

Abstract
Acute intermittent hypoxia (AIH) elicits a form of respiratory plasticity known as long-term facilitation (LTF). LTF is a progressive and sustained increase in respiratory motor output as expressed in phrenic and hypoglossal (XII) nerve activity. Since reactive oxygen species (ROS) play important roles in several forms of neuroplasticity, and ROS production is increased by intermittent hypoxia, we tested the hypothesis that ROS are necessary for phrenic and hypoglossal LTF following AIH. Urethane anesthetized, paralyzed, vagotomized and pump ventilated Sprague Dawley rats were exposed to AIH (11% O2, 3, 5 min episodes, 5 min intervals), and both phrenic and XII nerve activity were monitored for 60 min post-AIH. Although phrenic and XII LTF were observed in control rats, intravenous Manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP), a superoxide anion scavenger, attenuated both phrenic and XII LTF in a dose dependent manner. Localized application of MnTMPyP (5.5mM; 10µl) to the intrathecal space of the cervical spinal cord (C4) abolished phrenic, but not XII LTF. Thus, ROS are necessary for AIH-induced respiratory LTF, and the relevant ROS appear to be localized near respiratory motor nuclei since cervical MnTMPyP injections impaired phrenic (and not XII) LTF. Phrenic LTF is a novel form of ROS-dependent neuroplasticity since its ROS-dependence resides in the spinal cord.
Keywords: plasticity, spinal cord, control of breathing, superoxide anion
Acute intermittent hypoxia (AIH) elicits a form of respiratory plasticity known as long-term facilitation (LTF). LTF is a progressive increase in respiratory motor output that lasts hours following the final hypoxic episode (Fuller et al., 2000; Mitchell et al., 2001; Mitchell and Johnson, 2003; Feldman et al., 2003). Phrenic LTF requires serotonin-dependent protein synthesis in cervical spinal regions associated with the phrenic motor nucleus (Baker-Herman and Mitchell, 2002). In our working model (Mahamed and Mitchell, 2006; Baker-Herman et al., 2004), serotonin release during hypoxic episodes activates 5-HT2A receptors located on or near phrenic motor neurons and initiates signaling cascades that maintain phrenic LTF. Postulated signaling events downstream from 5-HT2A receptor activation include protein kinase C activation (McGuire and Ling, 2004), new synthesis of brain derived neurotrophic factor (BDNF), and subsequent activation of its high affinity receptor tyrosine kinase, TrkB (Baker-Herman et al., 2004). Relevant events downstream from TrkB activation are not clear at this time, but most likely involve activation of MAP kinases (e.g. ERK) or protein kinase B (Akt). On the other hand, phrenic LTF is subject to inhibitory constraints since okadaic acid-sensitive protein phosphatases suppress phrenic LTF following sustained hypoxia (Wilkerson et al., 2007). These putative cellular mechanisms of LTF share many common features with other forms of neuroplasticity such as sensitization of the gill-withdrawal reflex in Aplysia (Brunelli et al., 1976; Purcell et al., 2003; and see review by Barbas et al., 2003) and activity-dependent hippocampal LTP in rats (Poo, 2001; Kovalchuk et al., 2002; Huang and Reichardt, 2003).
Reactive oxygen species (ROS), such as the superoxide anion (•−O2) and hydrogen peroxide (H2O2) are necessary for activity-dependent synaptic plasticity in the mammalian hippocampus (Klann 1998; Klann et al. 1998). Consequently, mice over-expressing superoxide dismuatase have impaired hippocampal LTP (Thiels et al., 2000). Although the precise role of ROS in hippocampal LTP remains unknown, ROS affect the activity of both protein kinases and protein phosphatases (Klann et al. 1998; Klann and Thiels, 1999; Otani et al. 1993; Knapp and Klann, 2002). Indeed, protein kinase C, extracellular regulated kinase 2 (ERK2) and tyrosine kinase activities are enhanced, whereas protein phosphatases including serine/threonine (e.g. PP2A) and tyrosine phosphatases are inhibited by •−O2 and H2O2 (Klann and Thiels, 1999). ROS are necessary for long-term facilitation of carotid chemosensory neurons following AIH in rats preconditioned with chronic intermittent hypoxia (CIH) (Peng et al., 2003). Carotid sensory LTF is blocked by daily administration of the superoxide dismutase mimetic Manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP), thus implicating ROS in CIH-induced metaplasticity of sensory receptors that play an important role in the control of breathing (Peng et al., 2003).
Despite many similarities in cell-signaling mechanisms underlying multiple forms of neuroplasticity, the role of ROS in AIH-induced phrenic or XII LTF has not been explored. In support of such a role, brain ROS levels are increased during and following hypoxia (Fabian et al., 1995, Dirnagl et al., 1995; Fabian et al., 2004), particularly following intermittent hypoxia/re-oxygenation (Masaoka et al., 2005; Xu et al., 2004; Yuan et al., 2004). Further, BDNF release is greater in response to intermittent versus sustained hypoxia in PC-12 cells, a differential effect inhibited by the ROS scavenger N-acetyl-1-cysteine (Wang et al., 2005). Thus, the primary goal of this study was to test the hypothesis that ROS are necessary for phrenic and XII LTF, and that the relevant ROS are localized to the respective respiratory motor nuclei.
Experimental procedures
Experiments were performed on 3–4 month old male Sprague Dawley rats (Charles River, colony PO4). All experiments were approved by The Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin-Madison.
Surgical preparation
Anesthesia was induced using isoflurane, and the rats were tracheotomized and pump ventilated (Rodent Ventilator, model 683; Harvard Apparatus, South Natick, MA). Rats were maintained on isoflurane (3.5%) in 50% O2 (balance N2) for the duration of the surgery, and then slowly converted to urethane anesthesia (1.6–1.8 g/kg) via a tail vein catheter. Body temperature was maintained with a temperature controlled surgical table. Rectal temperature was monitored throughout the procedure using a calibrated temperature sensor (Fisher Scientific, USA). The depth of anesthesia was assessed periodically by testing blood pressure and phrenic nerve responses to toe pinch; supplemental doses of urethane were given as necessary. Inspired O2 concentration was monitored throughout the experiment using an O2 sensor (TED 60T, Teledyne Analytical Instruments, USA). One hour after beginning surgery, an intravenous infusion of a 1:11 solution of sodium bicarbonate (8.4%) and Standard Lactated Ringer's solution was initiated to maintain acid-base balance.
Following tracheotomy, rats were vagotomized and a catheter was inserted into the right femoral artery to enable measurements of arterial blood pressure (Gould, P23, USA). Arterial blood samples were drawn periodically and analysed for O2 (PO2) and CO2 (PCO2) partial pressures, pH and base excess using a blood gas analyser (ABL 500, Radiometer, Copenhagen, Denmark). The left phrenic and XII nerves were dissected using a dorsal approach, cut distally and desheathed. The nerves were submerged in mineral oil and placed on bipolar silver recording electrodes. The rats were paralyzed with pancuronium bromide once nerve signals appeared. Rats were allowed ~50min for electroneurograms and blood pressure to stabilize. Nerve activity was amplified (gain, 10,000; A-M systems, Everett, WA), bandpass-filtered (100 Hz to 10 kHz), and integrated (CWE 821 filter; Paynter, Ardmore, PA; time constant, 50 msec). The signal was digitized, recorded, and analyzed using a WINDAQ data acquisition system (DATAQ Instruments, Akron, OH).
To establish baseline nerve activity, rats were ventilated with hyperoxia (FIO2 ~ 0.50; PaO2> 180 mmHg), with CO2 added to the inspired gas. After the 50min stabilization period, the apneic threshold was determined by progressively lowering the inspired CO2. Steady state baseline nerve activity was established with Paco2 set 2–3 mmHg above the CO2 recruitment threshold (Bach and Mitchell, 1996). The drug or vehicle was injected 15 min after establishing baseline conditions. A further 20 min of baseline was recorded before starting the intermittent hypoxia protocol. In two animals, the apneic threshold was repeated after drug was injected; there was no apparent change due to drug administration.
End-tidal CO2 was monitored throughout the experiment using a flow-through capnograph (Novametrix, Wallingford, CT). Arterial blood gases and blood pressure were monitored and corrected as necessary to ensure that post-hypoxia values were maintained near baseline levels. PaCO2 was adjusted by manipulating inspired CO2; negative base excess values more severe than −3mEq/L were corrected with intravenous sodium bicarbonate, and progressive reductions in blood pressure were offset with intravenous injection of lactated Ringer's solution. Data from rats were included in the analysis only if they complied with the following criteria: (1) Pao2 during hypoxia was between 35 and 45 mmHg; (2) Pao2 during the hyperoxic baseline and recovery periods was >180 mmHg; and (3) Paco2 remained within 1 mmHg of baseline throughout the post-hypoxia protocol. Phrenic and XII nerve activity were recorded continuously. Blood samples (0.3 ml in a heparinized glass syringe) were drawn before, during the first hypoxic episode and 15, 30, and 60 min after the last hypoxic episode to ensure that blood gases met the criteria outlined above. Measurements of phrenic and XII nerve burst frequency and amplitude were evaluated for one minute immediately prior to each blood sample. At the end of the experiment, rats were humanely killed by exsanguination in accordance with ethical standards of the institution.
Protocol
All rats received AIH, which consisted of three, 5 min episodes of isocapnic hypoxia (Fio2, 0.11) with each episode separated by 5 min of moderate hyperoxia (see Fig. 1). To investigate the role of ROS in LTF, the superoxide anion scavenger, MnTMPyP (Sigma, USA) was injected intravenously at doses of either 5 (412 ± 9g; N = 8) or 10 mg kg-1 (379 ± 16g; N = 8). For each experiment, 10mg of MnTMPyP was dissolved in either 1 (10 mg kg−1) or 2 ml of saline (5mg kg−1 BW); the volume injected into each treatment group equated to just 1% of the body weight of the rat. In a third group (402 ±16g; N=7) rats were injected with vehicle only (saline).
Figure 1.
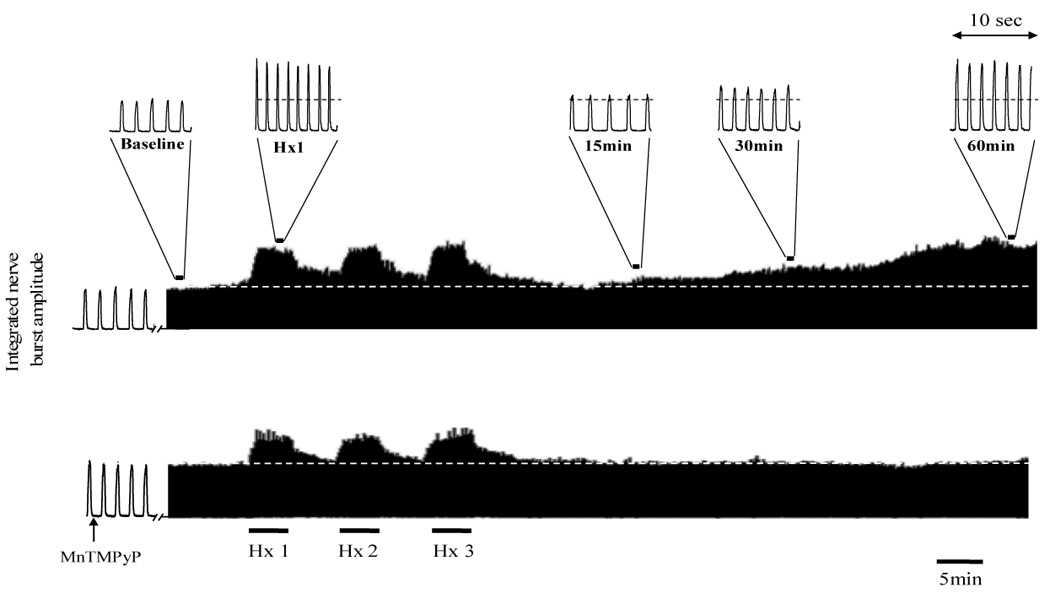
Representative neurograms recorded from the phrenic nerve of an anesthetized rat before, during and after acute intermittent hypoxia (AIH) and with vehicle (top trace) or 10mg kg−1 MnTMPyP injected intravenously (lower trace). Also indicated are magnified bins (10 seconds duration) depicting the various time points for data analysis and blood sampling throughout the protocol Dashed line indicates baseline amplitude. Note after the third and final episode of hypoxia (Hx 3) the gradual increase in phrenic nerve burst amplitude above pre-AIH levels (i.e. LTF, top trace) compared to MnTMPyP injected rats in which LTF was blocked (lower trace).
To localize the relevant ROS to the cervical spinal cord, a C2 laminectomy and durotomy were performed to allow localized injections of MnTMPyP within the vicinity of the phrenic motor nucleus (C4); this procedure has been described elsewhere (Baker-Herman and Mitchell, 2002). In brief, after C2 laminectomy, a small hole was made in the dura, and a silicone catheter (outer diameter, 0.6mm; Access Technologies) was advanced about 2mm caudally. The catheter was primed with 5.5mM MnTMPyP prior to positioning. Each rat (408 ± 13g; N=7) received a 10µl injection or artificial CSF (vehicle) (369 ±15g; N = 9) over ~5 min beginning 20 min prior to an AIH protocol.
Statistical analysis
Peak amplitude and frequency (bursts per minute) of phrenic and XII nerve activity were averaged in 1 min bins at each recorded data point (baseline, at the end of the first hypoxic exposure, and 15, 30, and 60 min post-AIH). Minute phrenic nerve activity (expressed as a % change from baseline) was determined by multiplying breath frequency and integrated nerve burst amplitude. At all time points, changes in nerve burst amplitude are normalized as a percentage of the baseline value, whereas burst frequency is expressed as absolute values. For both i.v. and i.t. treated rats, we compared phrenic and XII nerve responses of rats receiving vehicle (saline or aCSF) versus drug injections. Statistical comparisons were made for time and drug treatment effects using two-way, repeated measures ANOVA. The Bonferroni post hoc test was used to identify statistically significant individual comparisons. Differences were considered significant at p < 0.05. All values are expressed as mean ± 1 SEM.
Results
Neither PaCO2 nor pH were different from baseline values or between groups in any condition except during hypoxia in rats treated with 10mg kg−1 MnTMPyP. Despite maintenance of hypocapnia during AIH (">Table 1), a slight acidosis occurred during hypoxia in rats treated with 10mg kg−1 MnTMPyP; however, pH and base excess were restored to baseline values by 15 min post-AIH. As expected, both phrenic and XII nerve burst amplitude (Fig. 2A) and burst frequency (Fig. 2B) were significantly elevated above baseline levels during the first episode of isocapnic hypoxia (PaO2 = 38±2 mmHg). Significant hypotension was observed during hypoxia in all groups similar to previous studies on urethane anesthetized rats.
Table 1.
Body temperature (Tb) and blood gas values for i,v, treated groups. Values are means ± 1SEM.
| Group | Tb | PaO2 | PaCO2 | pH | MAP | |
|---|---|---|---|---|---|---|
| AIH + saline | 37.3 ± 0.1 | 266.5 ± 6.5 | 44.9 ± 0.8 | 7.399 ± 0.013 | 111.1 ± 5.9 | |
| baseline | AIH + 5mg kg−1 MnTMPyP | 37.4 ± 0.1 | 258.4 ± 10.3 | 44.1 ± 1.2 | 7.379 ± 0.012 | 105.5 ± 4.7 |
| AIH + 10mg kg−1 MnTMPyP | 37.3 ± 0.1 | 276.9 ± 8.0 | 46.4 ± 0.7 | 7.374 ± 0.005# | 103.3 ± 6.6 | |
| AIH + saline | 37.3 ± 0.1 | 37.7 ± 1.5* | 44.2 ± 1.0 | 7.397 ± 0.014 | 77.1 ± 8.5* | |
| Hypoxia | AIH + 5mg kg−1 MnTMPyP | 37.3 ± 0.1 | 40.2 ± 1.5* | 44.1 ± 1.1 | 7.371 ± 0.011 | 69.7 ± 5.4* |
| AIH + 10mg kg−1 MnTMPyP | 37.2 ± 0.1* | 38.7 ± 1.7* | 46.8 ± 1.0 | 7.354 ± 0.008# | 65.9 ± 7.0* | |
| AIH + saline | 37.3 ± 0.1 | 209.9 ± 14.8* | 44.7 ± 1.0 | 7.397 ± 0.018 | 112.3 ± 5.3 | |
| 15 mins | AIH + 5mg kg−1 MnTMPyP | 37.3 ± 0.1 | 196.6 ± 16.8* | 44.3 ± 1.4 | 7.373 ± 0.011 | 105.9 ± 6.4 |
| AIH + 10mg kg−1 MnTMPyP | 37.2 ± 0.1 | 200.1 ± 8.1* | 46.2 ± 1.1 | 7.372 ± 0.007 | 112.8 ± 4.7 | |
| AIH + saline | 37.3 ± 0.1 | 250.7 ± 9.6 | 45.0 ± 1.0 | 7.402 ± 0.015 | 104.2 ± 4.9 | |
| 30 mins | AIH + 5mg kg−1 MnTMPyP | 37.3 ± 0.0 | 238.6 ± 11.1 | 44.6 ± 1.4 | 7.376 ± 0.013 | 98.1 ± 7.9 |
| AIH + 10mg kg−1 MnTMPyP | 37.2 ± 0.1* | 254.9 ± 7.0 | 45.9 ± 0.9 | 7.383 ± 0.009 | 104.4 ± 5.2 | |
| AIH + saline | 37.4 ± 0.1 | 257.1 ± 10.1 | 45.2 ± 0.8 | 7.406 ± 0.015 | 97.5 ± 4.8* | |
| 60 mins | AIH + 5mg kg−1 MnTMPyP | 37.3 ± 0.1 | 246.5 ± 8.1 | 44.0 ± 1.2 | 7.389 ± 0.014 | 93.7 ± 6.3* |
| AIH + 10mg kg−1 MnTMPyP | 37.2 ± 0.1* | 266.0 ± 8.0 | 46.1 ± 0.5 | 7.388 ± 0.008* | 99.4 ± 4.5 | |
significant difference from baseline;
significant difference from control group (AIH+saline); P<0.05. Tb, body temperature; PaO2 and PaCO2, arterial pressure of oxygen and carbon dioxide, respectively; MAP, mean arterial pressure.
Figure 2.
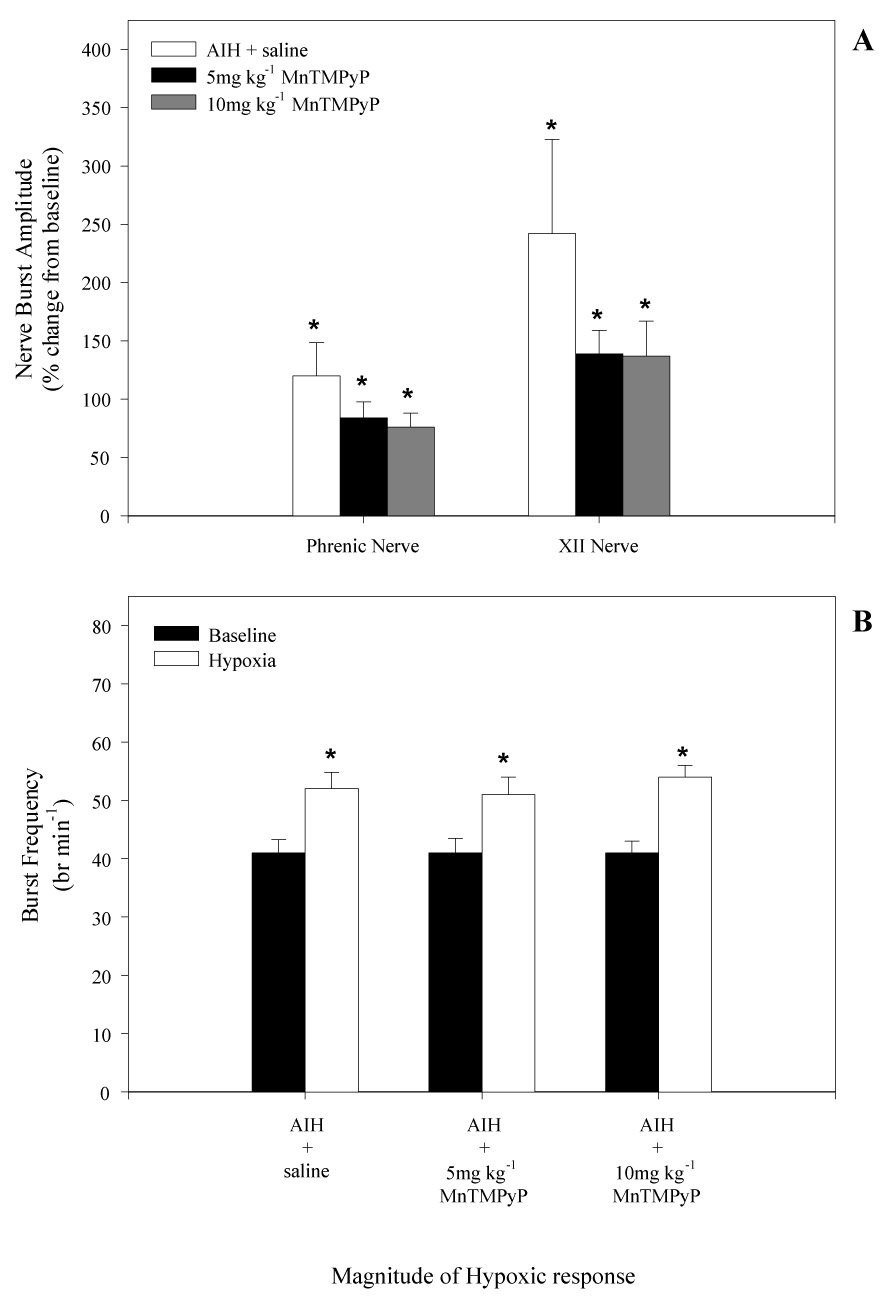
(A) Phrenic and XII nerve burst amplitude during the first hypoxic episode in rats that received i.v. injections of either saline or MnTMPyP (5 and 10mg•kg−1). Treatment groups are as indicated. Note, compared to saline injected rats, there was no effect of MnMTPyP injected i.v. on the magnitude of the hypoxic amplitude of phrenic or XII nerves. (B) Baseline breath frequency and hypoxic frequency response in saline, 5, and 10 mg•kg−1 treated rats. Note, there was no difference in baseline frequency between saline or MnTMPyP injected rats. MnTMPyP also had no effect on the magnitude of the hypoxic frequency response compared to saline treated rats. *indicates significant difference from baseline value (P<0.05).
In vehicle (saline) treated rats, AIH caused a progressive increase in phrenic and XII nerve burst amplitude characteristic of LTF (Fig. 1 and Fig 3). Burst amplitude had significantly increased above baseline at 30 and 60 min post-AIH. At 60 min post-AIH, phrenic and XII nerve burst amplitudes were 93 ± 22% and 70 ± 12% above baseline, respectively (Fig. 3A and 3B). Phrenic LTF was significantly reduced by i.v. injection of 5mg kg−1 MnTMPyP, whereas XII LTF was abolished at this same dose. At 10mg kg−1 MnTMPyP, both phrenic and XII LTF were abolished (Fig 3A and 3B, respectively).
Figure 3.
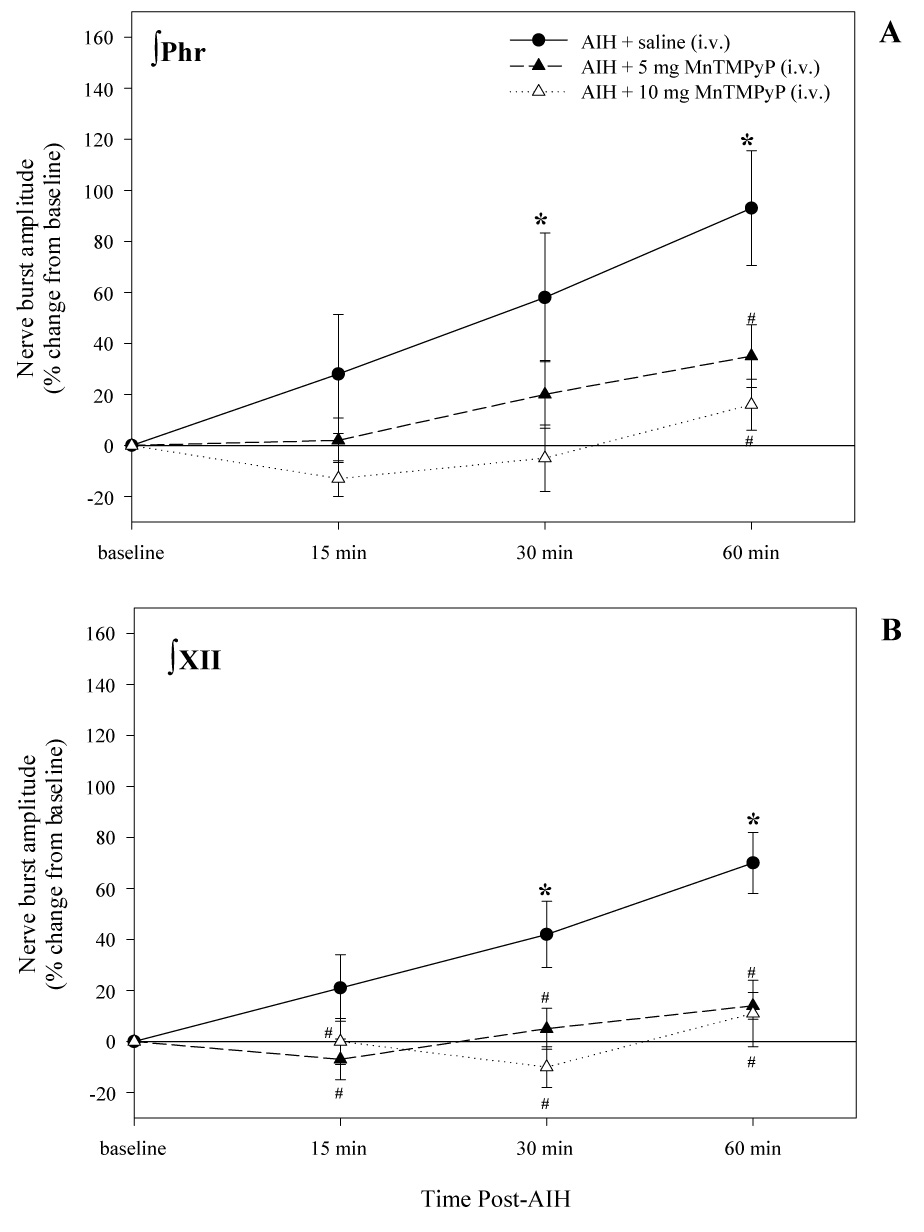
Changes in phrenic (A) and XII (B) nerve burst amplitude at 15, 30, and 60 minutes post-AIH for each treatment group. Values are means ± 1 SEM and are expressed as % change from baseline amplitude (i.e. 0%). *indicates significant difference from baseline value (P<0.05); #indicates significant difference from AIH treated groups at each experimental time point (P<0.05).
AIH also elicited a modest frequency LTF in the saline treated group, as indicated by a significant increase in nerve burst frequency 60 min post-AIH (Fig. 4A). This frequency LTF was also reduced (5mg kg−1) or abolished (10mg kg−1) by MnTMPyP, implying a drug effect on either central rhythm generating networks or on sensory pathways that modify respiratory rhythm such as the carotid body chemoreceptors (Peng et al., 2003). Minute phrenic and XII nerve activity also exhibited significant LTF in saline treated, but not MnTMPyP treated rats (Fig. 4B and 4C). MnTMPyP had no effects on phrenic or XII nerve activity during baseline conditions (data not shown) or during hypoxia (Fig 2).
Figure 4.
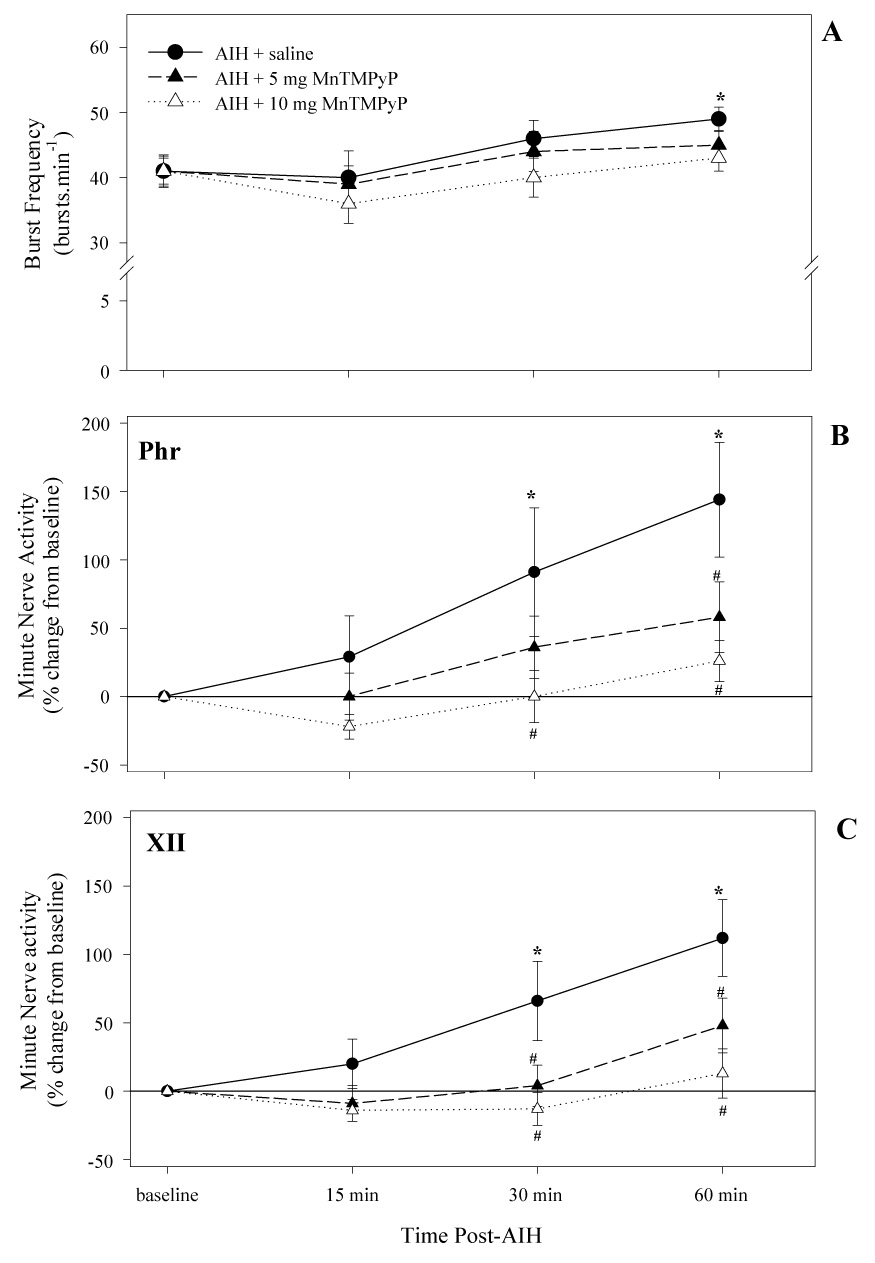
Frequency of respiratory motor output (A) and changes in minute neural activity (quantified as nerve burst amplitude (mV) × frequency (br min−1) as recorded from the phrenic (B) and XII (C) nerves at 15, 30, and 60 minutes post-AIH for each treatment group. Treatment groups are as indicated. Values are means ± 1 SEM and are expressed as a % change from baseline. *indicates significant difference from baseline value; #indicates a significant difference from vehicle injected rats at the corresponding time point. Values are means ± 1 SEM.
To test the hypothesis that MnTMPyP affects phrenic LTF by actions in the spinal cord, the drug was applied locally to the intrathecal space at C4 in an additional group of rats. Such an approach has been used previously to test the requirement for cervical spinal serotonin receptor activation in phrenic LTF (Baker-Herman and Mitchell, 2002). In these studies, XII nerve activity serves as an internal control for unintended drug distribution. Accordingly, intrathecal application of 5.5mM MnTMPyP at C4 abolished phrenic LTF (Fig. 5A). In these same animals, XII LTF was not different from vehicle control rats (Fig 5B), thereby demonstrating that effective MnTMPyP concentrations were restricted to the spinal cord.
Figure 5.
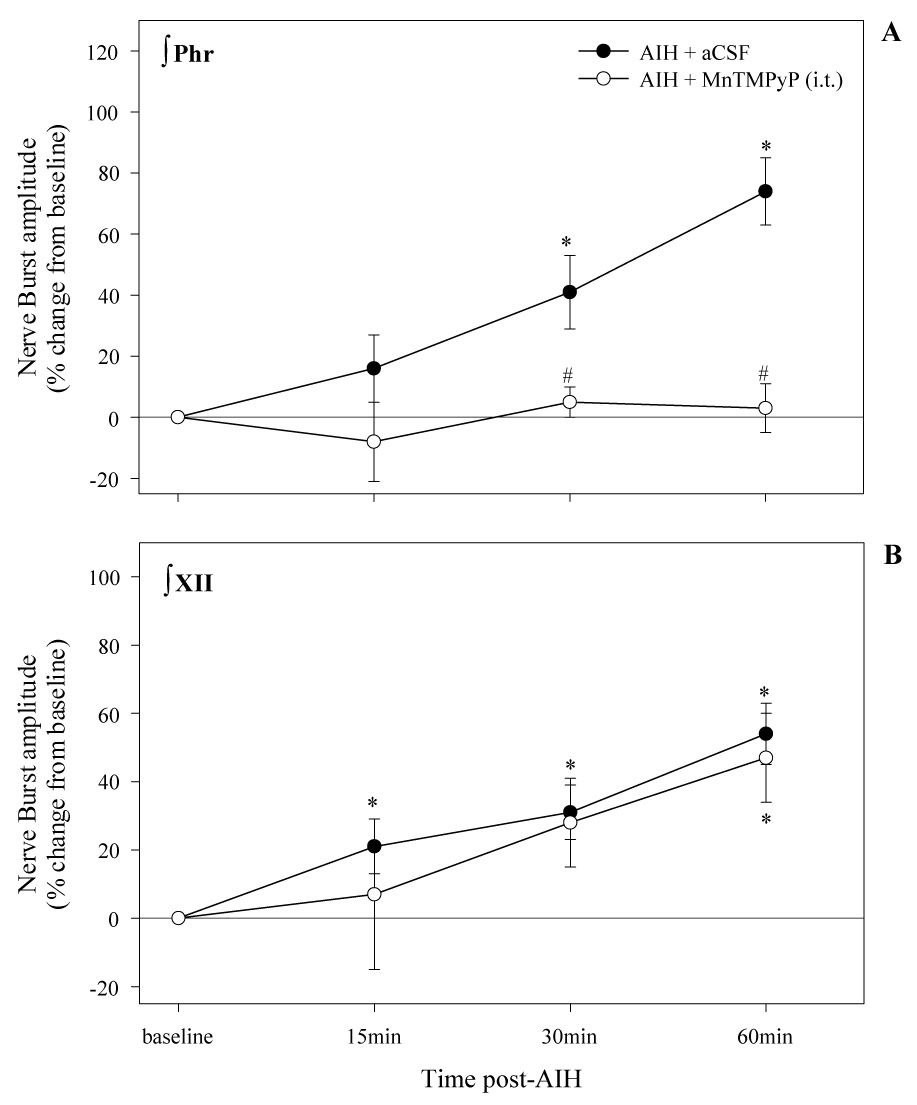
Phrenic and XII LTF in rats treated with an intrathecal injection of either aCSF (vehicle) or 5.5mM of the SOD mimetic, MnTMPyP. Note that application of the SOD mimetic to a localized region within the phrenic motor nuclei inhibited phrenic, but not XII LTF. Values are means ± 1 SEM. *signifies a significant difference from baseline; #indicates a significant difference from vehicle injected rats at the corresponding time point.
Discussion
The results demonstrate that ROS (particularly •−O2) are necessary for respiratory LTF following AIH in anesthetized rats. Further, the ROS relevant to phrenic LTF can be localized to the cervical spinal regions associated with the phrenic motor nucleus. Similarly, the lack of effect of intrathecal MnTMPyP on XII LTF, combined with the potent effect of intravenous MnTMPyP, suggest that XII LTF is also dependent on ROS, but localized near the XII motor nucleus in this case.
The superoxide dismutase (SOD) mimetic, MnTMPyP, is a cell permeable •−O2 scavenger that reduced both phrenic and XII LTF following systemic administration. Thus, •−O2 formation is necessary for the induction and/or maintenance of LTF in both motor outputs, thus suggesting a common mechanism. To our knowledge, this is the first example of ROS-dependent plasticity in the brainstem/spinal cord, although there is considerable evidence for ROS-dependence in other forms of neuroplasticity. For example, MnTMPyP (Klann 1998) and catalase (Klann et al., 1998) both block activity-dependent hippocampal LTP in CA1, suggesting possible roles for both •−O2 and HSO2 in its underlying mechanism. In transgenic mice over-expressing extracellular SOD, hippocampal LTP is impaired, an effect reversed by inhibition of copper-zinc SOD with a copper chelator (Thiels et al., 2000). ROS are also necessary for the mechanism whereby pretreatment with chronic intermittent hypoxia induces the capacity to express carotid chemosensory LTF following AIH (Peng et al. 2003). Carotid sensory LTF is a prolonged increase in chemoafferent neuron activity following AIH, but is observed only in rats pretreated with chronic intermittent hypoxia (Peng et al., 2003). Since sensory LTF is not elicited in rats receiving daily injections of MnTMPyP (5mg kg−1) during chronic intermittent hypoxia exposures (Peng et al., 2003), ROS are required in the mechanism whereby chronic intermittent hypoxia induces this mechanism. Carotid sensory LTF in CIH-treated rats might share similar ROS mechanisms to phrenic and XII LTF following AIH, although the time domains of these forms of plasticity are quite distinct.
Inhibition of phrenic and XII LTF via intravenous MnTMPyP does not exclude the possibility of a peripheral or non-spinal/brainstem site of action. The ROS-dependence of the small, AIH-induced frequency LTF (Fig. 4A) provides suggestive evidence for non-spinal mechanisms of action of MnTMPyP, although this result could equally be due to actions on intra-spinal synaptic pathways of sensory neurons projecting to the respiratory rhythm generating circuitry in the brainstem. Observations that MnTMPyP had no effects on baseline respiratory motor output, the CO2-apneic threshold or the magnitude of the short-term hypoxic phrenic nerve response provide evidence that the actions of MnTMPyP are highly specific to LTF, and thus do not reflect general actions of the drug on spontaneous neural activity or changes in afferent inputs such as the carotid bodies. Furthermore, differential responses of XII and phrenic LTF at low intravenous doses of MnTMPyP (5mg/kg) suggest differential sensitivity of the respective motor nuclei to ROS.
Intrathecal MnTMPyP administration to cervical spinal regions that contain the phrenic motor nucleus confirm that the ROS necessary for phrenic LTF are located in or near the phrenic motor nucleus and not at peripheral sites of action. This experimental approach has been used successfully to differentiate spinal versus brainstem or systemic effects in LTF (Baker-Herman and Mitchell, 2002). While an i.v. injection of MnTMPyP blocked both phrenic and XII LTF, it does not exclude the possibility that ROS scavenging affects phrenic and XII LTF through modifications occurring elsewhere, such as the carotid body or brainstem. Indeed, selective inhibition of phrenic, but not XII, LTF following i.t. injections of MnTMPyP suggests that the relevant ROS for phrenic LTF occur within spinal regions containing the phrenic motor nucleus versus non-specific effects via unintended drug distribution. Although AIH-induced phrenic LTF is distinguishable from carotid sensory LTF in CIH pre-treated rats, similar responses to MnTMPyP suggest at least some commonalities in their ROS-dependence.
Our data imply that superoxide anion formation in respiratory motor nuclei may be increased during or following AIH. During AIH protocols, ROS may be generated by increased neuronal activity, by ligand binding to receptors that generate ROS when activated (e.g. 5-HT2 and TrkB receptors), by the hypoxia itself, or by the swings in oxygen tension. Since hippocampal LTP requires activity-dependent ROS production (Klann, 1998; Knapp and Klann, 2002), increased motor neuron activity during hypoxia may induce ROS formation, possibly from the mitochondria; Peng and colleagues (2003) proposed that complex I of the mitochondrial respiratory chain is a significant source of ROS generation in carotid sensory LTF. If so, activity-dependent source of ROS is not sufficient to induce LTF since multiple receptor antagonists (e.g. the serotonin receptor antagonist methysergide) block LTF but have no effect on the magnitude of phrenic or XII nerve activity during hypoxia (Bach and Mitchell, 1996). Furthermore, whereas AIH induces respiratory LTF, the same cumulative duration of sustained hypoxia (25 min; Baker and Mitchell, 2000) or hypercapnia (Baker et al., 2001) does not, further supporting the hypothesis that activity-dependent ROS formation is not sufficient to induce LTF. Another promising candidate for the necessary ROS formation is NADPH oxidase activity, a major source of superoxide during hypoxia (Henderson and Chappell, 1996) and intermittent hypoxia (Hitomi et al., 2003; Zhan et al., 2005). Indeed, Peng and colleagues (2006) recently reported that NADPH oxidase contributes ROS necessary for carotid sensory LTF in PC12 cells following repetitive serotonin administration. On the other hand, we cannot exclude the possibility that the ROS necessary for AIH-induced LTF result from constitutive ROS production not specifically related to AIH; in this sense, ROS may be necessary but not sufficient for AIH-induced LTF.
Using a superoxide sensitive electrode in the cortex, Fabian and colleagues (2004) demonstrate that extra-cellular superoxide levels are unaffected by 20 minutes of hypoxia (10% inspired O2), but increase for a prolonged period (>20 min) following a transition from hypoxia to hyperoxia (100% inspired O2). If a similar pulse of superoxide production occurs at each of the three consecutive phases of re-oxygenation in our protocol, a cumulative effect similar to RC-integration might produce sufficient ROS to induce LTF during or following AIH, whereas sustained hypoxia might not. Greater ROS production during intermittent (versus sustained) hypoxia may account, at least part, for the pattern-sensitivity of respiratory LTF (Baker and Mitchell, 2000).
In recent years, fairly detailed cellular/synaptic mechanisms have been proposed to explain respiratory LTF (Mitchell et al., 2001; Feldman et al., 2003; Baker-Herman et al., 2004; Mahamed and Mitchell, 2006), but a possible role of ROS in respiratory LTF has not been previously explored. ROS may regulate respiratory LTF via actions on the kinase/phosphatase balance (Mitchell and Johnson, 2003), including both increased activation of kinases that enable LTF, and by inhibiting phosphatases that constrain LTF (Wilkerson et al., 2007). For example, intrathecal okadaic acid administration, a serine/threonine phosphatase inhibitor, reveals phrenic LTF following sustained hypoxia. Although the mechanisms leading to differential regulation of phosphatase activity during intermittent versus sustained hypoxia were not identified in these studies, ROS generated during and/or following AIH may activate relevant protein kinases (e.g. PKC) and/or inhibit relevant okadaic acid-sensitive protein phosphatases (e.g. PP2). As a provisional test of the latter possibility, we demonstrated that AIH-induced phrenic LTF is restored in rats treated with 10 mg/kg MnTMPyP (i.v.) when okadaic acid is administered intrathecally (MacFarlane and Mitchell, unpublished observation). Although not conclusive, these observations suggest that the failure of ROS-mediated phosphatase inhibition during AIH underlies the loss of LTF in rats treated with MnTMPyP, and that LTF is restored when another means is used to suppress those phosphatases (e.g. with okadaic acid).
Other possible mechanisms whereby ROS modulate LTF must be considered, including the regulation of intracellular calcium. AIH induces transient calcium spikes, causing auto-phosphorylation and sustained activation of calcium sensitive kinases such as ERK 1/2 (Putney, 1998). Indeed, ryanodine receptor inhibition blocks forms of hippocampal LTP (Martin and Buno 2003) and redox state influences intracellular Ca2+ release, thereby promoting synaptic plasticity via increased ERK phosphorylation (Hidalgo et al. 2005). Since ERK is one postulated “downstream” kinase from TrkB, indirect effects of calcium must be evaluated in future studies.
Conclusions
ROS formation during AIH is necessary for cellular events giving rise to brainstem-spinal cord mechanisms of respiratory LTF. Superoxide anions are likely to be the primary ROS species involved, primarily acting at sites localized in or near the respective motor nuclei (ie. phrenic and XII). One likely role of the superoxide anion is to regulate cellular phosphorylation state, thereby shifting the balance towards LTF-enabling processes. Overall, the results of this study significantly add to our understanding of the important signaling role played by spinal ROS formation in phrenic LTF. Further work is essential to determine the source of ROS and clarify its precise cellular and molecular targets.
Acknowledgements
Supported by NIH HL80209. We thank Brad Hodgeman for excellent assistance in the preparation of figures.
Abbreviations
- 5-HT
serotonin
- aCSF
artifical cerebral spinal fluid
- AIH
acute intermittent hypoxia
- BDNF
brain derived neurotrophic factor
- CIH
chronic intermittent hypoxia
- ERK
extracellular regulated kinase
- H2O2
hydrogen peroxide
- i.v.
intravenous
- i.t.
intrathecal
- LTF
long term facilitation
- •−O2
superoxide anion
- PaO2
arterial partial pressure of Oxygen
- PaCO2
arterial partial pressure of carbon dioxide
- ROS
reactive oxygen species
- TrkB
tyrosine receptor kinase
- XII
hypoglossal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
PM MacFarlane, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin, Madison, WI, USA 53706, Ph.: 608-263-5013, Fax: 608-263-3926, Email: macfarlanep@svm.vetmed.wisc.edu.
GS Mitchell, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin, Madison, WI, USA 53706, Ph.: 608-263-5878, Fax: 608-263-3926, Email: mitchell@svm.vetmed.wisc.edu.
References
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol (Lond) 2000;15:15–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker-Herman T, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22(14):6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7(1):48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Barbas D, DesGroseillers L, Castellucci VF, Carew TJ, Marinesco S. Multiple serotonergic mechanisms contributing to sensitization in aplysia: evidence of diverse serotonin receptor subtypes. Learn Mem. 2003;10(5):373–386. doi: 10.1101/lm.66103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelli M, Castellucci V, Kandel ER. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science. 1976;194(4270):1178–1181. doi: 10.1126/science.186870. [DOI] [PubMed] [Google Scholar]
- Carrasco MA, Jaimovich E, Kemmerling U, Hidalgo C. Signal transduction and gene expression regulation by calcium release from internal stores in excitable cells. Biol Res. 2004;37:701–712. doi: 10.4067/s0716-97602004000400028. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Lindauer U, Them A, Schreiber S, Pfister HW, Koedel U, Reszka R, Freyer D, Villringer A. Global cerebral ischemia in the rat: online monitoring of oxygen free radical production using chemiluminescence in vivo. J Cereb Blood Flow Metab. 1995;15(6):929–940. doi: 10.1038/jcbfm.1995.118. [DOI] [PubMed] [Google Scholar]
- Fabian RH, DeWitt DS, Kent TA. In vivo detection of superoxide anion production by the brain using a cytochrome c electrode. J Cereb Blood Flow Metab. 1995;15(2):242–247. doi: 10.1038/jcbfm.1995.30. [DOI] [PubMed] [Google Scholar]
- Fabian RH, Perez-Polo RJ, Kent TA. Extracellular superoxide concentration increases following cerebral hypoxia but does not affect cerebral blood flow. Int J Devl Neuroscience. 2004;22:225–230. doi: 10.1016/j.ijdevneu.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic nerve output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB. NADPH oxidase of neutrophils. Biochim. Biophys Acta. 1996;1273:87–107. doi: 10.1016/0005-2728(95)00140-9. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Bull R, Behrens MI, Donoso P. Redox regulation of RyR-mediated Ca2+ release in muscle and neurons. Biol Res. 2005;37:539–552. doi: 10.4067/s0716-97602004000400007. [DOI] [PubMed] [Google Scholar]
- Hitomi Y, Miyamura M, Mori S, Suzuki K, Kizaki T, Murakami K, Haga S, Ohno H. Intermittent hypobaric hypoxia increases the ability of neutrophils to generate superoxide anion in humans. Clin Exp Pharmacol Physiol. 2003;30:659–664. doi: 10.1046/j.1440-1681.2003.03891.x. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. TRK receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273(8):4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog. Neuropsychopharmacol Biol Psychiatry. 1999;23(3):359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci. 2002;22(3):674–683. doi: 10.1523/JNEUROSCI.22-03-00674.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic Induction of BDNF-Mediated Long-Term Potentiation. Science. 2002;295(5560):1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Mahamad S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnea? Exp Physiol. 2007;92(1):27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Martin ED, Buno W. Caffeine-mediated presynaptic long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2003;89:3029–3030. doi: 10.1152/jn.00601.2002. [DOI] [PubMed] [Google Scholar]
- Masaoka N, Nakajima Y, Hayakawa Y, Ohgame S, Hamano S, Nagaishi M, Yamamoto T. Transplacental effects of allopurinol on suppression of oxygen free radical production in chronically instrumented fetal lamb brains during intermittent umbilical cord occlusion. J Matern Fetal Neonatal Med. 2005;18(1):1–7. doi: 10.1080/14767050500127716. [DOI] [PubMed] [Google Scholar]
- McGuire M, Ling L. Activation of protein kinase C near/in phrenic motoneurons is required for phrenic Long-term facilitation in rats. Am J Respir Crit Care Med. 2004;169(7):A433. (abstr. suppl.) [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90(6):2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94(1):358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Otani S, Ben-Ari Y, Roisin-Lallemand M-P. Metabotropic receptor stimulation coupled to weak tetanus leads to long-term potentiation and a rapid elevation of cytosolic protein kinase C activity. Brain Res. 1993;613:1–9. doi: 10.1016/0006-8993(93)90446-t. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body of intermittent hypoxia: implications for recurrent apneas. PNAS. 2003;100(17):10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576(1):289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Purcell AL, Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Activation of a tyrosine kinase-MAPK cascade enhances the induction of long-term synaptic facilitation and long-term memory in Aplysia. Neuron. 2003;37:473–484. doi: 10.1016/s0896-6273(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Putney JW. Calcium signaling: up, down, up, down…what’s the point? Science. 1998;279:191–192. doi: 10.1126/science.279.5348.191. [DOI] [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Ourv TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ward N, Boswell M, Katz DM. Secretion of brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur J Neurosci. 2005;23(6):1665–1670. doi: 10.1111/j.1460-9568.2006.04682.x. [DOI] [PubMed] [Google Scholar]
- Wilkerson JER, Macfarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: roles of protein phosphatases and reactive oxygen species. Biochem Soc Trans. 2007;35(Pt 5):1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126(2):313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol. 2004;557(3):773–783. doi: 10.1113/jphysiol.2003.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Serrano F, Fenik P, Hsu R, Kong L, Pratico D, Klann E, Veasey SC. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005;172(7):921–929. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]