

Abstract
The retinal pigment epithelium (RPE) plays an essential role in maintaining the health of the retina. The RPE is also the site of pathologic processes in a wide variety of retinal disorders including monogenic retinal dystrophies, age-related macular degeneration, and retinal detachment. Despite intense interest in the RPE, little is known about its molecular response to ocular damage or disease. We have conducted a comprehensive analysis of changes in transcript abundance (the “genomic response”) in the murine RPE after light damage. Several dozen transcripts, many related to cell–cell signaling, show significant increases in abundance in response to bright light; transcripts encoding visual cycle proteins show a decrease in abundance. Similar changes are induced by retinal detachment. Environmental and genetic perturbations that modulate the RPE response to bright light suggest that this response is controlled by the retina. In contrast to the response to bright light, the RPE response to retinal detachment overrides these modulatory affects.
Keywords: retinal pigment epithelium, transcription, microarray, mouse, ocular disease, light damage, retinal detachment
Introduction
The retinal pigment epithelium (RPE) consists of a monolayer of flat polygonal cells sandwiched between the outer segments of retinal photoreceptors and the choroidal vasculature. The RPE performs a variety of essential functions to support the retina: it forms part of the blood–retina barrier and mediates exchange of metabolites between the retina and the choroid; it stores and recycles the visual pigment chromophore (the visual cycle); it engulfs and digests the tips of photoreceptor outer segments; and it maintains the ionic composition of the subretinal space (Marmor and Wolfensberger, 1998).
Not surprisingly, defects in RPE function or viability play a central role in a wide variety of ocular disorders. Monogenic defects in RPE proteins, including visual cycle enzymes [lecithin retinol acyltransferase (Lrat), retinoid isomerohydrolase (Rpe65), and retinol dehydrogenase-5 (RDH5)], the anion channel VMD2, tissue inhibitor of metalloproteinase 3, fibulin-like extracellular matrix protein-1, and the transmembrane tyrosine kinase receptor MERTK lead to congenital blindness, progressive retinal degeneration, or delayed visual adaptation (Retinal Information Network, http://www.sph.uth.tmc.edu/retnet/). A decrease in the adhesion between RPE and photoreceptors predisposes to retinal detachment, a common ophthalmologic emergency, and reattachment is frequently complicated by pathologic proliferation of the RPE at the site of detachment (proliferative vitreoretinopathy) (Pastor et al., 2002). A local loss of RPE cells (geographic atrophy) is the most common cause of impaired vision in patients with age-related macular degeneration (AMD), whereas disruption of the vascular barrier function of the RPE leads to choroidal neovascularization and severe vision loss in ∼10% of AMD patients (Rattner and Nathans, 2006).
Given the importance of the RPE in retinal health and disease, it would be of interest to fully delineate the molecular responses of RPE cells to injuries or diseases that affect the retina, the RPE, or both. To date, most investigations of RPE pathology have focused either on the anatomic effects of injury or disease in vivo (Anderson et al., 1981; Hafezi et al., 1997; Szurman et al., 2006) or on the cellular and molecular effects of chemical or photochemical stresses on cultured RPE cells and cell lines in vitro (Sparrow and Cai, 2001; Ganti et al., 2007). With the first approach, the molecular changes and the signaling pathways that control the response of the RPE to damage remain mostly undefined. With the second approach, the interpretation of the response of the RPE to damage is limited by differences in the environment of RPE cells in pure culture and in the intact eye, in particular by the absence of signals from photoreceptors or other ocular cells. However, one report clearly indicates that in the living eye the RPE exhibits a specific molecular response to damage, as judged by changes in the phosphorylation state of extracellular signal-regulated kinase and induction of c-Fos and c-Jun in the RPE after retinal detachment (Geller et al., 2001).
In the present study, we analyze changes in RPE transcripts (the “genomic response”) after damage in the living eye. After either bright light exposure or retinal detachment, abundance changes are found in substantially overlapping sets of RPE transcripts. These include a decrease in all RPE transcripts coding for components of the visual cycle, and a dramatic increase in the abundances of a small set of transcripts, many of which code for secreted proteins and cell surface receptors. Interestingly, the RPE response is strongly modulated by retinal damage or stress, implying the existence of a stress signal produced in the retina and sensed by the RPE.
Materials and Methods
cDNA probes.
cDNA probes for mouse matrix metalloproteinase 3 (Mmp3) (IMAGE ID 3962288) and mouse lymphocyte cytoplasmic protein-1 (Lcp1) (IMAGE ID 3486993) were obtained from Open Biosystems. Other mouse cDNA probes were PCR amplified using RPE or retina cDNA prepared with a Smart RACE cDNA amplification kit (BD Biosciences/Clontech). The identities of the probes were confirmed by sequencing.
Microarray hybridization and data analysis.
BALB/c mice were dark adapted for 1 week and then either used as controls or exposed to 6000 lux cool white light for 1.5 h as described previously (Rattner and Nathans, 2005). For RPE RNA isolation, the lens and the retina were removed, and RPE cells were dissociated by gentle agitation of the eyecups in calcium-free PBS for 15 min on ice and then collected by centrifugation at 3000 × g for 5 min at 4°C. For each hybridization reaction, total RNA was extracted from RPE cells collected from 20 mouse eyes using Trizol (Invitrogen) and an RNeasy kit (Qiagen). The Affymetrix small sample labeling protocol VII was used for probe production. Microarray experiments were performed using Affymetrix mouse genome 430 2.0 arrays. Data from triplicate hybridizations were preprocessed using robust multichip averaging (Irizarry et al., 2003), converted into a log2-transformed format, and a p value was calculated for each transcript using Student's t test statistics. Fold changes were calculated from the log2-transformed signal ratios.
RNA blotting.
RNA blotting was performed using standard methods with 32P-labeled probes isolated by PCR amplification from the indicated cDNA clones.
Mice.
BALB/c and rd1 −/− mice were obtained from the Jackson Laboratories. rd1 −/− mice were genotyped according to Hafezi et al. (1998). Rpe65 −/− mice (Redmond et al., 1998) were a gift from Dr. T. M. Redmond (National Institutes of Health, Bethesda, MD). Rod transducin α (Gnat1 −/−) mice (Calvert et al., 2000) were a gift from Dr. J. Lem (Tufts New England Medical Center, Boston, MA). Each mutation was crossed into a BALB/c background to obtain albino mice for light damage and/or retinal detachment experiments. BALB/c mice are homozygous for the L450 light-sensitive allele of Rpe65. The albino Gnat1 −/− mice were also all homozygous for this allele.
Retinal detachments.
Retinal detachments were performed as described previously (Rattner and Nathans, 2005). Briefly, mice were anesthetized with intraperitoneal ketamine and xylazine and by corneal application of 5 μl of 10% lidocaine in PBS. A 30-gauge needle was inserted at the limbus and withdrawn, and a blunt 33-gauge needle was inserted through the 30-gauge hole and advanced until it touched the back wall of the eye. To detach the retina, 1 μl of physiological saline was slowly expelled through the 33-gauge needle into the subretinal space. After the procedure, erythromycin ophthalmic ointment was applied to the corneal surface. To avoid stress caused by light exposure, retinal detachments in albino mice were conducted under dim light.
Antibodies.
Rabbit polyclonal antibodies against the 114 C-terminal amino acids of mouse oncostatin M receptor (OSMR) were generated by immunization with a T7 gene 10 fusion protein produced in Escherichia coli. Rabbit anti-β-tubulin polyclonal antibodies were purchased form Novus Biologicals.
Histology and immunocytochemistry.
Eyes were fresh frozen in optimal cutting temperature compound (Tissue-Tek), and 10 μm sections were cut and postfixed in PBS with 4% paraformaldehyde. Fixed sections were washed three times with PBS, blocked for 1 h with 10% normal goat serum in PBS with 0.3% Triton X-100, and incubated overnight at 4°C with anti-OSMR antiserum at 1:10,000 dilution in blocking solution. Bound anti-OSMR antibodies were detected with Alexa Flour 594-conjugated goat anti-rabbit IgG (Invitrogen).
Protein extracts from mouse retina and RPE.
For RPE extracts, posterior eyecups from which the lens and retina had been removed were prepared and solubilized in buffer containing 150 mm NaCl, 50 mm Tris, pH 7.5, 1 mm EDTA, 1% Nonidet P-40, and complete protease inhibitor mixture (Roche Applied Science). After a 15 min incubation on ice and a 15 s agitation by Vortex mixer, the lysate was cleared of nuclei and insoluble material by two centrifugations at 1600 × g for 5 min at 4°C. Retina extracts were prepared identically starting from dissected retinas. Protein concentration was determined using the Bio-Rad Protein Assay, and equal amounts of protein were resolved by 10% SDS-PAGE and transferred to a nitrocellulose filter for antibody binding.
In situ hybridization.
In situ hybridization was performed essentially as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993). Digoxigenin labeled riboprobes were transcribed with T7 RNA polymerase from the indicated cDNA clones. To facilitate detection of the chromogenic reaction product in pigmented RPE, hybridized sections were incubated overnight in PBS containing 3% hydrogen peroxide (Orchard, 1999) before the application of the alkaline phosphatase-conjugated anti-digoxigenin antibody. Images were captured on a Zeiss Imager Z1 microscope using Zeiss AxioVision 4.6 software.
Production and binding of AP-myc-OSM.
Myc-tagged human placental alkaline phosphatase (AP) fused to the mature mouse OSM polypeptide (amino acids 22–207) was secreted from transiently transfected 293 cells using a vector with a cytomegalovirus (CMV) immediate early gene enhancer and promoter; the AP-myc-OSM fusion protein was collected in DMEM/F-12 containing penicillin/streptomycin and 10% calf serum. Sections (10 μm) of fresh frozen eyes were air dried and stored at −80°C, hydrated with tissue culture medium for 1 h at 4°C, incubated for 1 h at 4°C in AP-myc-OSM conditioned medium with gentle rocking, and washed six times with ice-cold DMEM/F-12. The sections were then fixed with 60% acetone, 3% formaldehyde in 20 mm HEPES, pH 7.0, for 30 s, washed twice with Tris-buffered saline, and heated in a water bath at 65°C for 90 min to inactivate endogenous AP. Bound AP-myc-OSM was visualized with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate.
RPE eyecup explants and adenovirus infection.
Dissected mouse eyes were embedded in 3% low-melting-point agarose equilibrated to 37°C in serum-free DMEM/F-12 in a small plastic mold. After 5 min on ice, the mold was cut away from the solidified agarose block, and the front half of the eye was removed by cutting through the agarose and the embedded eye with a razor blade. After making a circular cut around the optic disc, the retina was gently removed from the posterior eyecup. The agarose block holding the posterior eyecup with the RPE now exposed was placed in DMEM/F-12 with penicillin/streptomycin and 10% calf serum and incubated at 37°C in 5% CO2. A stock of replication defective adenovirus type 5 harboring an E. coli β-galactosidase coding region under the control of a CMV promoter was purchased from Vector BioLabs. One microliter of the 1 × 1010 infectious unit per milliliter of virus sample was pipetted into the bowl formed by the eyecup and incubated for 30 min at 37°C. Tissue culture medium was then added to cover the surface of the eyecup and the agarose block in which it was embedded, and incubation was continued for an additional 2 d until the eyecup was fixed and processed for β-galactosidase histochemistry. Transmission electron microscopy was performed with eyecups fixed with 2% paraformaldehyde and 2% gluteraldehyde followed by osmication and embedding in Spurr's resin (Ted Pella).
Results
The response of the RPE to light damage
To profile changes in RPE transcript levels in response to retinal damage, we performed microarray hybridization with RNA from albino BALB/c mice that were either (1) dark adapted or (2) dark adapted, exposed to 6000 lux of cool fluorescent light for 1.5 h with dilated pupils, and allowed to recover for 24 h in darkness. Three independent pairwise comparisons were performed, with each sample containing RNA pooled from 20 eyes (Fig. 1 A). To harvest mouse RPE cells, we first removed the lens and retina, and then gently detached the RPE cells from the back wall of the eye and collected them by centrifugation. This procedure results in efficient enrichment of RPE cells as determined by the strength of the RNA blot hybridization signals obtained for the RPE-specific Rpe65, RPE-retinal G-protein-coupled receptor (Rgr), and Peropsin transcripts (Fig. 1 B). A low level of contamination of RPE by neural retina can by detected by RNA blot hybridization to rhodopsin (Rho) transcripts. Retina samples appear to be nearly free of RPE contamination as determined by hybridization to Rpe65 transcripts.
Figure 1.

Changes in RPE transcript levels in response to light damage. A, Scatterplot showing microarray hybridization intensity ratios and p values for three independent pairs of RPE samples from dark-adapted controls versus light-damaged BALB/c mice (20 eyes per sample). The scatterplot, including the subset of transcripts marked by red circles and blue squares (defined in the box in A), has not been corrected for redundancies in the Affymetrix M430 2.0 microarray annotation; thus, there are more data points than unique genes represented. B, RNA blot hybridization demonstrates increased or decreased abundances of different RPE transcripts 24 h after light damage. Left column, Transcripts induced by light damage, some of which are also induced in the retina. Right two columns, Transcripts encoding visual cycle proteins and RPE-specific G-protein-coupled receptors decrease in abundance after light damage. Gapdh and Rho transcripts serve, respectively, as (1) an internal control for equal loading of RNA and (2) retina contamination of the RPE sample. The Rpe65 blot demonstrates minimal contamination of the retina sample with RPE. C, Cellular localization of transcripts with altered abundances after light damage, as determined by in situ hybridization. Edn2, a transcript shown previously to accumulate in photoreceptors in response to bright light and other forms of damage, serves as a positive control. Vertical bars adjacent to each tissue section demarcate (from top to bottom) the RPE, outer nuclear layer, inner nuclear layer, and ganglion cell layer. ISH, In situ hybridization.
Of the 27,173 transcripts with signals above background on the Affymetrix M430 2.0 microarray, only 17 showed more than a fivefold and 146 more than a twofold increase in abundance with a p value less then 0.05, and only 22 transcripts showed more than a twofold decrease in abundance with a p value less then 0.05 (Fig. 1 A, supplemental Table 1, available at www.jneurosci.org as supplemental material) (in considering the numbers of transcripts interrogated by the microarray, we note that some of the entries in the Affymetrix spreadsheet are redundant). For a subset of these transcripts, we have more precisely assessed changes in abundance and anatomic localization by RNA blot and in situ hybridization (Fig. 1 B,C).
Six transcripts that were found to increase in abundance by microarray hybridization were tested by blot hybridization with retina and RPE RNA and by in situ hybridization to eye sections. In each case, the increase in abundance in the RPE was confirmed (Fig. 1 B,C and data not shown). Mmp3 and Serpin b1a transcripts were present (and induced) exclusively in the RPE. Cellular enhancer-binding protein δ (Cebpd) transcripts were also strongly induced in the inner nuclear layer of the retina (Fig. 1 C). Osmr, Serpin a3n, and Lcp1 transcripts were present at higher levels in the RPE compared with the retina and were induced in both tissues (Fig. 1 B). In the retina, Osmr transcripts localize to the inner nuclear layer, and Serpin a3n transcripts localize to both the inner nuclear layer and the ganglion cell layer (Fig. 1 C). The patterns of in situ hybridization in the inner nuclear layer and ganglion cell layer are most consistent with expression in Muller glia and astrocytes, respectively.
The microarray hybridizations also suggested that several RPE-specific transcripts exhibit a modest and, in most cases, statistically insignificant decline in abundance after light damage. However, microarray hybridization typically underestimates changes in transcript abundance because of the nonspecific background superimposed on the hybridization signal. This effect is most pronounced when the transcripts of interest are rare and/or the fold change is small. Therefore, we independently tested a set of RPE-specific transcripts by blot hybridization of retina and RPE RNA. The set includes transcripts coding for all of the known RPE components of the visual cycle: Rpe65, Lrat, cellular retinaldehyde-binding protein (Cralbp), retinol-binding protein 1 (Rbp1), and Rdh5 and Rdh10. We also tested two RPE-specific G-protein-coupled receptors, Rgr and peropsin (Rrh). In each case, a clear reduction in transcript levels was observed in the light-damaged RPE (Fig. 1 B), and this reduction was further confirmed for Rpe65, Rbp1, and Rdh10 by in situ hybridization (Fig. 1 C). Rdh10 and Rbp1 transcripts show a complex response to light damage in that their transcript abundances simultaneously decline in the RPE but increase in the inner nuclear layer of the retina. Among photoreceptor transcripts, Rho and Rdh12 decline and Rdh8 remains unchanged after light damage (Fig. 1 B). Retinal and RPE levels of glyceraldehyde phosphate dehydrogenase (Gapdh) transcripts remain unchanged.
Thus, the RPE responds to light damage by simultaneously increasing the abundance of one subset of transcripts and decreasing the abundance of a second subset. The observation that light damage produces a decline in the abundances of transcripts for every known RPE protein involved in the visual cycle supports the general idea that downregulating this cycle may be photoprotective (Danciger et al., 2000; Wenzel et al., 2001).
The response of the RPE to retinal detachment
In previous work, we observed that the set of transcripts that are induced in the neural retina by light damage substantially overlaps the sets that are induced by retinal detachment and genetically based photoreceptor degeneration, suggesting a generic retinal response to photoreceptor stress or damage (Rattner and Nathans, 2005). To determine whether the RPE also exhibits a common genomic response to diverse insults, small retinal detachments were induced in albino BALB/c eyes by subretinal injection of physiological saline. To avoid light damage to the albino eye, the procedure was conducted under dim light. Four days later, the light-damage-regulated transcripts described above were assayed for changes in abundance by in situ hybridization.
After retinal detachment, most of the transcripts analyzed showed a change in abundance in the RPE and/or retina that localized to the site of the detachment and closely matched the changes induced by light damage (compare Figs. 1 C, 2 A–H). As a control for the localized nature of detachment-induced stress, we observed an increase in Endothelin2 (Edn2) transcripts only in photoreceptors at the detachment site, as reported previously (Rattner and Nathans, 2005) (data not shown). Similarities between the retinal detachment and light-damage responses include: (1) the increase in Mmp3 transcripts exclusively in the RPE (Fig. 2 C), (2) the increase in Osmr and Serpin a3n transcripts in the RPE and the inner nuclear layer (Fig. 2 A,E), and (3) the reduction in Rdh10 and Rpe65 transcripts in the RPE (Fig. 2 D,H). As seen for the light-damage response in the retina, Rdh12 transcripts were reduced in photoreceptors at the detachment site (Fig. 2 F). In contrast, Serpin b1a and Cebpd transcripts did not exhibit changes in abundance resembling those induced by light damage. After detachment, Cebpd transcripts were locally induced in photoreceptors and in the inner nuclear layer but exhibited a low and variable degree of induction in the RPE (Fig. 2 B), and Serpin b1a transcripts exhibited no induction (Fig. 2 G).
Figure 2.
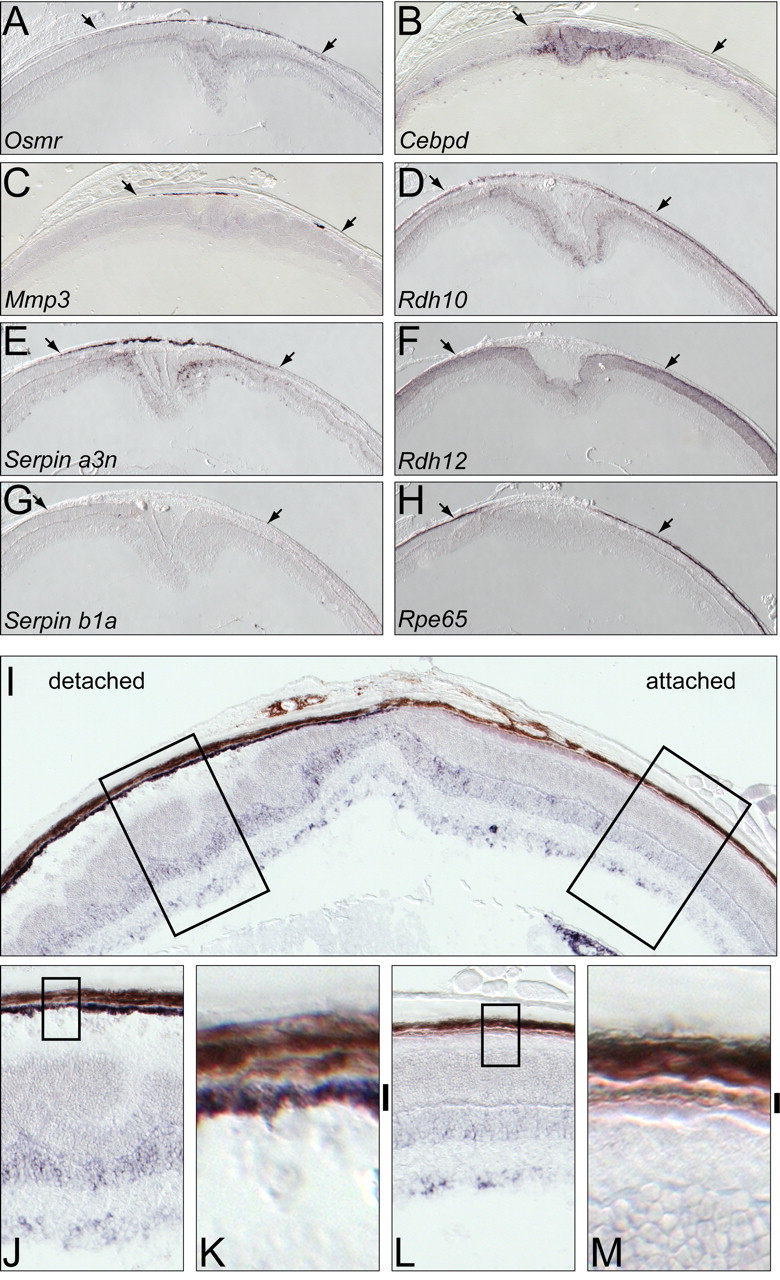
Retinal detachment produces localized changes in transcript abundance in the retina and RPE. A–H, In situ hybridization 4 d after localized retinal detachment in albino BALB/c mice. Arrows indicate the lateral extent of the detachment. Within the detached region, there are localized increases in RPE transcripts (A, C, E), localized decreases in RPE transcripts (D, H), localized increases in retina transcripts (B, D, E), and localized decreases in retina transcripts (F). I–M, In situ hybridization to Serpin a3n transcripts 4 d after localized retinal detachment in a pigmented C57BL/6 × 129 hybrid eye. The detachment is seen on the left side of the eye in I. The melanin was partially bleached by overnight incubation with 3% hydrogen peroxide (see Materials and Methods). The left and right boxed zones in I are enlarged in J and L, respectively, and the boxed zones within J and L are further enlarged in K and M, respectively. Vertical black bars to the right of K and M demarcate the RPE. In the detached RPE, the purple in situ hybridization signal is seen superimposed on the light brown melanin.
The experiments described above were conducted with albino mice in which the lack of ocular melanin is experimentally advantageous because it both enhances intraocular light exposure in light-damage experiments and facilitates detection of in situ hybridization signals in the depigmented RPE. To determine whether the RPE response to detachment is also observed in pigmented mice, we took advantage of the observation that a strong in situ hybridization signal can be detected in the RPE if the melanin is partially bleached with hydrogen peroxide (Orchard, 1999). As shown in Figure 2 I–M, 4 d after a pigmented C57BL/6 × 129 hybrid mouse was subject to a localized retinal detachment, Serpin a3n transcripts increased in abundance in the retina and RPE within the detached region. These data complement our previous observation that detachment induces a robust damage response in the retina in both pigmented and albino mice [Rattner and Nathans, (2005)]. These experiments imply that the protective role of ocular melanin in the context of bright light exposure simply reflects its function as a broad-band filter, and that pigmentation has little or no affect on the RPE and retinal damage responses per se.
Subcellular localization and the kinetics of OSMR accumulation
As described above, one of the transcripts induced in the retina and RPE by light damage and retinal detachment codes for OSMR. OSMR is a single-span transmembrane protein of ∼120 kDa that signals as a heterodimer with either glycoprotein-130 or the interleukin-31 receptor (IL-31R). A priori, OSMR could localize to the apical, the basolateral, or both faces of the RPE, with each localization having different implications with respect to the source of ligand. To define the subcellular localization of OSMR in the light-damaged RPE, we used two complementary approaches. First, polyclonal antibodies were produced against the 113 C-terminal amino acids of mouse OSMR. Using human embryonic kidney 293 and COS cells expressing recombinant mouse OSMR, the specificity of these antibodies was demonstrated by immunostaining and immunoblotting (Fig. 3 A). As a second approach, we produced a human placental AP-mouse OSM fusion and showed that this fusion protein recognizes mouse OSMR produced by transfected COS cells (Fig. 3 A).
Figure 3.

OSMR accumulation in the apical microvilli of the RPE after light damage. A, Specificity of OSMR-binding reagents. Left, Anti-OSMR immunoblot of control 293 cells (−) or 293 cells transfected with OSMR cDNA (+). Right, Anti-OSMR immunostaining (top) or AP-OSM binding followed by AP histochemistry (bottom) of control COS cells (−) or COS cells transfected with OSMR cDNA (+). B, Immunolocalization of OSMR (top) and binding of AP-OSM (bottom) in dark-adapted control (left) and light-damaged (right) BALB/c retinas. OSMR immunoreactivity and AP-OSM binding sites are induced 24 h after exposure to 1.5 h of bright light, and they localize to the apical face of the RPE and adjacent outer segment region. C, Immunoblots showing the time course of OSMR accumulation in the retina (top) and the RPE (bottom) after light damage to BALB/c mice. tub, Anti-β-tubulin control for sample loading; DAPI, 4′,6′-diamidino-2-phenylindole dihydrochloride; INL, inner nuclear layer; GCL, ganglion cell layer; ONL, outer nuclear layer; OS, outer segments; IS, inner segments.
In the dark-adapted eye, OSMR was below the limit of detection by immunostaining. However, 24 h after 1 h of light damage, OSMR was readily detected in the apical microvilli of the RPE (Fig. 3 B, top) where it colocalizes with peropsin (data not shown), a G-protein-coupled receptor that resides exclusively in these microvilli (Sun et al., 1997). The same damage induction and subcellular localization of OSMR was determined by AP-OSM binding (Fig. 3 B, bottom). The apical microvilli of the RPE interdigitate among the photoreceptor outer segments, and at the light-microscope level the two structures exhibit substantially overlapping distributions. Based on the apical localization of RPE OSMR, the OSMR ligand could plausibly be derived from photoreceptors, Muller glia, or macrophages or microglia that have invaded the subretinal space (Sanyal et al., 1984).
Immunoblotting of protein extracts from BALB/c retina and RPE prepared at various times after light damage shows a single polypeptide of the molecular weight expected for OSMR in extracts from light-damaged retina and RPE but not in control samples (Fig. 3 C). OSMR levels in the retina peak 1–3 d after light damage and return to basal levels 1–2 d later. OSMR levels in the RPE exhibit a sharper peak 1–2 d after light damage, returning to basal levels 1 d later. As judged by the immunoblot signal intensity, OSMR accumulates to similar levels in the light-damaged retina and RPE (Fig. 3 C). We suspect that the failure to detect OSMR in the retina by histologic methods (Fig. 3 B) reflects its broad distribution across all retinal layers, consistent with an assignment of the in situ hybridization signal of Osmr transcripts in the inner nuclear layer to Muller glia (Fig. 1 C).
Light preconditioning affects the RPE response to light damage
In albino rodents, preconditioning with low-level light decreases the retinal damage that would normally accompany exposure to bright light (Liu et al., 1998; Li et al., 2003). In previous work, we observed that this resistance is associated with the abrogation of bright light-induced increases in the abundance of various retinal transcripts (Rattner and Nathans, 2005). This pattern also applies to the retinal accumulation of three transcripts (Cebpd, Osmr, and Serpin a3n) that were identified in the present work as damage-induced in both the retina and RPE. In the retina, in situ hybridization showed that constant light preconditioning led to an accumulation of Serpin a3n transcripts in the inner nuclear and ganglion cell layers, Cebpd transcripts in the inner nuclear and photoreceptor layers, and Osmr transcripts in the inner nuclear layer, and, in each case, the levels were not further elevated by bright light exposure (Fig. 4 C and data not shown).
Figure 4.

The response of the retina and RPE to light damage is modified by constant light preconditioning. A, RNA blots showing Serpin a3n and Osmr transcript accumulation in BALB/c mouse retina either preconditioned for 5 d in constant light (CL; ∼100 lux) or maintained in darkness (DA; dark adapted) and then exposed for 1.5 h to 6000 lux with pupil dilation, followed by a 24 h recovery in darkness. B, Immunoblot showing OSMR accumulation in the retina and RPE of BALB/c mice treated as described in A. C, In situ hybridization to Serpin a3n, Cebpd, and Mmp3 transcripts in the RPE and/or retina 24 h after light damage to BALB/c mice that were either dark adapted or constant light preconditioned. Light-damage induction of RPE transcripts is suppressed by preconditioning. D, In situ hybridization to Serpin a3n and Osmr transcripts in constant light-preconditioned mice after retinal detachment. Induction of these transcripts in retina and RPE is not suppressed by preconditioning. Vertical bars to the right of each tissue section are described in Figure 1.
To assess the effect of constant light preconditioning on the RPE, we monitored OSMR protein levels by immunoblotting after bright light exposure, either with or without preconditioning. In the retina, preconditioning induced OSMR to nearly its damage-induced level, and this level was not further increased by bright light exposure (Fig. 4 B, left), consistent with the results of RNA blot hybridization (Fig. 4 A). In the RPE, preconditioning very modestly induced OSMR, and this level was not substantially increased by bright light exposure (Fig. 4 B, right). Similarly, in situ hybridization showed that preconditioning abrogates the bright light induction for Serpin a3n, Mmp3, Cebpd, and Osmr transcripts in the RPE (Fig. 4 C and data not shown). Whether preconditioning abrogates the RPE response because it (1) directly protects the RPE from the damaging affects of bright light or (2) protects the retina from damage, which secondarily suppresses the RPE response, is not distinguished by these experiments.
Interestingly, constant light preconditioning had little or no affect on the increases in RPE or retinal transcripts associated with detachment (Fig. 4 D). This observation suggests that the damage signal associated with retinal detachment is either qualitatively different from that associated with bright light exposure or of a magnitude that allows it to override the inhibition produced by preconditioning.
Consequences of genetic defects in retinal or RPE function on the RPE damage response
The experiments described thus far indicate that the RPE damage response can be activated by diverse stimuli (bright light and retinal detachment), but they do not define the site(s) of cellular damage that regulate the response. In particular, they do not assess the role of the retina in initiating, maintaining, or terminating the RPE damage response. As an initial approach to this question, we asked whether the damage response is altered in three genetic models of retinal dysfunction. For these experiments, each single gene defect was crossed into an albino background to increase its susceptibility to light damage.
The Gnat1 −/− mouse has a morphologically normal retina, but the rods, which constitute 97% of photoreceptors in the mouse retina, lack a physiologic light response (Calvert et al., 2000). Figure 5 A shows that the Gnat1 −/− light-damage response in the retina and RPE is indistinguishable from that seen in wild-type mice as judged by the regulation of OSMR protein levels. Similarly, in situ hybridization shows the wild-type pattern of light-damage regulation for Serpin a3n and Osmr transcripts (Fig. 5 D). These data indicate that light-dependent changes in rod phototransduction do not constitute a major regulatory signal for the RPE damage response, and they are consistent with the work of Hao et al. (2002) showing that in Gnat1 −/− mice the retina is not protected from light damage.
Figure 5.

The stress responses of the retina and RPE are modulated by genetically based retinopathies. A–C, Immunoblots showing OSMR levels in the retina and RPE of control and mutant albino mice that were dark adapted for 7 d and then exposed to 1.5 h of 6000 lux white fluorescent light, followed by a 24 h recovery in darkness. Comparisons are shown for Gnat1 +/− and Gnat1 −/− (A), RPE65 +/− and RPE65 −/− (B), and rd1 +/− and rd1 −/− (C). D, In situ hybridization to Serpin a3n and Osmr transcripts 24 h after light damage of dark-adapted albino Gnat1 −/− and RPE65 −/− mice. Light-damage induction of RPE transcripts is not affected in the Gnat1 −/− retina and RPE, but is suppressed in the RPE65 −/− retina and RPE. E, In situ hybridization showing Serpin a3n and Osmr transcript accumulation in the retina and RPE of albino RPE65 −/− mice 4 d after a local retinal detachment. Regions that are unaffected (left) or detached (right) are from the same eye. Retinal detachment leads to the accumulation of both Serpin a3n and Osmr transcripts in the RPE, and to an increase in the already elevated level of these transcripts in the retina. Vertical bars adjacent to each tissue section are described in Figure 1.
A more severe photoreceptor defect is observed in the Rpe65 −/− mouse in which visual cycle blockade results in unliganded visual pigment apoproteins in both rods and cones, a loss of the rod and cone light responses, and a slow photoreceptor degeneration (Redmond et al., 1998; Zhang et al., 2008). The experiments described here were performed with 4- to 5-week-old mice, an age at which the Rpe65 −/− retina still appears morphologically normal and rod photoreceptor loss is below the limit of detection. Interestingly, immunoblotting shows that in dark-adapted Rpe65 −/− mice the retina and RPE exhibit the same pattern of altered OSMR accumulation that is produced by constant light preconditioning in wild-type retinas (compare Figs. 4 B, 5 B), and bright light exposure produces no induction of Osmr or Serpin a3n transcripts in the RPE in previously dark-adapted Rpe65 −/− mice (Fig. 5 D). As seen for constant light preconditioning, the signal produced by retinal detachment over-rides the suppression of the RPE damage response caused by the Rpe65 −/− genetic background (Fig. 5 E).
Finally, we examined rd1 −/− mice, in which loss of the rod cGMP phosphodiesterase β subunit leads to a rapid and severe photoreceptor degeneration (Bowes et al., 1990). Although rd1 +/− mice were reported to exhibit a slow retinal degeneration, at 4–5 weeks of age, the rd1 +/− retina appears morphologically normal and glial fibrillary acidic protein (GFAP) is not elevated in Muller cells (data not shown), indicating little if any retinal stress. We therefore used rd1 +/− littermates as controls. Figure 5 C shows that at 4–5 weeks of age, when few photoreceptors remain in the rd1 −/− retina, the retina and RPE exhibit the same pattern of altered OSMR accumulation as produced by constant light preconditioning or loss of Rpe65.
If the RPE response to bright light was directly induced by photochemical or other types of damage to the RPE, then one would predict that this response would be unaffected in the rd1 −/− and Rpe65 −/− mutant backgrounds, in which the principal affects of mutation are to alter, either directly or indirectly, photoreceptor structure and function. Instead, the absence of a bright light response in the RPE of Rpe65 −/− and rd1 −/− mice suggests the alternate hypothesis that signals from the retina play an important role in regulating the RPE damage response. Although the exact manner in which the retinas of Rpe65 −/− and rd1 −/− mice might affect the RPE damage response is uncertain, two possibilities immediately suggest themselves: (1) because Rpe65 −/− and rd1 −/− retinas lack visual pigment, they are not susceptible to visual pigment-dependent light damage, a possible inducer of the RPE damage response, and (2) because Rpe65 −/− and rd1 −/− retinas exhibit constitutively elevated levels of GFAP in Muller glia, they may exist in a stressed state resembling that induced by constant light preconditioning, which is also characterized by GFAP accumulation in Muller glia (data not shown). We note that these two possibilities are not mutually exclusive.
Characterization of an RPE eyecup culture system
As described in the Introduction, studies of the response of primary RPE cultures and RPE cell lines do not reveal aspects of this response that involve interactions with other ocular cell types. To facilitate an ex vivo analysis of the RPE damage response that more faithfully recapitulates the situation in vivo, we describe here the initial characterization of a short-term eyecup culture system in which the RPE monolayer remains intact and attached to Bruch's membrane. In brief, mouse eyes are embedded in 3% low-melting-point agarose in culture medium, the globe is bisected with a razor cut at the equator, the anterior half of the eye is discarded, and the vitreous and retina are gently removed from the remaining eyecup. With the eyecup still embedded in agarose and the apical face of the RPE now in direct contact with cell culture medium, the sample is maintained in DMEM/F-12 with 10% bovine serum at 37 C. Because the scleral face of the eyecup remains embedded in the agarose block, the sample can be manipulated without directly touching any of the ocular tissues, thus preserving the shape of the eyecup and minimizing mechanical trauma to the RPE. Figure 6 A shows the appearance of the RPE and choroid from a mouse eyecup after 6 d in vitro. The RPE cells have maintained their intimate association with Bruch's membrane and their apical surfaces remain covered with microvilli.
Figure 6.
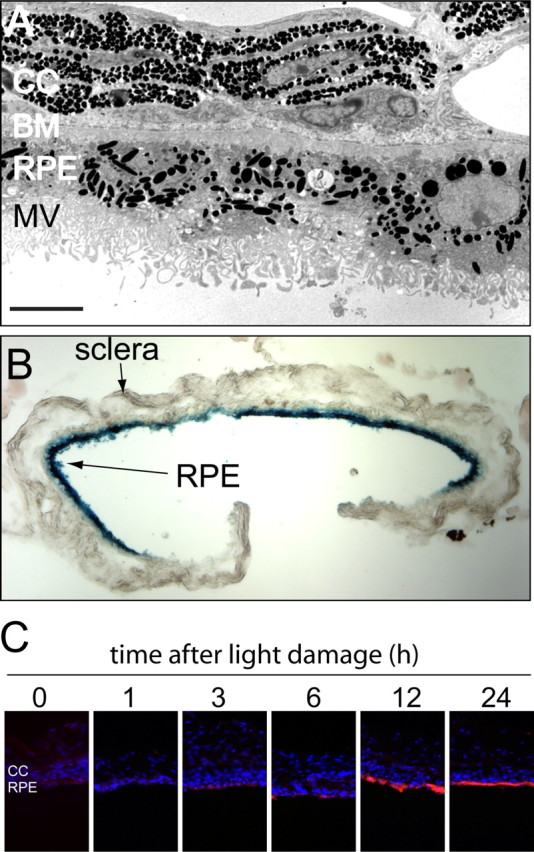
The RPE damage response is maintained in vitro in an eyecup explant. A, Transmission electron microscopy of the RPE and adjacent choroid from a C57BL/6 mouse after 6 d in vitro reveals relatively normal RPE morphology including apical microvilli. CC, Choriocapillaris; BM, Bruch's membrane; MV, microvilli. Size bar, 5 μm. B, RPE cells in an eyecup explant maintain transcriptional and translational competence. An eyecup from a BALB/c mouse was infected in vitro with a recombinant adenovirus vector carrying a β-galactosidase coding region under the control of a cytomegalovirus enhancer/promoter, incubated for 2 d, and then fixed and stained with X-gal. The blue X-gal reaction product is localized to the RPE. C, OSMR expression by RPE cells in eyecup explants prepared at different times after 1.5 h of light damage in vivo. Dark-adapted BALB/c mice were exposed to toxic light, RPE explants were prepared at the indicated times, and after 1 d in vitro the eyecups were immunostained for OSMR.
As a test of the viability of RPE cells within an eyecup, we infected each of a series of eyecups for 30 min with 107 infectious units of a recombinant adenovirus in which a β-galactosidase open reading frame is controlled by a CMV promoter. Two days later, X-gal staining revealed high levels of β-galactosidase activity throughout the RPE (Fig. 6 B). Little or no β-galactosidase activity was observed in uninfected eyecups (data not shown). This experiment shows that RPE cells in an eyecup maintain transcription and translation for at least 2 d of ex vivo incubation. They also demonstrate a high efficiency of viral transduction when a concentrated virus stock is briefly placed in the eyecup.
We next asked whether OSMR accumulates in response to the complete removal of the retina during eyecup preparation or in response to bright light at various times before eyecup preparation. Interestingly, the complete absence of the retina for either 24 or 48 h of eyecup culture did not result in detectable OSMR accumulation in the RPE (data not shown). This observation implies that the RPE does not sense a detachment by the absence of a diffusible molecule or binding interaction that is normally produced by the adjacent attached retina. Instead, the detached retina (or the vitreous that gains access to the subretinal space because of the retinal tear that accompanies the detachment) actively sends a signal that induces a damage response in the adjacent RPE. When eyecups were harvested 0, 1, 3, 6, 12, or 24 h after the standard 1 h of light damage and then cultured for an additional 24 h, we observed minimal OSMR accumulation unless the eyecups were prepared at least 12 h after light damage (Fig. 6 C). The simplest interpretation of this result is that signals generated elsewhere in the eye, most likely in the retina, are required to induce the RPE damage response, and that the requirement that the RPE remain within the intact eye for ∼12 h after the period of light damage reflects the time required to generate and respond to this signal.
Discussion
In this study, we present an analysis of the genomic response of the RPE to damage in the living eye. The principal observations are that (1) the genomic responses of the RPE to two different types of ocular damage, bright light and retinal detachment, are highly similar; (2) the genomic response of the RPE includes both increases and decreases in transcript levels, including large increases in the abundance of transcripts coding for Mmp3, Serpin a3n, Serpin b1a, and Osmr, and decreases in the abundance of all RPE transcripts coding for visual cycle components; and (3) the genomic response of the RPE is predominantly controlled by retinal stress or damage. Whereas many studies have focused on the retina's response to disease or injury (Hao et al., 2002; Rattner and Nathans, 2005; Zacks et al., 2006; Swiderski et al., 2007; Farjo et al., 2008), the present study is the first to systematically analyze the response of the RPE.
Downregulation of visual cycle genes as a generic damage response
A light-dependent decrease in rhodopsin and rod transducin α mRNA and protein levels, and a light-dependent increase in arrestin mRNA and protein levels occurs in the rodent retina, presumably because decreasing the physiologic light response confers protection from light toxicity (Farber et al., 1991; Organisciak et al., 1991). Our observation of a decrease in RPE transcripts coding for the visual cycle proteins Rpe65, Lrat, Cralbp, Rdh5, Rdh10, and Rbp1 suggests that slowing the light-dependent cycling of retinoids represents a second protective strategy. Independent support for this interpretation comes from the observation that mice carrying the hypomorphic L450M variant of Rpe65 exhibit enhanced resistance to light damage (Danciger et al., 2000; Wenzel et al., 2001; Redmond et al., 2007). The decreased abundance of visual cycle transcripts in the RPE after retinal detachment is surprising and suggests that downregulation of the visual cycle may also be protective in this context. Alternatively, downregulation of these transcripts could be part of the general RPE dedifferentiation response that is associated with detachment-induced RPE proliferation (Machemer and Laqua, 1975; Anderson et al., 1981), or it could simply reflect a limited repertoire of RPE stress responses.
Functional implications of RPE transcripts regulated by damage
The 22 unique entries corresponding to RPE transcripts that, by microarray hybridization, decrease in abundance by more than twofold after light-damage code for a highly diverse set of proteins (supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, among the 84 unique entries corresponding to RPE transcripts that increase >2.5-fold, 27 are known components of immune system function, and most of these are either cell-surface receptors or secreted signaling proteins (supplemental Table 1, available at www.jneurosci.org as supplemental material). An additional 6 of these 84 transcripts code either for proteases or protease inhibitors; this subset is of particular interest because it includes 5 of the 12 transcripts with the greatest fold increase. In total, 18 of the 84 most highly induced transcripts code for secreted proteins, and 27 of the 84 code for integral membrane proteins, most of which are known cell-surface receptors. These data indicate that the RPE damage response involves complex changes in cell–cell signaling that likely include a local modulation of the immune response and the acquisition by RPE cells of some of the characteristics of immune system cells.
Among the induced transcripts that we have examined in detail, the Osmr, Mmp3, Serpin a3n, and Serpin b1a transcripts conform to the general patterns of specialized function described in the preceding paragraph. Their known biological functions are briefly summarized here. OSMR plays a role in hematopoiesis and liver regeneration (Tanaka et al., 2003; Nakamura et al., 2004). As noted in Results, the localization of OSMR to the apical microvilli of the RPE suggests that its ligand, presumably a member of the OSM/leukemia inhibitory factor/IL-6 cytokine family, is derived from the retina or from immune cells that have infiltrated the retina. Mmp3 (stromelysin) is a widely expressed matrix metalloproteinase that plays a role in tissue remodeling in the context of bacterial infection and at the neuromuscular junction (Van Saun et al., 2003; Li et al., 2004). Serpin a3n is a secreted protease inhibitor that has been shown recently to inhibit the immune system protease granzyme B, an activity that likely plays a role in local immune privilege (Sipione et al., 2006). Interestingly, Serpin a3n is secreted by Sertoli cells, an epithelial-supporting cell that forms part of the blood–testis barrier. Because the testis and the eye are both sites of immune privilege and Sertoli and RPE cells both reside adjacent to a specialized basement membrane and form a blood-tissue barrier, it seems possible that Serpin a3n functions similarly in the two contexts. Serpin b1a is part of a distinct branch of the serpin superfamily characterized by the lack of a signal sequence and an intracellular localization. Recent work in Caenorhabditis elegans has shown that mutation of a Serpin b1a homolog leads to a dramatic increase in cell death in response to heat shock, oxidative stress, hypoxia, or ionic influx (Luke et al., 2007).
The damage response of the RPE shows many similarities to the damage response described previously for the retina (Rattner and Nathans, 2005). In particular, a fraction of the transcripts are induced in both tissues (e.g., Cebpd, Osmr, Serpin a3n) (Fig. 1), there is substantial overlap between the responses to light damage and retinal detachment, and many of the induced transcripts code for secreted or membrane proteins. Many of the transcripts induced in both the RPE and retina are induced in the inner nuclear layer of the retina, presumably in Muller glia, suggesting that there is a partial overlap in the biochemical and cell biological responses of the RPE and Muller glia to damage.
Communication from retina to RPE
The intimate anatomic and functional relationship between the retina and the RPE is consistent with the general idea that signals pass between these two tissues. Photoreceptors and Muller glia make a series of tight junctions that form the outer face of the retina and the inner border of the subretinal space. Thus, any signaling molecules transmitted from the retina to the RPE are likely to originate in one of these two cell types. One nonpathogenic example of signaling from retina to RPE relates to the daily cycle of outer segment shedding and phagocytosis, which appears to be controlled by a circadian clock located in photoreceptor cells (Green and Besharse, 2004).
We observed no RPE damage response after complete detachment of the retina in eyecup cultures, and no RPE response to light damage unless the eye remained intact for at least 12 h after illumination. We interpret both of these observations as reflecting a requirement for a retinal stress signal that activates the RPE damage response. The normal RPE damage response observed in Gnat1 −/− mice implies that altered rod phototransduction is unlikely to participate in generating the hypothesized stress signal. Interestingly, the data suggest that there are two distinguishable intensities or types of stress signal controlling the RPE response because the response to bright light can be suppressed by preconditioning, but the response to retinal detachment cannot. At present, the molecular identity of the signal that controls the RPE damage response remains to be determined.
Clinical implications
The role of the RPE in retinal disease has been a subject of long-standing interest. As noted in the Introduction, a variety of monogenic disorders of RPE function have been identified in patients with hereditary retinal dystrophies. Additionally, in some disorders in which the primary biochemical defect is not referable to the RPE the most significant pathophysiologic processes occur within or immediately adjacent to the RPE. For example, in Stargardt's recessive macular dystrophy, a visual cycle defect in the photoreceptor outer segment ultimately leads to the accumulation of toxic retinoid derivatives in the RPE (Mata et al., 2000). A second example is seen in the context of one of the principal genetic risk factors for AMD, allelic variation at the complement factor H locus, in which variation in complement concentration, structure, or function leads to an increased accumulation of sub-RPE deposits and loss of RPE cells (Francis et al., 2007; Boon et al., 2008). In addition to its role in disease processes, the RPE is of interest because it can be harvested and transplanted (da Cruz et al., 2007) and it is accessible to genetic modification by subretinal virus injection (Dinculescu et al., 2005). The viral gene transfer strategy has been used in Rpe65 deficient rodent and dogs (Bennicelli et al., 2008), and it is currently in human clinical trials (Bainbridge et al., 2008; Maguire et al., 2008).
The essential contributions of the RPE to retinal health and disease suggest that understanding the damage response of the RPE could improve both diagnosis and therapy. In particular, if the signals that control the RPE damage response can be defined, it may be possible to augment or suppress this response to improve the outcome of a broad spectrum of retinal diseases or injuries.
Footnotes
This work was supported by the Foundation Fighting Blindness, the National Eye Institute (A.R.), and the Howard Hughes Medical Institute (J.N.). We thank Hugh Cahill, Janice Lem, T. Michael Redmond, and Lelei Zhang for advice and/or materials, and Yanshu Wang and an anonymous reviewer for helpful comments on this manuscript.
References
- Anderson DH, Stern WH, Fisher SK, Erickson PA, Borgula GA. The onset of pigment epithelial proliferation after retinal detachment. Invest Ophthalmol Vis Sci. 1981;21:10–16. [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Bennicelli J, Wright JF, Komaromy A, Jacobs JB, Hauck B, Zelenaia O, Mingozzi F, Hui D, Chung D, Rex TS, Wei Z, Qu G, Zhou S, Zeiss C, Arruda VR, Acland GM, Dell'Osso LF, High KA, Maguire AM, Bennett J. Reversal of blindness in animal models of Leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther. 2008;16:458–465. doi: 10.1038/sj.mt.6300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon CJ, Klevering BJ, Hoyng CB, Zonneveld-Vrieling MN, Nabuurs SB, Blokland E, Cremers FP, den Hollander AI. Basal laminar drusen caused by compound heterozygous variants in the CFH gene. Am J Hum Genet. 2008;82:516–523. doi: 10.1016/j.ajhg.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicoló M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Jr, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci U S A. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz L, Chen FK, Ahmado A, Greenwood J, Coffey P. RPE transplantation and its role in retinal disease. Prog Retin Eye Res. 2007;26:598–635. doi: 10.1016/j.preteyeres.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Danciger M, Matthes MT, Yasamura D, Akhmedov NB, Rickabaugh T, Gentleman S, Redmond TM, La Vail MM, Farber DB. A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm Genome. 2000;11:422–427. doi: 10.1007/s003350010081. [DOI] [PubMed] [Google Scholar]
- Dinculescu A, Glushakova L, Min SH, Hauswirth WW. Adeno-associated virus-vectored gene therapy for retinal disease. Hum Gene Ther. 2005;16:649–663. doi: 10.1089/hum.2005.16.649. [DOI] [PubMed] [Google Scholar]
- Farber DB, Danciger JS, Organisciak DT. Levels of mRNA encoding proteins of the cGMP cascade as a function of light environment. Exp Eye Res. 1991;53:781–786. doi: 10.1016/0014-4835(91)90114-t. [DOI] [PubMed] [Google Scholar]
- Farjo R, Peterson WM, Naash MI. Expression profiling after retinal detachment and reattachment: a possible role for aquaporin-0. Invest Ophthalmol Vis Sci. 2008;49:511–521. doi: 10.1167/iovs.07-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PJ, Schultz DW, Hamon S, Ott J, Weleber RG, Klein ML. Haplotypes in the complement factor H (CFH) gene: associations with drusen and advanced age-related macular degeneration. PLoS ONE. 2007;2:e1197. doi: 10.1371/journal.pone.0001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganti R, Hunt RC, Parapuram SK, Hunt DM. Vitreous modulation of gene expression in low-passage human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2007;48:1853–1863. doi: 10.1167/iovs.06-0198. [DOI] [PubMed] [Google Scholar]
- Geller SF, Lewis P, Fisher SK. FGFR1, signaling, and AP-1 expression after retinal detachment: reactive Muller and RPE cells. Invest Ophthalmol Vis Sci. 2001;42:1363–1369. [PubMed] [Google Scholar]
- Green CB, Besharse JC. Retinal circadian clocks and control of retinal physiology. J Biol Rhythms. 2004;19:91–102. doi: 10.1177/0748730404263002. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Marti A, Munz K, Remé CE. Light-induced apoptosis: differential timing in the retina and pigment epithelium. Exp Eye Res. 1997;64:963–970. doi: 10.1006/exer.1997.0288. [DOI] [PubMed] [Google Scholar]
- Hafezi F, Abegg M, Grimm C, Wenzel A, Munz K, Stürmer J, Farber DB, Remé CE. Retinal degeneration in the rd mouse in the absence of c-fos. Invest Ophthalmol Vis Sci. 1998;39:2239–2244. [PubMed] [Google Scholar]
- Hao W, Wenzel A, Obin MS, Chen CK, Brill E, Krasnoperova NV, Eversole-Cire P, Kleyner Y, Taylor A, Simon MI, Grimm C, Remé CE, Lem J. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002;32:254–260. doi: 10.1038/ng984. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CK, Pender SL, Pickard KM, Chance V, Holloway JA, Huett A, Gonçalves NS, Mudgett JS, Dougan G, Frankel G, MacDonald TT. Impaired immunity to intestinal bacterial infection in stromelysin-1 (matrix metalloproteinase-3)-deficient mice. J Immunol. 2004;173:5171–5179. doi: 10.4049/jimmunol.173.8.5171. [DOI] [PubMed] [Google Scholar]
- Li F, Cao W, Anderson RE. Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci. 2003;44:4968–4975. doi: 10.1167/iovs.03-0140. [DOI] [PubMed] [Google Scholar]
- Liu C, Peng M, Laties AM, Wen R. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344. doi: 10.1523/JNEUROSCI.18-04-01337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke CJ, Pak SC, Askew YS, Naviglia TL, Askew DJ, Nobar SM, Vetica AC, Long OS, Watkins SC, Stolz DB, Barstead RJ, Moulder GL, Brömme D, Silverman GA. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell. 2007;130:1108–1119. doi: 10.1016/j.cell.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machemer R, Laqua H. Pigment epithelium proliferation in retinal detachment (massive periretinal proliferation) Am J Ophthalmol. 1975;80:1–23. doi: 10.1016/0002-9394(75)90862-4. [DOI] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF, Wolfensberger TJ. The retinal pigment epithelium: function and disease. New York: Oxford UP; 1998. [Google Scholar]
- Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000;97:7154–7159. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nonaka H, Saito H, Tanaka M, Miyajima A. Hepatocyte proliferation and tissue remodeling is impaired after liver injury in oncostatin M receptor knockout mice. Hepatology. 2004;39:635–644. doi: 10.1002/hep.20086. [DOI] [PubMed] [Google Scholar]
- Orchard GE. Heavily pigmented melanocytic neoplasms: comparison of two melanin-bleaching techniques and subsequent immunohistochemical staining. Br J Biomed Sci. 1999;56:188–193. [PubMed] [Google Scholar]
- Organisciak DT, Xie A, Wang HM, Jiang YL, Darrow RM, Donoso LA. Adaptive changes in visual cell transduction protein levels: effect of light. Exp Eye Res. 1991;53:773–779. doi: 10.1016/0014-4835(91)90113-s. [DOI] [PubMed] [Google Scholar]
- Pastor JC, de la Rúa ER, Martín F. Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res. 2002;21:127–144. doi: 10.1016/s1350-9462(01)00023-4. [DOI] [PubMed] [Google Scholar]
- Rattner A, Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J Neurosci. 2005;25:4540–4549. doi: 10.1523/JNEUROSCI.0492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Nathans J. Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci. 2006;7:860–872. doi: 10.1038/nrn2007. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Weber CH, Poliakov E, Yu S, Gentleman S. Effect of Leu/Met variation at residue 450 on isomerase activity and protein expression of RPE65 and its modulation by variation at other residues. Mol Vis. 2007;13:1813–1821. [PubMed] [Google Scholar]
- Sanyal S, De Ruiter A, Dees C. Light dependent accumulation of macrophages at the photoreceptor-pigment epithelial interface in the retina of albino mice. Experientia. 1984;40:852–854. doi: 10.1007/BF01951991. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxygenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Sipione S, Simmen KC, Lord SJ, Motyka B, Ewen C, Shostak I, Rayat GR, Dufour JM, Korbutt GS, Rajotte RV, Bleackley RC. Identification of a novel human granzyme B inhibitor secreted by cultured sertoli cells. J Immunol. 2006;177:5051–5058. doi: 10.4049/jimmunol.177.8.5051. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Cai B. Blue light-induced apoptosis of A2E-containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest Ophthalmol Vis Sci. 2001;42:1356–1362. [PubMed] [Google Scholar]
- Sun H, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Peropsin, a novel visual pigment-like protein located in the apical microvilli of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 1997;94:9893–9898. doi: 10.1073/pnas.94.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiderski RE, Nishimura DY, Mullins RF, Olvera MA, Ross JL, Huang J, Stone EM, Sheffield VC. Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest Ophthalmol Vis Sci. 2007;48:3329–3340. doi: 10.1167/iovs.06-1477. [DOI] [PubMed] [Google Scholar]
- Szurman P, Roters S, Grisanti S, Aisenbrey S, Schraermeyer U, Lüke M, Bartz-Schmidt KU, Thumann G. Ultrastructural changes after artificial retinal detachment with modified retinal adhesion. Invest Ophthalmol Vis Sci. 2006;47:4983–4989. doi: 10.1167/iovs.06-0491. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Hirabayashi Y, Sekiguchi T, Inoue T, Katsuki M, Miyajima A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood. 2003;102:3154–3162. doi: 10.1182/blood-2003-02-0367. [DOI] [PubMed] [Google Scholar]
- VanSaun M, Herrera AA, Werle MJ. Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J Neurocytol. 2003;32:1129–1142. doi: 10.1023/B:NEUR.0000021907.68461.9c. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001;21:53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacks DN, Han Y, Zeng Y, Swaroop A. Activation of signaling pathways and stress-response genes in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci. 2006;47:1691–1695. doi: 10.1167/iovs.05-1209. [DOI] [PubMed] [Google Scholar]
- Zhang H, Fan J, Li S, Karan S, Rohrer B, Palczewski K, Frederick JM, Crouch RK, Baehr W. Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J Neurosci. 2008;28:4008–4014. doi: 10.1523/JNEUROSCI.0317-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]