall SE, D’Alecy LG. Cardiopulmonary–cerebral resuscitation with 100% oxygen exacerbates neurological dysfunction following nine minutes of normothermic cardiac arrest in dogs. Resuscitation. 1994;27:159–70. doi: 10.1016/0300-9572(94)90009-4. (published erratum appears in Resuscitation 1994; 27(3):267) [DOI] [PubMed] [Google Scholar]