

SUMMARY
Almost two decades after identification of the CFTR gene, we lack answers to many questions about the pathogenesis of cystic fibrosis (CF), and it remains a lethal disease. Mice with a disrupted CFTR gene have greatly facilitated CF studies, but they fail to develop the characteristic pancreatic, lung, intestinal, liver, and other CF manifestations. Therefore, we produced pigs with a targeted disruption of both CFTR alleles. These animals exhibited defective chloride transport. They also developed meconium ileus, exocrine pancreatic destruction, and focal biliary cirrhosis, replicating abnormalities seen in newborn patients with CF. This swine model may provide opportunities to address persistent questions about CF pathogenesis and accelerate discovery of treatments and preventions.
Understanding human disease often requires animal models. Mice have been the overwhelming species of choice because methods for specifically altering their genome have been readily available. However in many cases, mice with targeted gene manipulations fail to replicate phenotypes observed in humans. One example is CF.
In 1938, Dorothy Anderson coined the term “cystic fibrosis of the pancreas” (1). Over the ensuing years, investigators learned that CF involved many other organs, including intestine, lung, sweat gland, liver, gallbladder, and male genital tract (2-4). We now know CF to be a common, autosomal recessive disease with a carrier rate of ∼5% in Caucasians. In 1989, the gene mutated in CF was identified and named cystic fibrosis transmembrane conductance regulator (CFTR) (5). Soon after scientists discovered that CFTR is a regulated anion channel that may also affect other transport processes (3).
Despite many advances, understanding of CF pathogenesis remains inadequate, and that has hindered development of treatments and preventions. Why have we not made more progress? While superb clinical research has guided thoughts about CF, interpretations about pathogenesis are often based on observations obtained long after the disease onset, and many studies cannot be done in humans. Although, cell-based models have proven quite valuable for research (6), CF involves a whole organism. In addition, gene-targeted mouse models have taught us much about CF, yet those mice do not develop pancreatic, airway, intestinal or liver disease typically found in humans (7-9>).
To develop a new CF model, we chose pigs because compared to mice their anatomy, biochemistry, physiology, size, lifespan and genetics are more similar to those of humans (10, 11). We used homologous recombination in fibroblasts of outbred domestic pigs to disrupt the CFTR gene and somatic cell nuclear transfer to generate CFTR+/− pigs (12).
At sexual maturity (∼6-7 months), female CFTR+/− pigs were bred to CFTR+/− males. Six litters produced 64 piglets. Genotyping (Fig. 1A) revealed 18 CFTR+/+, 26 CFTR+/−, and 20 CFTR−/− animals, a ratio not significantly different from the expected 1:2:1 (13). Figure 1B shows the first litter. Birth weights varied, but did not segregate by genotype (Fig. 1C). Piglets looked normal at birth, and genotype could not be discerned by appearance. A normal appearance is consistent with findings in humans.
Figure 1. CFTR−/− piglets appear normal at birth.
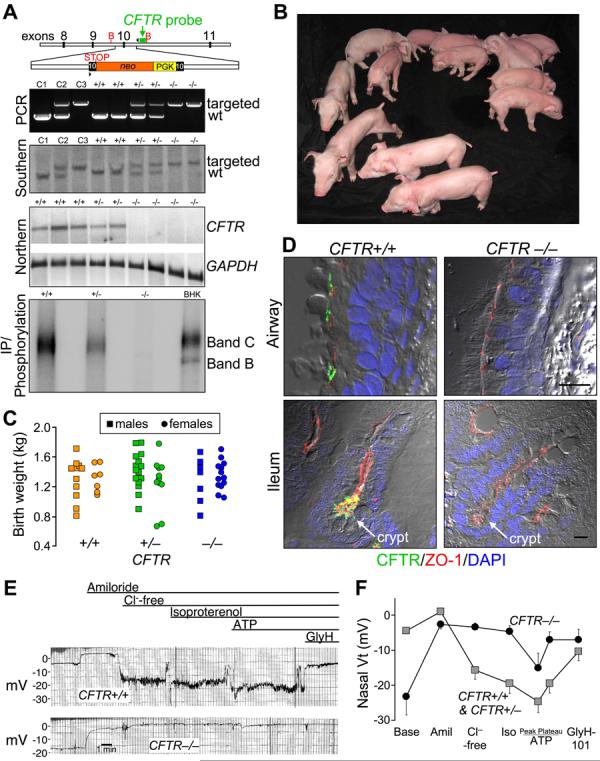
A) Upper panel depicts insertion into porcine CFTR exon 10 of a PGK promoter (yellow) driving a neomycin resistance cDNA (orange), and an engineered stop codon. Position of probe (green), PCR primers (arrowheads) and BglII sites (B) are indicated. Second and third panels show genotyping by PCR and Southern blot of genomic DNA. Lanes C1, C2, and C3 contain controls of CFTR+/+, +/− and −/− DNA. Fourth panel shows northern blot of ileal CFTR and GAPDH mRNA. Consistent with the northern blot, quantitative RT-PCR of exon 10, the targeted site, detected <0.1% of CFTR transcripts in CFTR −/− ileum relative to CFTR+/+ (n=6 and 4). Fifth panel shows immunoprecipitation and phosphorylation of CFTR plus recombinant CFTR in BHK cells. B) First litter containing piglets of all three genotypes. C) Birth weights. Mean±SD of weights: 1.31±0.24 kg for CFTR+/+, 1.35±0.28 kg CFTR+/−,and 1.31±0.23 kg CFTR−/−. D) Immunocytochemistry of CFTR in airway epithelia (top) and ileum (bottom). Figures are differential interference contrast with staining for ZO-1 (a component of tight junctions, red), CFTR (green), and nuclei (DAPI, blue). See also Fig. S1. Bars, 10 μm. E) Tracings of in vivo nasal voltage (Vt) measured in newborn piglets. After baseline measurements, the following agents/solutions were sequentially added to the epithelial perfusate: amiloride (100 μM), Cl−-free solution, isoproterenol (10 μM), ATP (100 μM), and GlyH-101 (100 μM). F) Average nasal Vt measurements as indicated in panel E. Data from 4 CFTR+/+ and 4 CFTR+/− piglets were not statistically different and were combined and compared to data from 5 CFTR−/− piglets. Values of baseline nasal Vt for CFTR−/− piglets differed from the controls, as did the changes in Vt induced by adding amiloride, a Cl−-free solution, and GlyH-101 (all P<0.05). Data are mean±SEM.
Northern blot and quantitative RT-PCR did not detect normal CFTR transcripts (Fig. 1A). Immunoprecipitation detected no normal CFTR protein. Like human CFTR (14, 15), the porcine protein localized apically in airway epithelia and ileal crypts (Fig. 1D).
As in humans, we assessed CFTR function in vivo by measuring transepithelial voltage (Vt) across nasal epithelia (16) (Fig. 1E,F). Like humans with CF, baseline Vt was hyperpolarized in CFTR−/− piglets. Amiloride, which inhibits ENaC Na+ channels, reduced Vt in all genotypes. To test for CFTR channel activity, we perfused the apical surface with a Cl−-free solution and added isoproterenol to increase cellular levels of cAMP; these interventions hyperpolarized nasal Vt in wild-type and heterozygous, but not CFTR−/− animals. Perfusion with ATP to activate P2Y2 receptors and Ca2+-activated Cl− channels (17) further hyperpolarized Vt, and the response did not differ significantly between genotypes. Perfusion with the CFTR inhibitor GlyH-101 (18) depolarized Vt in controls, but not CFTR−/− piglets. These data reveal loss of CFTR Cl− channel activity in newborn CFTR−/− pigs. While lack of data from newborn humans precludes direct comparison, the data qualitatively match those from adults and children with CF (16).
What phenotypes would be expected if newborn CFTR−/− piglets model human disease? Figure 2A shows some human CF phenotypes and the time range when they become clinically apparent (3, 19). The earliest manifestation (hours to 2 days) is meconium ileus, an intestinal obstruction occurring in ∼15% of CF infants (2, 3, 19, 20). Obstruction can occur throughout the small intestine or colon, but most often occurs near the ileocecal junction. Distal to the obstruction, the bowel is small and atretic (microcolon). Intestinal perforation in utero or postnatally occurs in some infants.
Figure 2. CFTR−/− piglets develop meconium ileus.
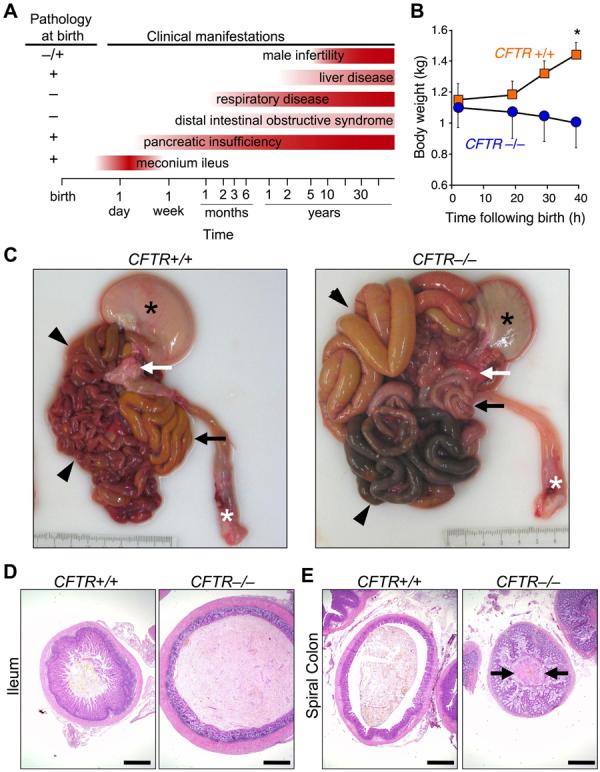
A) Schematic shows some clinical and histopathological CF manifestations. Note that pathological abnormalities are present before clinical disease becomes apparent. B) Weight following birth. Animals were fed colostrum and milk-replacer. n = 7 CFTR+/+ and 4 CFTR−/−. Data are mean±SEM. *P<0.05. C) Gross appearance of gastrointestinal tract. Piglets were fed colostrum and milk-replacer for 30-40 h and then euthanized. Stomach (black *), small intestine (arrowheads), pancreas (white arrow), rectum (white *), and spiral colon (black arrow). Of 16 CFTR−/− piglets, the obstruction occurred in small intestine in 7 and spiral colon in 9. D, E) Microscopic appearance of the ileum (D) and colon (E). H&E stain. Bars, 1 mm. Images are representative of severe meconium ileus occurring in 16 of 16 CFTR−/− piglets.
Following birth, CFTR−/− piglets failed to pass feces or gain weight (Fig. 2B). By 24-40 h, they stopped eating, developed abdominal distension, and had bile-stained emesis. These are clinical signs of intestinal obstruction. We examined histopathology between birth and 12 h in piglets that had not eaten and between 24 and 40 h in pigs fed colostrum and milk replacer. Except as noted, the pathologic changes refer to the early time period. After 30-40 h, CFTR−/− piglets had stomachs containing small amounts of green, bile-stained milk (Fig. 2C). The proximal small intestine was dilated by small amounts of milk and abundant gas. The site of obstruction ranged from mid-distal small intestine to proximal spiral colon, the anatomical equivalent of the human ascending colon. Perforation and peritonitis occurred in some piglets. Dark green meconium distended the CFTR−/− intestine and adjacent villi showed degeneration and atrophy, whereas CFTR+/+ ileum had long villi (Fig. 2D). Distal to the meconium, luminal diameter was reduced with mild to severe mucinous hyperplasia including mucoid luminal “plugs” (Fig. 2E). These changes replicate those in humans with CF (3, 19).
The penetrance of meconium ileus was 100% in CFTR−/− piglets vs. ∼15% in human CF. Potential explanations for the difference include a restricted genetic background in our pigs vs. humans, a null mutation in the pigs vs. mutations in humans that might yield tiny amounts of protein function, and anatomical or physiological differences (F1, 13).
Exocrine pancreatic insufficiency afflicts 90-95% of patients with CF (1-4, 19, 21, 22). The porcine CFTR−/− pancreas was small (Fig. 3A). Microscopic examination revealed small, degenerative lobules with increased loose adipose and myxomatous tissue and scattered to moderate cellular inflammation (Fig. 3B,C). Residual acini had diminished amounts of eosinophilic zymogen granules (Fig. 3D). Centroacinar spaces, ductules, and ducts were variably dilated and obstructed by eosinophilic material plus infrequent neutrophils and macrophages mixed with cellular debris (Fig. 3E). Ducts and ductules had foci of mucinous metaplasia. Pancreatic endocrine tissue was spared (Fig. 3C). These changes reflect those originally described by Anderson and others (1, 19, 21).
Figure 3. CFTR−/− piglets have exocrine pancreatic destruction and liver and gallbladder abnormalities.
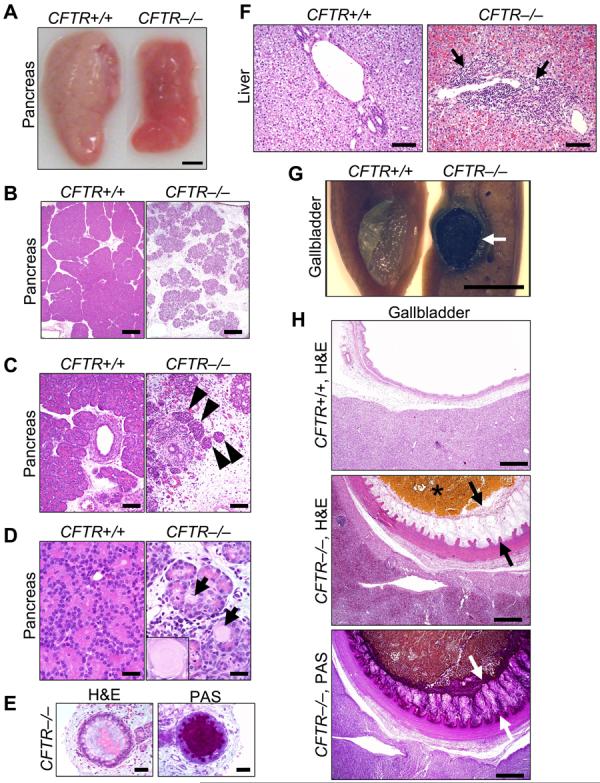
A) Gross appearance of pancreas. Bar, 0.5 cm. B) Loss of parenchyma in the CFTR−/− pancreas. H&E stain. Bars, 500 µm. C) Pancreatic ducts and islets of Langerhans (arrowheads). Bars, 100 µm. D) CFTR−/− ductules and acini dilated by eosinophilic inspissated material that formed concentrically lamellar concretions (arrows and insert). H&E stain. Bars, 33 µm. E) Ducts within the CFTR−/− pancreas. H&E stain, left; PAS stain, right. Bars, 50 µm. F) Microscopic appearance of liver. H&E stain. Arrows indicate focal expansion of portal areas by chronic cellular inflammation. Bars, 100 µm. G) Gross appearance of gallbladder. When the CFTR+/+ gallbladder was sectioned, bile drained away rapidly with collapse of the mucosal wall. CFTR−/− bile was congealed (arrow) and retained in the lumen of a smaller gallbladder. Bar, 0.5 cm. H) Microscopic appearance of gallbladder. CFTR−/− gallbladders had congealed, inspissated bile with variable mucus production (arrows, H&E stain) highlighted as a magenta color in periodic acid-Schiff (PAS) stained tissue. Bars, 500 µm. Images are representative of severe pancreatic lesions (15/15 CFTR−/− piglets), mild to moderate liver lesions (3/15), and mild to severe gall bladder/duct lesions (15/15).
Humans often require surgery to relieve meconium ileus (3). Therefore, 1 CFTR+/+ and 3 CFTR−/− piglets had an ileostomy. Because of technical problems in post-operative care, only 1 piglet recovered, a CFTR−/−. Once newborns recover from meconium ileus, their disease resembles that in other patients. Likewise, the piglet grew and had pancreatic insufficiency. At 10 weeks, he had an episode like “distal intestinal obstruction syndrome” that was successfully treated as in humans (3, F2, 13). While these observations come from a single animal, they copy human CF to a remarkable degree.
Focal biliary cirrhosis is the second most common cause of CF mortality (3, 20, 23). The porcine CFTR−/− liver revealed infrequent, mild to moderate hepatic lesions (Fig. 3F). Chronic cellular inflammation, ductular hyperplasia, and mild fibrosis were typical of focal biliary cirrhosis. Gallbladder abnormalities, including gallstones, occur in 15-30% of patients, and a small gallbladder is a common autopsy finding (3). Similarly, porcine CFTR−/− gallbladders were small and often filled with congealed bile and mucus (Fig. 3G,H). Epithelia showed diffuse mucinous changes with folds extending into the lumen.
Approximately 97% of males with CF are infertile (3); the vas deferens is often normal at birth, and obstruction is thought to cause progressive deterioration (F3, 13). In all piglets the vas deferens appeared intact. Paranasal sinus abnormalities occur in most children and adults with CF (3). Although CFTR−/− porcine paranasal sinuses showed no abnormalities, this negative result is difficult to interpret because it is unclear when sinus disease develops in humans. Salivary glands, nasal cavity, esophageal glands, kidney, heart, striated muscle, spleen, adrenals, eyes, brain, skin, and a few eccrine sweat glands on the snout revealed no abnormalities in CFTR−/− piglets. In all tissues, we observed no differences between wild-type and CFTR+/− animals.
Lung disease is currently the major cause of CF morbidity and mortality (2-4). The onset of clinical respiratory manifestations varies with some patients developing symptoms a few months after birth and others after several years. Eventually, most patients develop chronic airway infection and inflammation that destroy the lung. The lungs of neonatal CFTR−/− piglets appeared the same as their littermates. CFTR−/− lungs lacked evidence of cellular inflammation in airways or parenchyma (Fig. 4A). Airway epithelia and submucosal glands appeared similar in all three genotypes, and we found no evidence of dilated or plugged submucosal gland ducts (F4, 13) (Fig. 4B). Bronchoalveolar lavage 6-12 h after birth showed no evidence of infection, and there were no significant differences between cell counts or levels of IL-8 across genotypes (Fig. S2, S3, 13).
Figure 4. The lungs of newborn CFTR−/− and CFTR+/+ piglets appear normal.
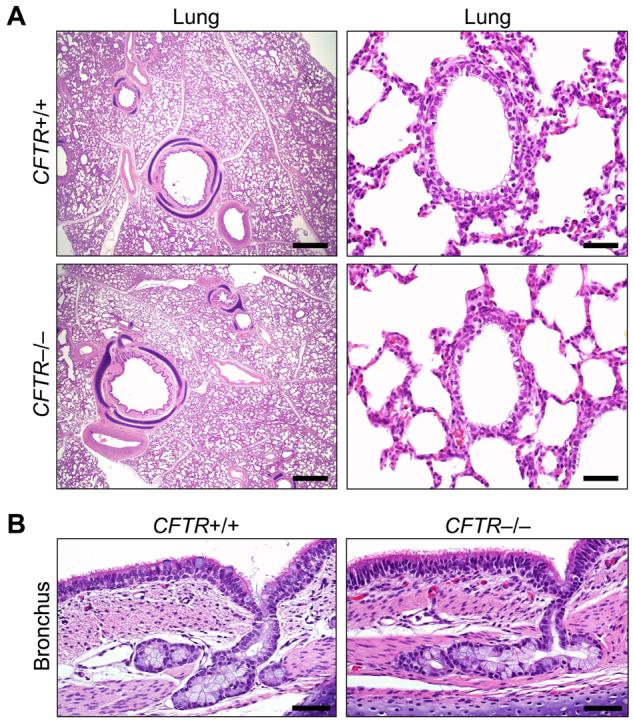
A) Microscopic appearance of lung from piglets <12 hr old. H&E staining. Bars, 1 mm (left) and 50 µm (right). B) Bronchial epithelia and submucosal glands. H&E staining. Bars, 50 µm. Images are representative of lack of lesions in 15 of 15 CFTR−/−.
The “chicken and egg” conundrum about whether CF airway inflammation occurs before infection or whether infection precedes inflammation remains a persistent question. Studies of bronchoalveolar lavage in infants and young children have both supported and argued against the presence of inflammation (increased IL-8 and neutrophilia) without infection (24, 25). In vitro airway epithelial models have also given conflicting results (26, 27). Studies of human fetal trachea transplanted into mice suggested inflammation might occur in developing CF airways (28). While our data do not resolve this controversy, we had the advantage of studying lungs between birth and 12 h, and we found no evidence of abnormal infection or inflammation. Tracking the lungs as CFTR−/− piglets are exposed to additional environmental challenges should inform understanding of how respiratory disease develops during childhood and in young adults.
The clinical, electrophysiological, and pathological findings in newborn CFTR−/− pigs were remarkably similar to those in human neonates with CF (Table S1, 13). Abdominal lesions dominate the initial presentation in both, with identical appearance of meconium ileus and exocrine pancreatic destruction. In addition, both have hepatic changes consistent with early focal biliary cirrhosis and abnormalities of the gallbladder and bile ducts. The lack of abnormalities in vas deferens and lungs at birth is another similarity. Finding that the phenotype of newborn CFTR−/− piglets copies that of human newborns with CF suggests this model should provide investigators with new opportunities to understand the disease and develop novel prevention and treatment strategies. In addition, there are many other human diseases in which knock-out and knock-in mice fail to reproduce typical human phenotypes and fail to predict responses to therapeutics. This development of a gene-targeted, mammalian disease model, other than a mouse, also suggests strategies to circumvent research bottlenecks presented by limitations of mice.
Supplementary Material
Acknowledgments
We thank A Arias, E Bagnall, J Bartlett, B Bauer, T Bohnert, E Burnight, K Dobbs, C Dohrn, L Dowell, L Gakhar, D Guilliams, R Hanfland, Yanhong Hao, SC Isom, S Jones, S Korte, B Kussman, J Launspach, M Linville, E Mahan, T Mayhew, C McHughes, K Munson, M Parker, C Randak, S Ramachandran, J Ross, A Small, L Spate, D Vermeer, E Walters, and J Whyte for excellent assistance. We thank Cystic Fibrosis Foundation Therapeutics and Dr. Robert Bridges for the gift of GlyH-101. CSR, RSP, MJW, and the University of Iowa Foundation have applied for a patent related to this work, and CSR and MJW are founders of Exemplar Genetics, which is licensing materials and technology reported here. This work was supported by the National Heart Lung and Blood Institute (HL51670), the National Institute of Diabetes and Digestive and Kidney Diseases (DK54759), Food for the 21st Century, Cystic Fibrosis Foundation, and HHMI. CSR was supported by NIH Training Grant HL07638. DAS is a Parker B. Francis Fellow and was supported by the NIAID (AI076671). MJW is an Investigator of the HHMI.
REFERENCES
- 1.Andersen DH. Am. J. Dis. Child. 1938;56:344. [Google Scholar]
- 2.Quinton P. Physiol. Rev. 1999;79:S3. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- 3.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver CR, et al., editors. McGraw-Hill; New York: 2001. pp. 5121–5189. [Google Scholar]
- 4.Rowe SM, Miller S, Sorscher EJ. N. Engl. J. Med. 2005 May 12;352:1992. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 5.Riordan JR, et al. Science. 1989;245:1066. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 6.Karp PH, et al. In: Epithelial Cell Culture Protocols. Wise C, editor. Vol. 188. Humana Press, Inc.; Totowa, NJ: 2002. pp. 115–137. [Google Scholar]
- 7.Grubb BR, Boucher RC. Physiol. Rev. 1999;79:S193. doi: 10.1152/physrev.1999.79.1.S193. [DOI] [PubMed] [Google Scholar]
- 8.Guilbault C, Saeed Z, Downey GP, Radzioch D. Am. J. Respir. Cell Mol. Biol. 2007 Jan;36:1. doi: 10.1165/rcmb.2006-0184TR. [DOI] [PubMed] [Google Scholar]
- 9.Clarke LL, Gawenis LR, Franklin CL, Harline MC. Lab Anim Sci. 1996 Dec;46:612. [PubMed] [Google Scholar]
- 10.Ibrahim Z, et al. Xenotransplantation. 2006 Nov;13:488. doi: 10.1111/j.1399-3089.2006.00346.x. [DOI] [PubMed] [Google Scholar]
- 11.Rogers CS, et al. Am J Physiol Lung Cell Mol Physiol. 2008 May 16; doi: 10.1152/ajplung.90203.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers CS, et al. J. Clin. Invest. 2008:1571. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Supplemental material in Science Online.
- 14.Crawford I, et al. Proc.Natl. Acad.Sci.U.S.A. 1991;88:9262. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denning GM, Ostedgaard LS, Cheng SH, Smith AE, Welsh MJ. J. Clin. Invest. 1992;89:339. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Standaert TA, et al. Pediatr. Pulmonol. 2004 May;37:385. doi: 10.1002/ppul.10448. [DOI] [PubMed] [Google Scholar]
- 17.Mason SJ, Paradiso AM, Boucher RC. Br. J. Pharmacol. 1991 Jul;103:1649. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muanprasat C, et al. J. Gen. Physiol. 2004 Aug;124:125. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oppenheimer EH, Esterly JR. Perspect. Pediatr. Pathol. 1975;2:241. [PubMed] [Google Scholar]
- 20.Wilschanski M, Durie PR. J. R. Soc. Med. 1998;91(Suppl 34):40. doi: 10.1177/014107689809134s07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imrie JR, Fagan DG, Sturgess JM. Am. J. Pathol. 1979 Jun;95:697. [PMC free article] [PubMed] [Google Scholar]
- 22.Blackman SM, et al. Gastroenterology. 2006 Oct;131:1030. doi: 10.1053/j.gastro.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oppenheimer EH, Esterly JR. J. Pediatr. 1975 May;86:683. doi: 10.1016/s0022-3476(75)80351-9. [DOI] [PubMed] [Google Scholar]
- 24.Khan TZ, et al. Am. J. Respir. Crit. Care Med. 1995;151:1075. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong DS, et al. Pediatr. Pulmonol. 2005 Dec;40:500. doi: 10.1002/ppul.20294. [DOI] [PubMed] [Google Scholar]
- 26.Stecenko AA, et al. Inflammation. 2001 Jun;25:145. doi: 10.1023/a:1011080229374. [DOI] [PubMed] [Google Scholar]
- 27.Aldallal N, et al. Am.J.Respir. Crit. Care Med. 2002 Nov 1;166:1248. doi: 10.1164/rccm.200206-627OC. [DOI] [PubMed] [Google Scholar]
- 28.Tirouvanziam R, et al. Am. J. Respir. Cell Mol. Biol. 2000 Aug;23:121. doi: 10.1165/ajrcmb.23.2.4214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.