

Abstract
The largest kindred with inherited prion disease P102L, historically Gerstmann-Sträussler-Scheinker syndrome, originates from central England, with émigrés now resident in various parts of the English-speaking world. We have collected data from 84 patients in the large UK kindred and numerous small unrelated pedigrees to investigate phenotypic heterogeneity and modifying factors. This collection represents by far the largest series of P102L patients so far reported. Microsatellite and genealogical analyses of eight separate European kindreds support multiple distinct mutational events at a cytosine-phosphate diester-guanidine dinucleotide mutation hot spot. All of the smaller P102L kindreds were linked to polymorphic human prion protein gene codon 129M and were not connected by genealogy or microsatellite haplotype background to the large kindred or each other. While many present with classical Gerstmann-Sträussler-Scheinker syndrome, a slowly progressive cerebellar ataxia with later onset cognitive impairment, there is remarkable heterogeneity. A subset of patients present with prominent cognitive and psychiatric features and some have met diagnostic criteria for sporadic Creutzfeldt-Jakob disease. We show that polymorphic human prion protein gene codon 129 modifies age at onset: the earliest eight clinical onsets were all MM homozygotes and overall age at onset was 7 years earlier for MM compared with MV heterozygotes (P = 0.02). Unexpectedly, apolipoprotein E4 carriers have a delayed age of onset by 10 years (P = 0.02). We found a preponderance of female patients compared with males (54 females versus 30 males, P = 0.01), which probably relates to ascertainment bias. However, these modifiers had no impact on a semi-quantitative pathological phenotype in 10 autopsied patients. These data allow an appreciation of the range of clinical phenotype, modern imaging and molecular investigation and should inform genetic counselling of at-risk individuals, with the identification of two genetic modifiers.
Keywords: P102L, sCJD, early-onset dementia, Gerstmann-Sträussler-Scheinker syndrome, prion disease
Introduction
Prion diseases occur uniquely in sporadic, acquired and inherited forms. Inherited prion disease (IPD) accounts for around 15% of all human prion disease (Collinge, 2005). IPD also forms a significant proportion of familial young onset dementia (Finckh et al., 2000). The longer average duration of IPD, and need for genetic counselling of at-risk family members, results in a larger impact on health service and social care providers when compared with other forms of prion disease. IPD is associated with point mutations in the human prion protein gene (PRNP), premature stop codon mutations and alteration in the number of octapeptide repeat insertions (OPRI) in the unstructured region of the N-terminal domain (Mead, 2006).
The P102L mutation has classically been associated with Gerstmann-Sträussler-Scheinker syndrome (GSS). This is characterized clinicopathologically by onset of cerebellar ataxia with dementia occurring much later and is associated with multicentric PrP amyloid plaques in the cerebellum and cerebral cortex. There is considerable heterogeneity, however, even between first-degree relatives, which has led to delayed diagnosis in those not presenting with typical GSS. The heterogeneity in age at onset, clinical symptoms and pathological findings raises questions about modifying factors that may play a role in other aetiologic categories of prion diseases and other forms of neurodegenerative disease. Phenotypic variability even within the same IPD P102L family does not always reflect the classical symptom pattern or pathological changes (Hainfellner et al., 1995; Piccardo et al., 1998; Arata et al., 2006). It is for this reason that recent descriptions have favoured the use of a molecular genetic classification rather than the traditional clinicopathological ones (Collinge et al., 1992, 2005). Explanations for this heterogeneity have begun to emerge, although the reasons for it remain largely unexplained (Parchi et al., 1998; Wadsworth et al., 2006).
The large family from central England, originally referred to as the ‘W’ kindred, was first reported 30 years ago with an unusual inherited neurodegenerative disease (Cameron and Crawford, 1974). While autosomal dominant transmission was recognized early on, an atypical form of Huntington's disease was suspected to be responsible. The observation that a member of the family presented with the classical clinical and neuropathological features of sporadic Creutzfeldt-Jakob disease (sCJD) was confused when other affected family members fitted neither clinical nor pathological criteria for this disease (Rosenthal et al., 1976). An inherited susceptibility to neurological disease of different types including sCJD was therefore postulated (Rosenthal et al., 1976; Adam et al., 1982). Following the demonstration that these heritable disorders may also be transmissible to experimental animals (Masters et al., 1981), the possibility of an agent originating in the genome, and not requiring exogenous aetiological factors for its development, was proposed (Baker et al., 1985). In 1989, linkage of the P102L PRNP mutation to GSS in members of this kindred was reported (Hsiao et al., 1989), only the second reported mutation in a neurodegenerative disease [the earlier being the report of a six OPRI PRNP mutation in another kindred (Owen et al., 1989)].
In this article, we present genetic and genealogical investigation of multiple international P102L pedigrees that we have identified over 17 years of PRNP diagnostic testing in London. Microsatellite and genealogical analyses have allowed us to build an updated and expanded family tree for the ‘W’ kindred, the largest P102L kindred reported to date. Additionally, we present genealogical and molecular evidence of multiple independent P102L mutations from the United Kingdom and elsewhere in the English-speaking world. This large collection of a rare inherited disease documents the modern neurological and molecular investigations and marked phenotypic variability of P102L prion disease. A genetic correlation with clinical and pathological parameters has also been made, identifying two previously uncharacterized modifiers of the phenotype of this mutation.
Materials and Methods
Clinical information
For recent patients, a standardized history and examination was performed either in hospital, where additional investigations such as MRI and EEG were carried out, or on domiciliary visits. Peripheral neurophysiological examinations were performed if clinically indicated by dysaesthesiae or lower motor neuron signs. Neuropsychological assessments were also carried out where patients consented to them. Routine investigations and investigations into other potential causes of neurological deterioration had often been carried out before diagnosis was reached and the results of these were sought. For historical patients, clinical histories and examination details were obtained where possible from medical notes completed at the time of clinical review. In some patients, notes had been destroyed prior to the beginning of this study and information on presentation and cause of death was sought from relatives and death certificates. Historical patients in whose cases tissue did not survive for DNA analysis have been considered affected if there is evidence of a neurological disease causing paralysis, cerebellar features and/or dementia occurring before the age of 65 years without an alternative cause being established. Patients were classified based on current neurological assessment or medical records, according to their presenting features. Individuals who presented with prominent cognitive or psychiatric features were contrasted with those without such features, who tended to present with unsteadiness and weakness in the absence of frank cognitive or psychiatric features.
Genealogical investigations
Genealogical work was carried out on all P102L patients from the English-speaking world with tissue samples and/or clinical information donated to the MRC Prion Unit, London. This was initially limited to family tree information from the informant (patient or relative). Further work was carried out where a geographical proximity to a known patient from the ‘W’ kindred was established and later where microsatellite haplotyping suggested a common haplotype to an extant patient. Information was gathered from census returns; birth, marriage and death certification as well as parish records; and work-house, asylum and hospital records, where these could be traced. The results of clinical investigations were sought where hospital notes survived. Pathological reports have been sought where no tissue survives for examination using modern techniques.
Molecular analysis and investigations
DNA was extracted from peripheral blood lymphocytes, and in some patients from frozen post-mortem brain tissue, using Nucleon Blood DNA kit or a modified phenol–chloroform extraction. The open reading frame was screened for the presence of deletions or insertions in PRNP using size fractionation of a PCR amplicon. The PRNP open reading frame was sequenced using an ABI-377 automated DNA sequencer. All previously identified P102L patients with DNA stored in the MRC Prion Unit where research consent had been given underwent PRNP locus haplotype analysis using 12 microsatellites on chromosome 20, 3–6 Mb (NCBI Entrez-gene mapviewer, http://www.ncbi.nlm.nih.gov/sites/entrez). PCR using fluorescent primers specific for the microsatellite regions were carried out followed by size discrimination on an acrylamide gel using MegaBace sequence analyser 3.0 software (Amersham Biosciences, 2001). Microsatellite haplotypes were determined empirically from pedigree information. Statistical work was carried out using SPSS version 12.0.1 for Windows (SPSS.com) and Excel software (Microsoft). Estimates of effect size were made by linear regression analysis. Statistical genetic analysis used PLINK software (version 0.99, http://pngu.mgh.harvard.edu/purcell/plink/).
Neuropathology
Five of the 10 patients described here have had neuropathology reported previously, primarily before the availability of PrP immunohistochemistry (Rosenthal et al., 1976; Adam et al., 1982). Samples were taken from formalin-fixed brain, where available. Cortex (frontal and occipital), basal ganglia and cerebellum were sampled. Tissue was fixed in 10% buffered formal saline, followed by incubation in 98% formic acid for 1 h. Following further washing for 24 h in 10% buffered formal saline, tissue samples were processed and embedded in paraffin wax. Sections were cut at a nominal thickness of 4 µm, treated with 98% formic acid for 5 min and then boiled in a low ionic strength buffer (2.1 mM Tris, 1.3 mM EDTA, 1.1 mM sodium citrate, pH 7.8) for 20 min. All blocks were stained with haematoxylin and eosin and with immunohistochemistry using anti-PrP monoclonal antibodies ICSM18 (1:10 000 dilution of a 1 mg/ml solution) which labels mutant and wild-type PrP and ICSM35 (1:3000 dilution of a 3 mg/ml solution) which labels only mutant PrP (D-Gen, London) and were incubated at 42°C for 32 min. Haematoxylin was used as the counterstain. Haematoxylin and eosin staining of serial sections was done using conventional methods. Appropriate controls were used throughout.
Samples were examined by the same pathologist (S.B.), blinded to clinical details, and comparisons made between them. A semi-quantitative scale was used to grade spongiform change and PrP deposition (Fig. 1). Diffuse and plaque-related PrP deposition was documented and contrasted. Clinicopathological comparisons were made following this, with note being made of phenotype, PRNP codon 129 and apolipoprotein E (APOE) genotypes.
Fig. 1.

Representative histology from P102L patients showing appearances graded semi-quantitatively. (A, C and E) show progressively worsening degrees of spongiform change on haematoxylin and eosin stain of brain slices. (B, D and F) show increasing levels of plaque deposition, as demonstrated by immunohistochemistry with anti-PrP antibody ICSM 35. Views in the left-hand column do not necessarily correspond to the same region as the right column. A, C and D are from Patient 2.VII.2. B and E are from Patient VII.4, while panel F is from Patient 3.VIII.1. The scaling bar shown is 100 µM.
Results
Genealogy and genetic studies of shared ancestry
Eighty-four patients were identified from what were initially considered to be 15 separate kindreds. Sixty of these patients presented for the first time, while six are patients who were reported previously but in whose cases significant additional information has been obtained and is presented here. Twenty-two of the total 84 patients, including members of 14 kindreds, had been seen at the National Prion Clinic or on domiciliary visits by clinic staff. Eight were seen and examined by T.W. Of the remaining 62 patients, 5 are contemporary patients seen at other neurological centres and where DNA or tissue samples and clinical data has been supplied with patients’ consent and ethics committee approval. Fifty-seven patients are historical, typically being the ancestors of contemporarily identified patients, in whom a diagnosis of young onset neurological disease had been made or had been reported by immediate descendents (see Materials and methods section). Among the historical patients, a variety of clinical diagnoses had been applied and were obtained from hospital notes and death certificates (Table 1). While diagnostic labels have changed over time, those used still provide information about predominant symptoms and clinical impressions of physicians at the time.
Table 1.
Listed causes of death from certificates for historical patients of P102L IPD
| Diagnosis | Number of patients |
|---|---|
| Disseminated sclerosis | 6 |
| Cerebellar degeneration | 4 |
| Paralysis | 3 |
| Cerebral degeneration | 1 |
| Cerebral and cerebellar cortical atrophy | 1 |
| Spinal cerebellar atrophy | 1 |
| Progressive cerebellar ataxia | 1 |
| Hereditary progressive ataxia | 1 |
| Olivo-ponto-cerebellar degeneration | 1 |
| Huntington's chorea | 1 |
| Heredo-familial muscular dystrophy | 1 |
| Neurofibromatosis | 1 |
| Intracranial haemorrhage | 1 |
| Binswanger's encephalitis | 1 |
| Multiple cerebrovascular accidents | 1 |
| Senile dementia | 1 |
| Schizophrenia | 1 |
These are listed by frequency. For the many examples with only one individual each given this diagnosis, they have been listed loosely by type in the following order: cerebellar lesion implicated, genetic causes, vascular causes, cognitive/psychiatric and other.
A mutation-associated microsatellite haplotype for the UK IPD P102L ‘W’ kindred was determined (Fig. 2). Five smaller unrelated kindreds shared this haplotype, supporting the existence of a recent common ancestor for all of these patients (shown as kindred 1 in Fig. 3). Further, IPD P102L patients did not share a haplotype with either the large kindred or with each other, suggesting that separate mutational events had been responsible for their occurrence (kindreds 2–9, Fig. 4). A final kindred where no DNA was obtainable for haplotyping is not included in Fig. 4.
Fig. 2.

Microsatellite genotypes for P102L IPD patients at 13 loci linked to codon 129. Shown left to right 5′–3′ with their physical distance from codon 129 below. PRNP is situated between D20S97 and D20S895. VI.14 is a member of the original ‘W’ kindred. Below are representative individuals from the five kindreds sharing a microsatellite background. The disease-associated haplotype is shown in bold. The bottom half of the diagram shows the microsatellite genotypes of representatives of the other eight P102L kindreds. Although it has not been possible to derive a mutation-associated haplotype for these, none appear to share a haplotype with the ‘W’ kindred or with each other.
Fig. 3.

Pedigree of the original ‘W’ kindred (in grey) with newly identified and linked kindreds (in black). Only presumed affected patients (filled symbols) or those thought to have transmitted the mutation but not known to be affected (empty symbols) have been included for the sake of simplicity and confidentiality. All of the new patients have been linked genealogically to the original family with the exception of the two small pedigrees on the right of the diagram where possible links are represented with a question mark. Members of these kindreds both share a microsatellite background with the ‘W’ kindred and have ancestors closely located geographically to them. Possible reasons for the inability to uncover an exact link genealogically are discussed in the text.
Fig. 4.

Eight further family trees (six from the UK, two from elsewhere in Europe). Microsatellite haplotype analysis from these patients suggests that none is related to the ‘W’ kindred or to each other. Separate mutational events are therefore likely to have been responsible.
Following the identification of a common mutation-associated haplotype, genealogical work in three patients directly established links by common ancestor with the ‘W’ kindred. Two other families, which shared a haplotype, could not be linked directly to the ‘W’ kindred. In both patients, ancestors were identified in the same or neighbouring villages to the original ‘W’ kindred. In one family, an illegitimate ancestor provides a likely link; his mother was living adjacent to a known member of the ‘W’ kindred, according to contemporary census entries. The inability to link in the final family genealogically may relate to illegitimacy or alternatively may be explained by a common ancestor prior to the end of the 18th century, when the earliest identified shared relatives for the ‘W’ kindred were living. Emigration by family members accounts for the identification of related individuals in other parts of the English-speaking world: Canada, the United States and Australia. Migration to other parts of the United Kingdom explains the lack of geographical proximity to known patients in recent generations and the failure up to now to recognize the common ancestry without supportive genetic evidence of common microsatellite haplotype. The genealogical work performed suggests an earliest common ancestor for the presented patients born in the mid-eighteenth century. However, the mutation itself may have originated before this time.
Clinical findings
Following microsatellite and genealogical studies, 40 new patients were identified from the ‘W’ kindred, likely to be associated with the P102L PRNP mutation. Twenty-eight further patients from nine separate European kindreds apparently not related to each other or to the ‘W’ kindred are also presented. A full list of the major clinical findings is provided in Table 2. Details on each patient are also provided in Appendix 1 of Supplementary data, except in cases of patients previously reported or where patients are still living and details have not been included for the sake of confidentiality.
Table 2.
Clinical features from P102L IPD patients reported
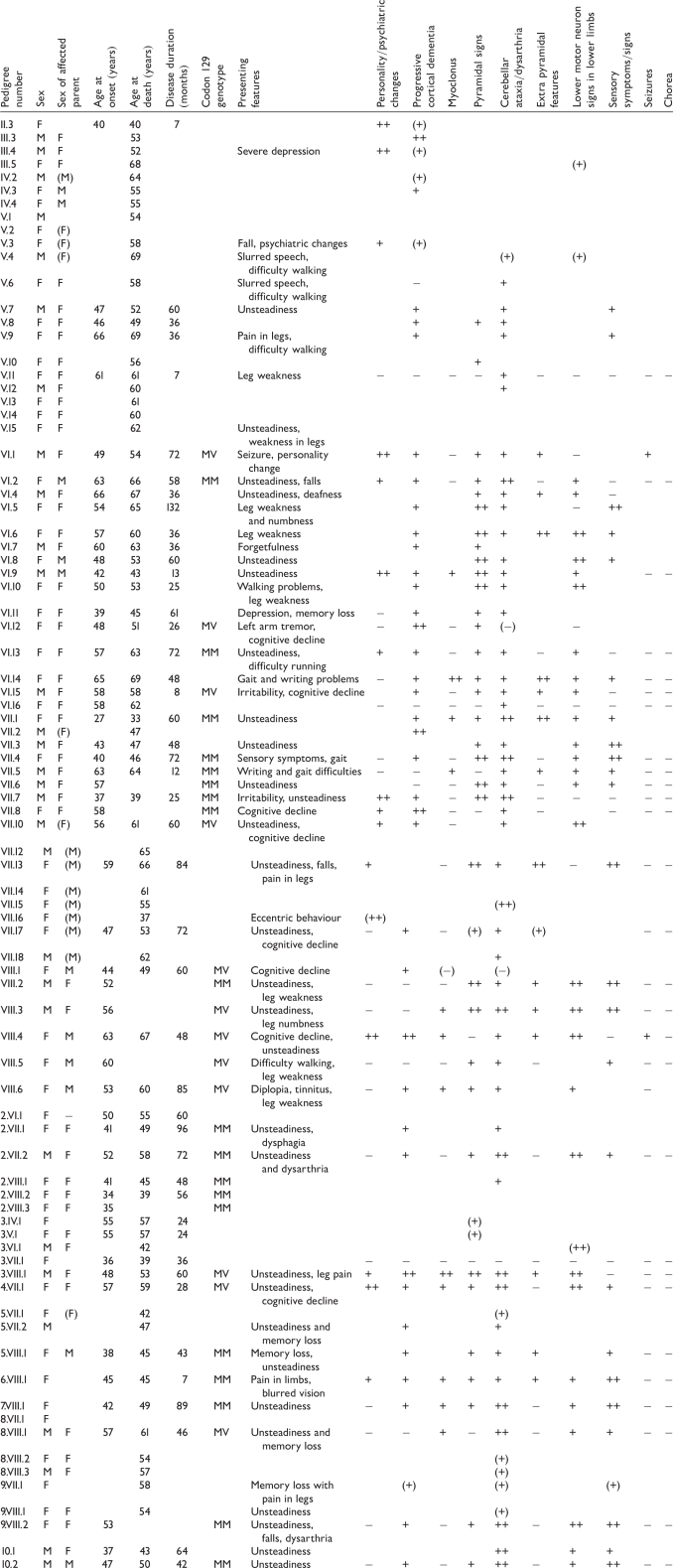 |
‘+’ denotes record of a feature being present. ‘++’ is used where clinicians have recorded findings as ‘severe’ or ‘marked’ or where deficit has interfered with independence. ‘−’ denotes a feature mentioned as being absent by clinicians. Missing data is represented by a blank. Data relying on inference from death certificate details or family memories are surrounded by brackets.
The most commonly occurring primary presenting features were unsteadiness (present in 72% of patients), cognitive symptoms (28%) and leg weakness (22%). Less common were leg pains or dysaesthesiae (16%) and severe psychiatric features (10%). New onset deafness, epileptic seizures, parkinsonism and diplopia occurred as presenting features in individual patients (2% of patients where data were available) (Fig. 5).
Fig. 5.

Clinical features on presentation in P102L IPD. The relative frequency of cognitive symptoms and signs on presentation is notable, along with the commonly occurring peripheral limb symptoms and psychiatric features.
The commonest clinical syndrome was progressive cerebellar ataxia associated with leg weakness and areflexia, similar, but not identical to, the classical GSS phenotype. Indeed, cerebellar signs were present in nearly all of the patients where information could be obtained at some point in the illness (98%). Cognitive features were usually mild initially and developed later in the illness course. Cognitive features were eventually present, however, in the large majority of patients at some point during the disease where information was available (81%), although in many patients these were mild. Lower motor neuron pattern leg signs with areflexia, often associated with objective evidence of muscle weakness and myopathic gait, was present in the majority of patients (79%) (Fig. 6). In one patient (4.VII.1), frank lower limb fasiculations were observed. Sensory symptoms in the legs were also common, with dysaesthesiae and hyperaesthesiae being frequently described by affected patients (69%). Extrapyramidal features, especially parkinsonism and severe psychiatric symptoms such as personality change, delusions, paranoia and visual hallucinations, occurred in around half of patients (48 and 47% of patients, respectively). Apraxia, however, in contrast with patients with PRNP insertional mutations (Collinge et al., 1992, 2005; Mead et al., 2006), was recorded in only a single individual. Myoclonus and seizures occurred in a minority of patients, although still relatively common (36 and 7%, respectively). One patient (VII.2) presented with a prominent generalized dystonia in addition to a cerebellar ataxia. This has not been reported previously in association with IPD P102L and the unusual phenotype in this patient delayed diagnosis until post-mortem examination of brain tissue. No reports of choreic movement disorders or of the dyspraxia and premorbid personality changes commonly reported in some other forms of IPD were obtained (Collinge et al., 1992; Mead et al., 2006).
Fig. 6.

Clinical features reported during course of P102L IPD. The high percentage of patients where cognitive deficits were apparent, even if only in later stages, is notable as is the frequency of sensory signs and lower motor neuron signs. Chorea was not reported or observed in any of the patients presented.
There was a considerable heterogeneity regarding clinical features. Although a majority of patients presented with the phenotype of progressive ataxia with a later onset of cognitive involvement, a significant subset presented with predominantly psychiatric and cognitive features that often remained the major clinical feature, in spite of the later development of cerebellar and other physical signs. A still smaller subset presented with a dementing illness having a rapid and more global pattern of deficits reminiscent of sCJD. In five of the patients presented here, where rapid onset and uncertain or absent family history complicated the clinical picture (VI.15, VI.9, VII.7, 4.VII.1 and 6.VIII.1), the diagnosis of sCJD was considered; indeed, one patient (VI.9) met World Health Organisation's diagnostic criteria for probable and the others possible sCJD.
Mean age at onset was 50 years (range 27–66 years, SD 9.4) while mean age at death was 55 years (range 33–69 years, SD 8.7). Mean duration of illness was 49 months but the range was very wide (7–132 months, SD 26.1). The mutation appears to have occurred in all the patients presented, linked to the commoner PRNP codon 129 methionine allele. As a result, all the patients where genotyping was performed were methionine homozygotic (MM) or methionine/valine heterozygotic (MV). Evidence from some other forms of IPD (but not previously for P102L) shows that codon 129 homozygotes have an earlier age at onset and death than 129 heterozygotes (Collinge et al., 1992; Dlouhy et al., 1992; Mead et al., 2006). Among the P102L patients reported here, MM homozygotes have an earlier age at onset compared with MV heterozygotes (47.3 ± 10.2 versus 54 ± 5.7, P = 0.02, t-test heteroscedastic). When individuals with known codon 129 genotype were ranked by age at onset, the youngest eight (n = 31) were codon 129MM. In addition, the standard deviation in age at onset and death for codon 129MM was much wider than that for codon 129MV. There was no significant difference in duration of illness between the two codon 129 genotype groups.
Intriguingly, the data presented here relates to 54 female affected patients but only 30 males (P = 0.01, binomial test given a predicted proportion of 0.5). As a consequence of this discrepancy, individuals included here were much more likely to have inherited the mutation from their mothers than from their fathers, where such information was available (54 affected mothers, 19 affected fathers, P = 1 × 10−4). The 30 affected males transmitted the mutation to 4 sons and 13 daughters. The 54 affected females transmitted the mutation to 24 sons and 31 daughters. This sex distortion was not significant. A similar effect was also reported in 6-OPRI IPD (53 affected females versus only 33 males, P = 0.008, binomial test) (Mead et al., 2006) and when combined with these data, the overall significance was more marked (109 affected females versus 65 males, P = 0.001). In addition, the proportion of females to males with both mutations was similar (1.6 : 1 in 6-OPRI, 1.9 : 1 in P102L). No differences were found in terms of age at onset or death when comparing male and female affected patients or indeed when comparing those who inherited the mutation from their mothers versus those who inherited it from their fathers.
Neither age at onset nor duration of illness was significantly different between patients with a predominantly cognitive or psychiatric versus those with predominantly cerebellar or motor onset. There was no correlation of codon 129 genotype with these subgroups of cognitive/psychiatric compared with the peripheral/cerebellar onset groups, although numbers were small and in many patients the codon 129 was unavailable (n = 22/44 with missing genotype data).
Electroencephalography
A total of 16 individuals had EEG performed after disease onset and 13 of these demonstrated non-specific abnormalities. All of these abnormal studies were in the context of significant cognitive impairment or psychiatric symptoms and were performed between 6 months and 3 years after onset. One of these individuals also had widespread epileptiform discharges with no apparent clinical correlates. Three further studies were reported as normal. These were 2–3 years after symptoms developed, although none of these patients demonstrated significant cognitive or psychiatric symptoms or signs at the time of the study, each having a predominantly cerebellar syndrome. In one patient (VII.8) an EEG was performed many years before symptom onset when the neurological examination was unremarkable. This was also a normal study, although a repeat 1 year after onset of predominantly cognitive symptoms was mildly, though non-specifically, abnormal, with no periodic sharp wave complexes. In a single patient already reported, periodic sharp wave complexes were found in the context of a sCJD-like clinical picture (Rosenthal et al., 1976).
Electromyography and nerve conduction studies
Ten of the newly reported patients underwent peripheral neurophysiological studies, eight having nerve conduction studies (NCS) in combination with EMG, two having NCS only. In all patients the investigations were indicated by painful sensory symptoms, sometimes accompanied by areflexia and leg weakness at the time of the study. Of the 10 patients who had NCS, 2 had evidence of a mild axonal sensorimotor neuropathy. In the other eight, no abnormalities were picked up on NCS in spite of symptoms and often signs suggestive of peripheral neuropathy. A single patient had EMG evidence of muscle denervation in combination with an axonal neuropathy. EMG was normal in seven others, at least two of whom had objective weakness of the lower limbs on clinical examination. A single patient without NCS or EMG abnormalities in spite of painful sensory symptoms and leg weakness underwent thermal threshold testing. This demonstrated a defect in the lower limbs but not in the upper, consistent with either small fibre neuropathy or central pathway involvement. The patient was cognitively intact at the time of the investigation and therefore able to report the sensations accurately. Overall, there was not a consistent peripheral nerve deficit to account for sensory symptoms or areflexia.
Neuroimaging
Sixteen of the newly presented patients had neuroimaging. Fifteen had MRI performed while a further two also had CT scans. One patient, before the era of routine MRI, had only a CT. Three of the MRIs demonstrated a degree of generalized cerebral atrophy out of keeping with the age of the patient. In one patient, localized cerebellar atrophy was reported. These were all performed in patients with significant cognitive and or psychiatric symptoms. Five had normal MRIs between 1 and 3 years after onset of symptoms. In all of these patients, either cognitive or psychiatric features were absent (four patients) or there were only subtle cognitive and psychiatric symptoms reported (one patient).
Interestingly, four patients who had MRI performed were found to have multiple white matter lesions. In one patient, the combination of clinical signs and MRI appearances led to a diagnosis of Binswanger's disease being made before the correct diagnosis was reached. All of the CT scans performed were within normal limits. None of the MRI scans reported showed any changes associated with prion disease of other subtypes: the ‘pulvinar’ sign or cortical or basal ganglia high signal (Fig. 7) (Macfarlane et al., 2007).
Fig. 7.

MRI findings in P102L IPD. (A) Sagittal T1-weighted image (of 2.VII.2) showing cerebellar atrophy. (B) and (C) Axial T2-weighted images (of VI.2) showing multiple white matter lesions in the basal ganglia. Similar findings were found in two other patients leading in one to a diagnosis of Binswanger's disease being made in combination with the clinical picture. These findings are probably incidental but the possibility of a link to P102L IPD remains.
Neuropsychology
A total of 11 patients not previously reported had formal neuropsychological assessment after symptom onset, exploring two or more cognitive domains. In nine of these, evidence for cognitive deficits with decline from predicted pre-morbid functioning was elicited, even where bedside assessment suggested normal cognition. Although two further patients had no deficits on assessment in spite of clearly symptomatic disease, no formal assessment of executive function or attention had been performed in one and full verbal and performance IQ had not been completed in the other. Deficits detected on neuropsychological assessment were widespread, including frontal, subcortical and attentional abnormalities. However, these deficits were patchy, with no clear consistent pattern emerging. Three patients with a predominantly psychiatric or cognitive onset (VII.8, VIII.2 and 5.VIII.1) demonstrated evidence of frontal dysfunction. Patients with a predominantly cerebellar onset performed better on these tests, providing objective evidence for the distinction.
Cerebrospinal fluid
Nine individuals had CSF taken for analysis. In one patient there was borderline elevation of protein. In this patient oligoclonal bands were reported although no record of a serum sample for comparison has been found. In all other patients where CSF was examined, the samples were acellular with normal protein and glucose levels. In only three patients was CSF examined for the presence of the 14-3-3 protein, a non-specific marker of neuronal cell death, which is nonetheless useful in the diagnosis of sCJD (Sanchez-Juan et al., 2006). 14-3-3 was positive and s100b was elevated in two of these 14-3-3-positive patients, while in the third, s100b and neuron-specific enolase were elevated but 14-3-3 protein was not detected. The P102L IPD patients with positive 14-3-3 were clinically heterogeneous, one being a rapid onset patient where sCJD was considered in the differential, the other being a slowly progressive cerebellar syndrome.
Non-PRNP genetic modifiers
The APOE genotype was ascertained in 22 patients. Surprisingly, this demonstrated a later onset of disease in individuals having at least one E4 allele, compared with those with no E4 (linear regression analysis P = 0.015). The APOE and PRNP codon 129 genotype modify phenotype independently of each other and together contribute to around 37% of the variability of P102L IPD age at onset (eta-squared of ANOVA).
Two single nucleotide polymorphisms, known as 1368 and 34296, have been shown to be over-represented in sCJD patients compared with controls (Mead et al., 2001; Vollmert et al., 2006). These loci were genotyped in patients with available DNA to examine for a potential effect on phenotype in IPD P102L. No evidence for genotype modifications of age at onset was found.
Neuropathology
A total of 11 patients were available for neuropathological examination. Varying degrees of atrophy were observed macroscopically, with brain weights varying from 1200 to 1740 g. Histopathological examination demonstrated spongiform change, astroctyosis and PrP deposition. These three features, however, were very variable between individuals and brain regions. Spongiform change was present in some brain regions in the majority, but not all, of the patients examined (7 out of 10 patients) and varied from mild to severe. PrP deposition was most commonly found in the context of multicentric amyloid plaques. PrP antibodies ICSM 18 and ICSM 35 positively labelled these plaques. Multicentric plaques were commonly found in the molecular layer of the cerebellum but also less frequently in the granular layer. Similar plaques were found in the cortex in most patients examined (9 out of 10 patients) (Fig. 8). In a single patient (VI.12), the only definite abnormality seen was small numbers of plaques positively stained with ICSM 18 in the basal ganglia, although the tissue examined was 20 years old. More diffuse PrP deposition was demonstrated in the majority of patients available (7 out of 10). A single patient (VIII.1) showed very atypical diffuse staining that appeared to follow cortical tracts and was similar to other IPD patients but not to previously described P102L pathology.
Fig. 9.

Photograph of original 1871 asylum entry relating to admission and care of III.3 from Fig. 2. His condition is described as hereditary and reference is made to his relatives’ care in the same institution. He is reported as being ‘scarcely able to move one leg in front of another’ and his countenance is described as ‘imbecile’. He appears to have had dementia as well as weakness: ‘Can only answer in monosyllables & then incoherently; is much paralysed’. He was treated with daily doses of brandy until he died. ‘Paralysis’ was listed as the cause on the death certificate.
Fig. 8.

Deposition of abnormal prion protein in cortex and cerebellum. The upper panels show characteristic multicentric PrP plaques, stained with the anti-PrP monoclonal antibody, ICSM35 (A+B from Patient 2. VII. 2). The lower panel shows synaptic deposition of abnormal PrP (C+D from Patient 7. VIII. 1), demonstrating significant pathological heterogeneity. The scaling bar shown in 100 μM.
Clinicopathological comparison demonstrated no clear association of clinical subtype (cerebellar onset or cognitive onset), disease duration or codon 129 genotype with severity of spongiform change and degree or type of plaque deposition. Six PRNP codon 129 MM homozygotes and four codon 129 MV heterozygotes were included. All the patients with available post-mortem tissue were of the common 3,3 APOE genotype. A single atypical patient (VII.1) with very young onset and prominent dystonia showed prominent PrP plaque deposition in all areas examined (including the basal ganglia), with no clear spongiform change. Findings were not dissimilar from other, later onset patients. Unfortunately, the atypical histological patient (VIII.1) had only limited clinical information available. Two muscle biopsies were also examined. The findings were non-specific on conventional stains and no PrP immunoreactivity was demonstrated.
Discussion
The patients presented here constitute by far the largest study of the classical GSS-associated PRNP mutation, P102L. Modern investigation, molecular genetic techniques and advances in pathological examination have allowed us to revisit a historical British pedigree and other smaller kindreds. We find evidence of a mutation hot spot at codon 102, a subgroup presenting with rapid cognitive decline and the significant modification of phenotype by two single nucleotide polymorphisms.
The microsatellite haplotyping presented here demonstrates the existence of multiple separate and worldwide P102L kindreds of UK origin, confirmed in most patients by established genealogical links that had not been identified previously. However, the identification of a further six UK P102L kindreds along with two others of European origin suggests that multiple mutational events have occurred in the recent past. This might be supported by the identification of at least one of these patients (6.VIII.1) in an individual without suggestive family history or of known P102L patients living nearby, suggesting that this might represent a novel mutation. Given the identification of IPD patients in otherwise typical sCJD presentations, as well as in patients without a family history of neurodegeneration, novel mutational events may occur more commonly than previously thought. The commonest point mutations associated with IPD occur at cytosine-phosphate diester-guanidine dinucleotide sites hypothesized to be mutation ‘hot spots’ in human DNA (Vollmert et al., 2006). These hot spots are related to the spontaneous demethylation of cytosine to thymidine. Cytosine-phosphate diester-guanidine dinucleotide sites are associated with E200K, D178N and P102L point mutations, which between them account for the bulk of IPD patients worldwide (Dagvadorj et al., 2002; Mead, 2006). An alternative possibility to multiple separate mutational events being responsible for these apparently unconnected P102L IPD kindreds is that these small pedigrees do share a common ancestor with the large pedigree but that this was long enough in the past for linkage to the disease haplotype to have broken down. We estimate a probability of recombination of the 3MB microsatellite haplotype as about 10% per generation (assuming a genetic:physical ratio of 3.7).
The unexpected finding that significantly greater numbers of females were identified with P102L IPD and the consequently higher number of individuals having an affected mother rather than father as the parent from whom the mutation was inherited is puzzling. This discrepancy has been reported once previously in this family, albeit with smaller numbers of individuals and without available genetic techniques, allowing the linkage of kindreds not known to share ancestry (Baker et al., 1985). It has also been observed in the large 6-OPRI IPD kindred from the South-East of England (Mead et al., 2006). In the earlier study in P102L, the finding was explained by postulating that affected females had significantly more children than their unaffected siblings and that greater than half these were daughters. Such an explanation is intriguing, though hard to explain. Alternatively, because of possible illegitimacy, we suspected that genealogical research might more readily identify affected mothers than affected fathers. In order to consider the possibility of ascertainment bias, the gender of members of the kindred identified from parish records or census entries, but who have not been identified as having suffered from IPD P102L, were collected. An excess of males in these untraced individuals was indeed identified. When these were added to the presumed affected male and female individuals, an excess of females to males remained (82–68), although this was not significantly different from an expected proportion of 0.5. It seems likely that ascertainment bias is responsible for this gender difference, although the intriguing possibility of a biological explanation remains.
While the majority of P102L IPD patients here present with progressive ataxia accompanied by mild or absent cognitive symptoms and signs, a subset present with a predominantly cognitive and or psychiatric onset, with mild or absent cerebellar signs initially. Psychiatric involvement has been severe enough to necessitate antipsychotic medication in four patients (VII.1, VII.8, VIII.4 and 3.VIII.1) and inpatient treatment in two (VII.1 and VII.8). The existence of these distinct phenotypes (cognitive and psychiatric versus cerebellar onset) is supported by the finding of early prominent frontal executive impairment on neuropsychology in these patients not seen in other patients so tested. Neuropsychological assessment is now routine at the National Prion Clinic but in the past this was more usually performed only when clinical evidence of cognitive impairment existed. In addition to these neuropsychological differences, there is a suggestion that the cognitive and psychiatric presentations have an earlier age at onset and death. Neuropsychology also highlights, however, that most if not all patients have cognitive deficits when psychology is performed, even if the findings are subtle, although selection bias may limit this conclusion. No pathological correlates of these two syndrome types have been identified, although numbers compared are small. Kuru and growth hormone-associated prion disease are notable for their onset with cerebellar symptoms (Collinge, 2001), which prompts speculation that cerebellar onset in P102L might result from the onset of prion replication outside the central nervous system. The range of tissue available did not permit the thorough testing of this hypothesis.
The high degree of clinical heterogeneity and the lack of characteristic findings on commonly available clinical neurological investigations make correct diagnosis of P102L IPD challenging. The finding of positive CSF 14-3-3 combined with the albeit unusual occurrence of periodic sharp wave complexes on EEG and an sCJD-like phenotype in some individuals raises the possibility of missed diagnosis. It also supports our clinical practice of advising PRNP analysis routinely in all those presenting with otherwise undiagnosed pre-senile dementing or ataxic illnesses.
Peripheral sensory symptoms and muscle weakness appear almost universal during the course of P102L IPD. The demonstration of muscle denervation and axonal sensorimotor neuropathy suggest peripheral neurological pathology is responsible. Experimental mice over-expressing wild-type PrP have shown demyelinating peripheral nerve pathology (Westaway et al., 1994). However, evidence of peripheral neuropathy in P102L IPD was inconsistent and where present was axonal, a form that is not uncommon in the population from unrelated causes. No prion protein (scrapie isoform) positivity could be demonstrated on two muscle biopsies examined here and although an indirect pathological process could still be responsible, peripheral findings in P102L IPD may be incidental, while sensory symptoms could be centrally rather than peripherally driven.
Presented for the first time here is the finding that P102L IPD patients with methionine homozygosity at codon 129 present significantly earlier than codon 129 heterozygotes. Earlier age at onset is well recognized in codon 129 homozygotes in other IPD mutations (Collinge et al., 1992; Poulter et al., 1992; Mead et al., 2006), although the association with IPD associated with point mutations rather than OPRIs is less clear (Dlouhy et al., 1992). The less strong effect in P102L with respect to 6-OPRI may explain why this observation has not been made before where smaller numbers of patients have been examined (Hainfellner et al., 1995; Barbanti et al., 1996). There are several possible mechanisms of action of codon 129 genotype on clinical phenotype. It has long been known that the interaction between prion protein, scrapie isoform (PrPSc) and prion protein, normal cellular isoform (PrPc) occurs most efficiently when the proteins have an identical primary structure (Palmer et al., 1991); therefore, prion replication may occur more rapidly and clinical onset earlier in 129MM individuals. As a further complexity, the primary structure of PrP determines the permissible conformations of PrPSc. The extent to which a pathogenic 102L-129M PrP conformer is permitted by wild-type PrPc with 129V or 129M may also be important [see Collinge, 2007 for a recent review (Collinge and Clarke, 2007)]. 102L-129M PrP may be able to adopt several different pathogenic conformations, which may be permissible to a greater or lesser extent by the wild-type protein (Parchi et al., 1998; Hill et al., 2006). The involvement of wild-type protein is known to be a variable phenomenon in P102L and a possible determinant of phenotype (Wadsworth et al., 2006).
The identification of the P102L mutation on a methionine allele in all of the patients presented here with adequate haplotype data probably relates simply to the frequency of this allele in the background population. P102L patients existing on a codon 129 valine allele have been reported, apparently occurring in the context of a distinct phenotype with prominent psychiatric features and seizures, different from classical P102L codon 129 methionine patients (Young et al., 1997; Bianca et al., 2003). Such a phenotypic difference could relate to different prion strain propagation originating from the valine allele.
Genotype–phenotype correlations presented here, corrected for codon 129, demonstrate an effect of APOE genotype on age at onset, with individuals carrying the E4 allele having a significantly later age at onset than those without. While this evidence appears in contrast to the strong association of E4 with risk of Alzheimer's disease (Corder et al., 1993) and to published work on the possible impact of APOE polymorphisms in sCJD showing an overrepresentation of the E4 allele in sCJD patients (Amouyel et al., 1994; Van Everbroeck et al., 2001), such findings have not been replicated in all published studies (Nakagawa et al., 1995; Zerr et al., 1996) nor in our own observations (387 sCJD patients versus unaffected controls, unpublished data). Recent reports present a similar result in frontotemporalobar degeneration, with later disease onset in those with the APOE E4 genotype in the context of progranulin mutations (Gass et al., 2006; Beck et al., 2008). APOE may thus have contrasting effects in the context of different neurodegenerative disease types.
Most of the P102L IPD patients shared common features on pathological analysis. Spongiform change, astrocytosis and PrP deposition, both as multicentric plaques and synaptic deposits, were seen in the majority. However, the degree of severity of spongiform change and plaque deposition were very variable. This did not seem to correlate with age at onset or duration of illness. Neither did predominantly cerebellar or cognitive clinical presentations seem to correlate with pathological findings. PRNP codon 129 MM homozygotes and MV heterozygotes were equally represented in the pathology series but no significant differences were seen between these two groups. However, the small numbers in each subgroup and the age of some of the samples examined (with tissue up to 30 years old) may prevent correlations from being made. A single highly atypical clinical patient showed no significant histological differences from the rest of the series, while an atypical pathological patient has limited clinical data, although what is known about this individual is not obviously different from the other patients examined. These results suggest no clear clinicopathological correlations, but sample size was necessarily small.
Immunoblots of proteinase K-digested brain homogenate from P102L patients demonstrate a spectrum of involvement of protease-resistant wild-type PrP in P102L IPD (Wadsworth et al., 2006). Four of the seven P102L IPD patients examined in this study were negative for prion protein (scrapie isoform), which most probably relates to sampling issues and the differing density of PrP deposition in tissue samples both within and between patients. Our large study therefore does not expand the existing data and we were unable to test whether protease-resistant PrP diversity might contribute to clinical heterogeneity. This remains a plausible concept in IPD that requires a large series of fresh frozen brain tissue and molecular analysis of PrP type following partial protease digestion.
The need for public health control measures, together with the evident diagnostic challenges that IPD heterogeneity causes, make a strong argument for including PRNP gene analysis in the list of investigations for suspected prion disease of any type and indeed of all undiagnosed familial dementia or cerebellar syndromes. Successful diagnosis allows clinicians to provide more accurate prognostic information to patients, to allow participation in the clinical trials and reduce the risk of iatrogenic transmission of disease. As a consequence of these geographically highly mobile ancestors, and the large number of untraced individuals in the nineteenth century who were clearly at risk of inheriting the P102L mutation, it remains likely that further patients and at-risk individuals exist who have yet to be identified. It is hoped that the data presented here will help to raise awareness of P102L IPD and its associated presentations.
Funding
Medical Research Council; Department of Health's NIHR Biomedical Research Centres, Department of Health.
Supplementary Material
Acknowledgements
We are grateful to all the patients and their families for generously contributing to this report and to the neurology colleagues who referred patients to the National Prion Clinic. Prof. James Ironside at the National CJD surveillance Unit and the London MRC Neurodegenerative Diseases Brain Bank, Institute of Psychiatry, were involved in providing tissue for neuropathological assessment. Ray Young assisted with illustrations.
Glossary
Abbreviations:
- APOE
apolipoprotein E
- GSS
Gerstmann-Sträussler-Scheinker syndrome
- IPD
inherited prion disease
- NCS
nerve conduction studies
- OPRI
octapeptide repeat insertion
- PRNP
human prion protein gene
- PrPc
prion protein (cellular isoform)
- PrPSc
prion protein (scrapie isoform)
- sCJD
sporadic Creutzfeldt-Jakob disease
References
- Adam J, Crow TJ, Duchen LW, Scaravilli F, Spokes E. Familial cerebral amyloidosis and spongiform encephalopathy. J Neurol Neurosurg Psychiatry. 1982;45:37–45. doi: 10.1136/jnnp.45.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amouyel P, Vidal O, Launay JM, Laplanche JL. The apolipoprotein E alleles as major susceptibility factors for Creutzfeldt-Jakob disease. Lancet. 1994;344:1315–18. doi: 10.1016/s0140-6736(94)90691-2. [DOI] [PubMed] [Google Scholar]
- Arata H, Takashima H, Hirano R, Tomimitsu H, Machigashira K, Izumi K, et al. Early clinical signs and imaging findings in Gerstmann-Straussler-Scheinker syndrome (Pro102Leu) Neurology. 2006;66:1672–8. doi: 10.1212/01.wnl.0000218211.85675.18. [DOI] [PubMed] [Google Scholar]
- Baker HF, Ridley RM, Crow TJ. Experimental transmission of an autosomal dominant spongiform encephalopathy: does the infectious agent originate in the human genome? Br Med J. 1985;291:299–302. doi: 10.1136/bmj.291.6491.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbanti P, Fabbrini G, Salvatore M, Petraroli R, Cardone F, Maras B, et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Straussler-Scheinker disease (PrP-P102L mutation) Neurology. 1996;47:734–41. doi: 10.1212/wnl.47.3.734. [DOI] [PubMed] [Google Scholar]
- Beck J, Rohrer J, Campbell T, Isaacs A, Morrison K, Goodall E, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. 2008;131:706–20. doi: 10.1093/brain/awm320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianca M, Bianca S, Vecchio I, Raffaele R, Ingegnosi C, Nicoletti F. Gerstmann-Straussler-Scheinker disease with P102L-V129 mutation: a case with psychiatric manifestations at onset. Ann Genet. 2003;46:467–9. doi: 10.1016/s0003-3995(03)00017-0. [DOI] [PubMed] [Google Scholar]
- Cameron E, Crawford AD. A familial neurological disease complex in a Bedfordshire community. J R Coll Gen Pract. 1974;24:435–6. [PMC free article] [PubMed] [Google Scholar]
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry. 2005;76:906–19. doi: 10.1136/jnnp.2004.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, Baker H, et al. Inherited prion disease with 144 base pair gene insertion: II: clinical and pathological features. Brain. 1992;115:687–710. doi: 10.1093/brain/115.3.687. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke A. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene Dose of apolipoprotein-e type-4 allele and the risk of alzheimers-disease in late-onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Petersen RB, Lee HS, Cervenakova L, Shatunov A, Budka H, et al. Spontaneous mutations in the prion protein gene causing transmissible spongiform encephalopathy. Ann Neurol. 2002;52:355–9. doi: 10.1002/ana.10267. [DOI] [PubMed] [Google Scholar]
- Dlouhy SR, Hsiao K, Farlow MR, Foroud T, Conneally PM, Johnson P, et al. Linkage of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease to the prion protein gene. Nat Genet. 1992;1:64–7. doi: 10.1038/ng0492-64. [DOI] [PubMed] [Google Scholar]
- Finckh U, Muller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66:110–7. doi: 10.1086/302702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J, et al. The original Gerstmann-Straussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995;5:201–11. doi: 10.1111/j.1750-3639.1995.tb00596.x. [DOI] [PubMed] [Google Scholar]
- Hill AF, Joiner S, Beck J, Campbell TA, Dickinson A, Poulter M, et al. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain. 2006;129:676–85. doi: 10.1093/brain/awl013. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338:342–5. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- Macfarlane RG, Wroe SJ, Collinge J, Yousry TA, Jager R. Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry. 2007;78:664–70. doi: 10.1136/jnnp.2006.094821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Gajdusek DC, Gibbs CJ., Jr Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain. 1981;104:559–88. doi: 10.1093/brain/104.3.559. [DOI] [PubMed] [Google Scholar]
- Mead S. Prion disease genetics. Eur J Human Genet. 2006;14:273–81. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- Mead S, Mahal SP, Beck J, Campbell T, Farrall M, Fisher E, et al. Sporadic - but not variant - Creutzfeldt-Jakob disease is associated with polymorphisms upstream of PRNP Exon 1. Am J Hum Genet. 2001;69:1225–35. doi: 10.1086/324710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, Poulter M, Beck J, Webb T, Campbell T, Linehan J, et al. Inherited prion disease with six octapeptide repeat insertional mutation–molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297–317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Kitamoto T, Furukawa H, Ogomori K, Tateishi J. Allelic variation of apolipoprotein E in Japanese sporadic Creutzfeldt-Jakob disease patients. Neurosci Lett. 1995;187:209–11. doi: 10.1016/0304-3940(95)11366-5. [DOI] [PubMed] [Google Scholar]
- Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, et al. Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet. 1989;1:51–2. doi: 10.1016/s0140-6736(89)91713-3. [DOI] [PubMed] [Google Scholar]
- Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–2. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci USA. 1998;95:8322–7. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccardo P, Dlouhy SR, Lievens PMJ, Young K, Bird TD, Nochlin D, et al. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–88. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- Poulter M, Baker HF, Frith CD, Leach M, Lofthouse R, Ridley RM, et al. Inherited prion disease with 144 base pair gene insertion: I: genealogical and molecular studies. Brain. 1992;115:675–85. doi: 10.1093/brain/115.3.675. [DOI] [PubMed] [Google Scholar]
- Rosenthal NP, Keesey J, Crandall B, Brown WJ. Familial neurological disease associated with spongiform encephalopathy. Arch Neurol. 1976;33:252–9. doi: 10.1001/archneur.1976.00500040036005. [DOI] [PubMed] [Google Scholar]
- Sanchez-Juan P, Green A, Ladogana A, Cuadrado-Corrales N, Saanchez-Valle R, Mitrovaa E, et al. CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2006;67:637–43. doi: 10.1212/01.wnl.0000230159.67128.00. [DOI] [PubMed] [Google Scholar]
- Van Everbroeck B, Croes EA, Pals P, Dermaut B, Jansen G, van Duijn CM, et al. Influence of the prion protein and the apolipoprotein E genotype on the Creutzfeldt-Jakob Disease phenotype. Neurosci Lett. 2001;313:69–72. doi: 10.1016/s0304-3940(01)02264-9. [DOI] [PubMed] [Google Scholar]
- Vollmert C, Windl O, Xiang W, Rosenberger A, Zerr I, Wichmann HE, et al. Significant association of a M129V independent polymorphism in the 5' UTR of the PRNP gene with sporadic Creutzfeldt-Jakob disease in a large German case-control study. J Med Genet. 2006;43:e53. doi: 10.1136/jmg.2006.040931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth J, Joiner S, Linehan J, Cooper S, Powell C, Mallinson G, et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129:1557–69. doi: 10.1093/brain/awl076. [DOI] [PubMed] [Google Scholar]
- Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang S-L, et al. Degeneration of skeletal muscle, peripheral nerves and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell. 1994;76:117–29. doi: 10.1016/0092-8674(94)90177-5. [DOI] [PubMed] [Google Scholar]
- Young K, Clark HB, Piccardo P, Dlouhy SR, Ghetti B. Gerstmann-Straussler-Scheinker disease with the PRNP P102L mutation and valine at codon 129. Mol Brain Res. 1997;44:147–50. doi: 10.1016/s0169-328x(96)00251-3. [DOI] [PubMed] [Google Scholar]
- Zerr I, Helmhold M, Poser S, Armstrong VW, Weber T. Apolipoprotein E phenotype frequency and cerebrospinal fluid concentration are not associated with Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:1233–8. doi: 10.1001/archneur.1996.00550120041014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.