

Abstract
To investigate the effect of phosphorylation on the interactions of phospholamban (PLB) with itself and its regulatory target, SERCA, we measured FRET from CFP-SERCA or CFP-PLB to YFP-PLB in live AAV-293 cells. Phosphorylation of PLB was mimicked by mutations S16E (PKA site) or S16E/T17E (PKA+CaMKII sites). FRET increased with protein concentration up to a maximum (FRETmax) that was taken to represent the intrinsic FRET of the bound complex. The concentration dependence of FRET yielded dissociation constants (KD) for the PLB-PLB and PLB-SERCA interactions. PLB-PLB FRET data suggest pseudo-phosphorylation of PLB increased oligomerization of PLB but did not alter PLB pentamer quaternary structure. PLB-SERCA FRET experiments showed an apparent decrease in binding of PLB to SERCA and an increase in the apparent PLB-SERCA binding cooperativity. It is likely that these changes are secondary effects of increased oligomerization of PLB; a change in the inherent affinity of monomeric PLB for SERCA was not detected. In addition, PLB-SERCA complex FRETmax was reduced by phosphomimetic mutations, suggesting the conformation of the regulatory complex is significantly altered by PLB phosphorylation.
Phospholamban (PLB)2 is a 52-amino acid peptide localized primarily to the sarcoplasmic reticulum of cardiac muscle cells. The peptide comprises two α-helical domains connected by a flexible linker. NMR studies suggest an L-shaped tertiary structure; the PLB cytoplasmic domain N-terminal α-helix (domain IA) is significantly parallel to the bilayer surface, while the transmembrane helix (domain II) is roughly normal to the membrane (1, 2). The monomeric form of PLB binds and inhibits the sarco(endo)plasmic reticulum calcium ATPase (SERCA) (3). Inhibition is partially relieved by phosphorylation of PLB by PKA (4) and CaMKII (5), and PLB is a major effector of adrenergic signaling (6). PLB also binds to itself, forming pentamers (7). Most evidence suggests that the pentamer is unable to bind or regulate SERCA (8, 9). NMR and EPR (1) and FRET (10, 11) data suggest the pentamer is a “pinwheel” quaternary complex of L-shaped subunits, however, other diverse structures have been proposed (12–14).
Phosphorylation has been proposed to alter the architecture of PLB. Wegener and Jones (7) observed a significant shift in the PLB pentamer electrophoretic mobility in SDS gels, and postulated that mobility was changed as a result of altered quaternary conformation of the pentamer. Specifically, they envisioned PLB cytoplasmic domains assuming a more compact conformation after phosphorylation (15). Consistent with such a structural transition is a recent NMR study showing that phosphorylation decreases the membrane association of the phospholamban cytoplasmic domain (16). Furthermore, Karim et al. (17) found that phosphorylation influenced the proportion of a structurally dynamic form of PLB. To test whether phosphorylation stabilizes a compact pentamer conformation, we obtained distance constraints by measuring fluorescence resonance energy transfer (FRET) between N-terminal fluorescent protein fusion tags. In this study, we mimicked phosphorylation of PLB by PKA and CaMKII with glutamate substitutions at positions 16 and 17 (18, 19). The extent to which glutamate substitutions recapitulate phosphorylation of PLB is not known. However, the S16E mutation has been shown to ameliorate the disease process in a heart failure animal model (18), making it attractive as an experimental tool and potential therapy.
In addition, we investigated the effect of phosphorylation on the structure and affinity of the PLB regulatory complex with SERCA. Several groups have provided evidence that the interaction of PLB with SERCA is altered by PLB phosphorylation, either by partial dissociation (20), or by a change in PLB-SERCA tertiary/quaternary structure (17, 21–23). The question of whether PLB dissociation from SERCA is a primary or secondary effect is complicated by the linkage of this binding interaction to the equilibrium of PLB oligomeric forms. For example, the PLB pentamer-destabilizing I40A mutation shifts the pentamer-monomer equilibrium toward the monomer, resulting in more binding of monomeric PLB to SERCA (9, 10) Underscoring the functional interplay of these coupled equilibria is the observation that the I40A mutant is a “superinhibitor” of the pump (9). Inversely, shifting PLB to the pentamer is expected to partially deplete the monomer pool, and by mass action reduce binding of PLB to SERCA. A shift toward the pentamer form could be accomplished by phosphorylation of PLB, which has been shown to increase oligomerization in vitro (24). To address whether both binding equilibria are directly and independently regulated, it is necessary to measure the energetic consequence of phosphorylation on PLB oligomerization in the biologically relevant environment of the cell membrane. In the present study, we endeavor to quantify pseudo-phosphorylation-induced equilibrium shifts in live cells using FRET to detect binding of CFP-SERCA or CFP-PLB to YFP-PLB. Our findings address unanswered questions relating to phosphorylation effects on the structure of the PLB pentamer and regulatory complex and the affinity of the PLB-PLB and PLB-SERCA interactions. We propose an integrated model in which these features determine the functional regulation of calcium handling in the heart.
EXPERIMENTAL PROCEDURES
Molecular Biology and Cell Culture—PLB was mutated using the QuikChange mutagenesis kit (Statagene, La Jolla, CA) to create pseudo-phosphorylations at the PKA site (S16E) or PKA/CaMKII sites (S16E/T17E). Mutants were compared with a PKA-nonphosphorylatable (S16A) control. CFP-PLB or CFP-SERCA and YFP-PLB were co-expressed in AAV-293 cells as previously described (10, 25). The calcium pump with an N-terminal CFP fusion tag retains normal calcium sensitivity and 40–50% catalytic activity, indicating this labeling method does not prevent enzymatic cycling (26). After transfection, cells were trypsinized and re-plated on poly-d-lysine-coated glass bottom culture dishes and allowed to adhere to the surface for 2 h. This treatment resulted in well-separated cells, reducing cross-talk of fluorescence from adjacent cells. In addition, the short time of adherence yielded cells with uniform spherical morphology, which facilitated analysis of fluorescence images.
FRET Measurement—Cells were imaged with an inverted microscope (Nikon TE2000-U) equipped with a metal halide lamp, an APO 60× 1.49 NA objective, and a back-thinned CCD camera (iXon 887; Andor Technology, Belfast, Northern Ireland). The CCD camera was cooled to –100 °C, using a recirculating liquid coolant system (Koolance, Inc., Auburn, WA). The illumination was introduced through an excitation filter wheel equipped with 427/10 nm (for CFP) and 504/12 nm (for YFP) bandpass filters and a stationary multiple band dichroic mirror (Semrock, Rochester NY). Emission was detected through the same dichroic mirror and an emission filter wheel equipped with 472/30 nm (for CFP) and 542/27 nm (for YFP) filters. The system's relative sensitivity to CFP and YFP was calibrated by imaging drops of purified CFP and YFP at a series of known concentrations. FRET between CFP- and YFP-labeled proteins was quantified by progressive acceptor-selective photobleaching of the entire microscopic field of observation, as previously described (10). Image acquisition and acceptor photobleaching was automated with custom software macros in Meta-Morph (Molecular Devices Corp., Downingtown, PA) that controlled motorized excitation/emission filter wheels (Sutter Instrument Co., Novato, CA). The progressive photobleaching protocol was as follows: 100-ms acquisition of CFP image and 40-ms acquisition of YFP image, followed by 10-s exposure to YFP-selective photobleaching (504/12 nm). The intensity of this YFP photobleaching excitation was 230 micro-watts measured at the sample. At this power level, YFP fluorescence is reduced by more than 95% in 10 min, but CFP fluorescence is preserved. This protocol has been validated with standard samples (10). For intrapentameric FRET experiments, the relative protein expression of each cell was assessed as a sum of the starting YFP fluorescence (prebleach) and the final CFP fluorescence after FRET was abolished (postbleach). A limitation of this retrospective analysis is that it does not yield absolute concentrations in units of protein molar fraction or species per unit area (e.g. mol/m2). However, this method offers the advantage of not being restricted to reconstituted systems of defined lipid/protein ratio. Thus it is broadly applicable to quantifying relative protein concentrations in living cells.
The observed FRET was calculated for each cell from the extent of donor fluorescence enhancement after acceptor photobleaching according to E = 1 – (Fprebleach/Fpostbleach). Observed FRET was compared cell-by-cell to starting YFP fluorescence, which was taken as an index of protein concentration. CFP-PLB to YFP-PLB FRET concentration dependence was fit to a hyperbolic curve of the form y = (FRETmax)X/ (KD1 + X), where X = protein concentration in arbitrary units (AU). The parameter FRETmax was taken to represent the intrinsic FRET of the pentamer. KD1 is the dissociation constant (in arbitrary units) of the pentamer. Fitting of SERCA-S16A-PLB FRET data were performed as above, but the concentration dependence of FRET to S16E and S16E/T17E pseudo-phosphorylated mutants was best described by a Hill function of the form y = (FRETmax)Xn/((KD2)n + Xn) where n is the Hill coefficient. Values of n greater than 1 indicate positive cooperativity of binding.
Distance Measurement—Regulatory complex probe separation distance was calculated from intrinsic FRET efficiency (FRETmax) according to the relationship r = R0[(1/FRETmax) – 11/6](27). Fig. 1A is the simulation of FRET as a function of distance with a Foster radius (R0) of 49.2 Å for CFP-YFP energy transfer (28). The distance between fluorescent protein probes in pentameric PLB was calculated using a model of FRET within a ring-shaped oligomer (29) as previously described (10). This model assumes random mixing of donor/acceptors and a symmetric ring-shaped assembly of n PLB subunits. The fluorescence intensity decay of the n-mer is given by Equation 1,
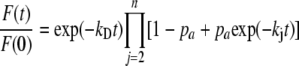 |
(Eq. 1) |
where kD is the decay rate of the donor alone, pa is the molar fraction of acceptor, 1 – pa is the molar fraction of donor. The term kj is given by Equation 2,
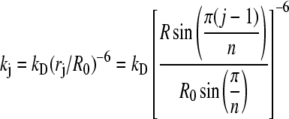 |
(Eq. 2) |
where R0 is the Förster distance, rj is the distance between the donor and the acceptor on subunit j in the n-mer ring, and R is the distance between fluorophores on adjacent subunits. If oligomers are in equilibrium with a molar fraction X of monomers, and assuming that E = 0 for these monomeric donors, the observed steady-state energy transfer efficiency is given by Equation 3.
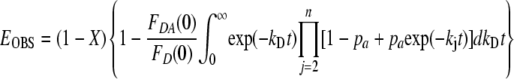 |
(Eq. 3) |
Because the measured FRETmax value represents the intrinsic FRET of the complex, monomer fraction X = 0. The mol fraction acceptor is empirically determined from the starting fluorescence of YFP and the final fluorescence of CFP. A contour plot of the simulated dependence of FRET efficiency on acceptor mol fraction and probe separation distance for a pentamer is shown in Fig. 1B. A MatLab simulation that implements Equation 2 was used to analyze PLB-PLB FRET data, using a pentamer as the major oligomeric species. Specifically, the proportion of dimers is assumed to be low. PAGE and cross-linking experiments (11, 25) indicated that the proportion of dimers is low at equilibrium. Regarding higher-order oligomers, the relative populations of oligomeric forms of 3+ subunits do not significantly affect the distance measurements. For protein complexes with transfer distances larger than R0, the intrinsic FRET efficiency of the phospholamban oligomer does not depend strongly on the number of subunits (29). For example, we have previously determined that the distance estimated for a pentamer (n = 5) changed by 0.8 Å or 0.2 Å if oligomers were actually n = 3 or n = 7 subunits, respectively (10).
FIGURE 1.

Theoretical models for interpretation of FRET. A, simulated dependence of FRET on probe separation distance for the PLB-SERCA regulatory complex. B, simulated dependence of FRET (contours, %) on probe separation distance and acceptor mol fraction for the PLB pentamer.
Computational Modeling of PLB Pentamer Electrostatics—To investigate the charge effects of PKA phosphorylation of PLB Ser-16 and CaMKII phosphorylation of Thr-17, we performed Poisson-Boltzmann electrostatics calculations (30) using predicted pKa values (31) for the pinwheel structure of the PLB pentamer (1XNU) (11) and modeled the results in Pymol with the APBS plugin. Conditions for the model were as follows: protein dielectric 2.0, solvent dielectric 80.0, ion radius 2.0, solvent radius 1.4, temperature 310 K, 0.15 m monovalent salt. Charges were visualized on a solvent-accessible surface, encoding +1 to –1 kT/e from blue to red.
RESULTS
Intrapentameric FRET—For AAV-293 cells expressing non-phosphorylatable CFP-S16A-PLB and YFP-S16A-PLB, we measured a population average FRET efficiency of 33 ± 1.1%. Overall FRET progressively increased with phosphorylation-mimicking glutamate substitutions at the PKA (Ser-16) and CaMKII (Thr-17) sites (Fig. 2A). The average intrapentameric FRET efficiency of S16E-PLB was (36 ± 1.1%), and the average FRET efficiency of the double phosphorylation mimic S16E/T17E-PLB was (41 ± 1.6%). To determine whether this change in overall FRET resulted from increased oligomerization or a pentamer conformational change, we compared the measured FRET efficiency of each cell with the brightness of its YFP emission. This fluorescence intensity was taken as an index of protein concentration (10). Fig. 2B shows that FRET efficiency increased with protein concentration, up to a maximum value. A hyperbolic fit of the FRET versus [Protein] relationship (Fig. 2B) yielded estimates of relative KD and FRETmax. The data in Fig. 2B are pooled for clarity of presentation. Examples of the raw data from which KD1 and FRETmax parameters were obtained are presented as supplemental information. Average parameter values from several such experiments (n = 4–5) are summarized in Fig. 2C and Table 1. Compared with the non-phosphorylatable S16A-PLB, the pseudo-phosphorylation mutants S16E-PLB and S16E/T17E-PLB showed decreased KD1. The data indicate an increased oligomerization affinity with pseudo-phosphorylation. The FRETmax parameter of the hyperbolic fit was taken to indicate the intrinsic FRET efficiency of the pentamer complex and was 47.1 ± 1.3%, 46.9 ± 1.7%, and 46.8 ± 0.5% for S16A, S16E, and S16E/T17E, respectively.
FIGURE 2.

Effects of phosphomimetic mutations on PLB-PLB FRET. A, mean intrapentameric FRET increases with pseudo-phosphorylation. B, concentration dependence of FRET for S16A (black squares), S16E (red circles), and S16E/T17E (blue triangles). C, mean KD1 and FRETmax parameters obtained by hyperbolic fitting of data as in B. * indicates p < 0.05 versus S16A.
TABLE 1.
Summary of effects of phosphomimetic mutations
| S16A | S16E | S16E/T17E | |
|---|---|---|---|
| PLB Pentamer | |||
| Mean FRET (%) | 33.2 ± 1.1 | 35.9 ± 1.1 | 40.7 ± 1.6 |
| FRETmax (%) | 47.1 ± 1.3 | 46.9 ± 1.7 | 46.8 ± 0.5 |
| Acceptor mol fraction | 0.93 ± 0.02 | 0.91 ± 0.01 | 0.89 ± 0.01 |
| Probe distance (Å) | 58.7 ± 0.4 | 58.5 ± 0.6 | 58.3 ± 0.2 |
| KD1 (AU) | 2.2 ± 0.2 | 1.5 ± 0.4 | 1.0 ± 0.3 |
| Sample sizea | 338 | 442 | 231 |
| PLB-SERCA Complex | |||
| Mean FRET (%) | 25.1 ± 1.2 | 14.3 ± 1.3 | 15.9 ± 0.8 |
| FRETmax (%) | 29.5 ± 1.4 | 26.2 ± 1.4b | 22.3 ± 0.2b |
| Probe distance (Å) | 58.3 ± 0.7 | 60.1 ± 0.8b | 62.5 ± 0.03b |
| Apparent KD2 (AU) | 4.5 ± 0.4 | 7.7 ± 0.9b | 8.7 ± 0.6b |
| Hill coefficient | NDc | 1.6 ± 0.3b | 1.6 ± 0.2b |
| Sample size | 343 | 304 | 257 |
Sample size indicates the number of cells analyzed. Fit parameters are reported as mean ± S.E. for 3-5 independent data sets.
Value was obtained by Hill fit.
ND indicates value was not determined.
Regulatory Complex FRET—To investigate the effect of pseudo-phosphorylation mutations on the structure and apparent affinity of the regulatory complex, we measured FRET between CFP-SERCA and PLB mutants labeled with YFP. Of the three PLB variants, S16A showed the highest overall regulatory complex FRET efficiency 25 ± 1.2%. FRET from CFP-SERCA to YFP-PLB decreased with pseudo-phosphorylation mutations; 14 ± 1.3% for S16E-PLB and 16 ± 0.8% for S16E/T17E (Fig. 3A). FRET increased with protein concentration to a maximum (Fig. 3B). As with PLB oligomerization (Fig. 2B), this relationship is approximately hyperbolic (10). However, the phosphomimetic mutants showed apparent cooperative binding to SERCA and were best described by a Hill function of the form y = (FRETmx)Xn/((KD2)n + Xn) where n is the Hill coefficient. An interpretation of this result is offered under “Discussion.” The data in Fig. 3B are pooled for clarity. Parameters KD2 and FRETmax were obtained from raw data (supplemental information). The apparent KD2 and FRETmax values obtained by hyperbolic regression of S16A or Hill fits of S16E and S16E/T17E are summarized in Fig. 3C and Table 1 (n = 3–5). The data suggest that PLB binding to SERCA is reduced by phosphomimetic mutations (increased KD2).
FIGURE 3.

Effects of phosphomimetic mutations on SERCA-PLB FRET. A, regulatory complex mean FRET decreases with pseudo-phosphorylation. B, concentration dependence of FRET for S16A (black squares), S16E (red circles), and S16E/T17E (blue triangles). C, mean KD2 and FRETmax parameters obtained by hyperbolic fits of S16A data or Hill function regression of S16E and S16E/T17E. * indicates p < 0.05 versus S16A.
Compared with the non-phosphorylatable PLB (S16A), the PKA site mutant (S16E) and PKA/CaMKII sites mutant (S16E/T17E) showed progressively decreased FRETmax (Table 1). The change in FRETmax is consistent with a change in the conformation of the regulatory complex with phosphorylation.
DISCUSSION
The present observations relating to PLB structure and affinity provide insight into the mechanism of kinase regulation of SERCA inhibition by PLB. Fig. 4 shows a conceptual model of SERCA regulation, highlighting possible effects of phosphorylation on reversible regulatory transitions. In this scheme, the “inhibited” complex of PLB and SERCA is depicted in blue (Fig. 4, low Ca2+ affinity). For clarity, phosphorylation is shown as having a stimulatory effect on rates or paths leading away from the low Ca2+ affinity inhibitory complex (Fig. 4, A–D), though one could posit other mechanisms.
FIGURE 4.

A scheme for regulation of SERCA by PLB. Proposed effects of phosphorylation on PLB pentamer structure (A), oligomerization affinity (B), affinity for SERCA (C), and regulatory complex structure (D). Effects shown in B and D are supported by the present data. The compact conformation of PLB5 was not detected, nor was there evidence for a direct effect on the affinity of PLB1 for SERCA.
PLB Pentamer Structure and Equilibrium—We did not detect a pentamer conformational change (Fig. 4A) with phosphomimetic mutations. The PLB-PLB FRETmax parameter, a measure of the probe separation distance, did not change with pseudo-phosphorylation indicating the quaternary structure was unaffected. These results are consistent with a recent NMR study that observed no significant changes to short-range distance constraints with the S16E mutation (19). According to a computational model of intrapentameric energy transfer based on ring-oligomer FRET theory (29), the average nearest-neighbor probe separation distance was 58.7 ± 0.4 Å for S16A-PLB, 58.5 ± 0.6 Å for S16E-PLB, and 58.3 ± 0.2Å for S16E/T17E-PLB. Notably, these probe separation distances are in very good agreement with the distance of 58.7 ± 0.5 Å we previously reported for WT-PLB (10). We conclude that the phosphorylation-induced changes in PLB structure reported in the literature (16, 22, 32–34) do not result in a large-scale transition to a compact pentamer conformation, as was suspected from gel mobility shift experiments (7). The strong dependence of FRET efficiency on distance makes this structural conclusion robust; a 1% difference in probe separation distance yields a 3% difference in FRET.
The fraction of oligomeric PLB is probably a more salient regulatory indicator than the architecture of the pentamer. Indeed, the data suggest the monomer-pentamer equilibrium is strongly regulated by phosphorylation (Fig. 4B). We found that phosphomimetic mutations increase PLB oligomerization in the membranes of live cells by decreasing the measured KD1 from 2.21 ± 0.24 for S16A to 1.05 ± 0.27 for S16E/T17E-PLB (Fig. 2C and Table 1). Thus, the observed increase in overall pentamer FRET (Fig. 2A) must arise from the increased oligomerization of phosphorylation-mimicking mutations, rather than a change in quaternary conformation.
To determine the effect of phosphorylation-mimicking mutations on PLB charge distribution we performed electrostatics calculations on PLB phosphomimetic mutants. Fig. 5 shows calculated surface potential represented from–1 (red) to +1 kT/e (blue). The WT pentamer (Fig. 5A) is characterized by significantly positive cytoplasmic domains (Fig. 5B) except for a small region of negative charge conferred by Glu-19 near the hub of the pinwheel. S16E (Fig. 5C) and S16E/T17E (Fig. 5D) substitutions expand and intensify this negative region, but do not significantly increase regions of neutral potential (white), nor abolish the positive charges at the extremities of the PLB cytoplasmic domains. This model predicts that putative electrostatic forces repelling PLB cytoplasmic domains away from normal (7) would not be alleviated by phosphorylation, as repellant positive charges are replaced by repellent negative charges. Moreover, it is unlikely that long-range charge repulsion is a major structural determinant since such charges are well-screened at physiological salt concentrations and are not expected to project significant force over many tens of angstroms. Instead, short-range electrostatic interactions may dominate the prevailing pentamer structure, such as between PLB and lipid headgroups (16, 35). Short-range interactions between PLB subunits may also stabilize the phosphorylated pentamer. Computational modeling suggests that phosphomimetic mutations create oppositely charged helix faces near the hub of the pinwheel (Fig. 5E). We speculate that interactions between positive and negative faces would stabilize the pentamer complex, providing additional oligomerization binding energy and decreasing KD1 (Fig. 2C). Furthermore, such bipolar charge distributions may reduce the binding activation energy barrier and orient incoming PLB subunits during oligomerization.
FIGURE 5.

Computational modeling of PLB electrostatic potential. A, PLB pentamer structure 1XNU. B, surface potentials of WT-PLB showing –1 (red) and +1 kT/e (blue) charges. C, S16E. D, S16E/T17E. The model suggests that charges are well localized at physiological salt concentrations, and electrostatic repulsion between cytoplasmic domains is unlikely to define the quaternary conformation of the pentamer. E, an exploded view of two adjacent S16E/T17E pentamer subunits shows a bipolar charge distribution on S16E/T17E-PLB cytoplasmic domains that could contribute to oligomerization.
PLB-SERCA Structure and Equilibrium—The present FRET data show that pseudo-phosphorylation did not abolish the interaction of PLB with SERCA, which is consistent with other reports (21, 23). All PLB variants bound saturably to SERCA (Fig. 3B). However, overall FRET between CFP-SERCA and YFP-labeled pseudo-phosphorylated PLB was reduced compared with S16A (Fig. 3A). This reduction in overall FRET is due to a right-shifted concentration dependence combined with a decreased maximal FRET efficiency for S16E and S16E/T17E (Fig. 3B). The FRET versus [Protein] relationship was quantified for S16A using the hyperbolic regression used for PLB-PLB FRET data (Fig. 3). However, unlike S16A-PLB or the previously investigated WT- or I40A-PLB (10), the pseudo-phosphorylated regulatory complex FRET data were not well-described by a hyperbola of the form y = ax/(b + x). Regression was significantly improved by using a Hill function of the form y = (FRETmax)Xn/((KD2)n + Xn), in which n is a measure of apparent cooperativity. Fig. 3B shows regulatory complex FRET data with a hyperbolic fit of S16A and Hill fits of S16E and S16E/T17E data. Because we cannot distinguish between the monomeric and pentameric forms of YFP-PLB in the CFP-SERCA/YFP-PLB FRET assay, the PLB1-SERCA equilibrium cannot be observed in isolation from the PLB5-PLB1 equilibrium. This has several practical consequences for interpreting SERCA-PLB FRET concentration dependence. First, the KD2 fit parameter is not an absolute indicator of the affinity of PLB for SERCA and the apparent increase in KD2 with pseudo-phosphorylation does not necessarily imply a decrease in the intrinsic affinity of monomeric PLB for SERCA (17, 20) (Fig. 4C). Rather, reduced PLB1 binding to SERCA is simply due to increased oligomerization; the observed 1.9-fold increase in KD2 (Fig. 3) can be accounted for by the 2.1-fold decrease in KD1 (Fig. 2). Second, the apparent cooperativity observed for S16E and S16E/T17E mutants does not necessarily reflect multiple binding sites for PLB on SERCA. The concentration of monomeric PLB is depleted by oligomerization, reducing its availability to SERCA and shifting the onset of SERCA binding to higher concentrations. Once the concentration of protein exceeds both KD1 and KD2, binding to SERCA increases markedly. Inaccessibility of PLB at low concentrations (near KD1) combined with saturable binding at high concentration (above KD2) makes the binding curve steep and apparently cooperative. This effect is most pronounced when the disparity between KD1 and KD2 is large, as it is for pseudo-phosphorylated PLB.
Also obtained from the SERCA-PLB FRET concentration dependence is the maximal regulatory complex FRET (FRETmax), which gives the intrinsic FRET efficiency of the complex. As far as can be detected by FRET, CFP-SERCA and YFP-PLB comprise a bimolecular complex (10). Therefore, the probe separation distance can be obtained from FRETmax by the relationship r = R0[(1/FRETmax – 1)1/6] (27). Assuming random relative dipolar orientations (κ2 = 2/3), and accounting for 3% nonspecific energy transfer (10), the measured intrinsic FRET efficiencies correspond to probe separation distances of 58.3 ± 0.7, 60.1 ± 0.8 and 62.5 ± 0.03Å for S16A, S16E, and S16E/T17E, respectively (Table 1). The 58.3 ± 0.7-Å distance of the S16A-PLB-SERCA complex is consistent with previous measurements of SERCA bound to WT- and I40A-PLB, and is compatible with models of the regulatory complex in which the n-termini of PLB and the pump are on opposite sides of the complex (36, 37). The measured decrease in FRETmax suggests an increase in probe separation distance with phosphorylation. This implies a rearrangement of the PLB-SERCA complex that further separates the protein N termini, without loss of binding. Other alternative models for the regulation of SERCA by calcium and PLB phosphorylation have also been proposed (17, 20, 23, 38, 39).
Functional Implications and Physiological Significance—It is likely that the observed structural rearrangement inferred from the observed decrease in intrinsic regulatory complex FRET is important for regulating SERCA function. We envision a simple regulatory scheme in which both the phosphorylated and unphosphorylated PLB-SERCA complexes are catalytically active but characterized by different quaternary structures and different effective calcium affinities (Fig. 4). The conformations resolved in the present study (Fig. 3) may be useful for screening small molecule drug libraries to identify candidates that stabilize the high calcium affinity (low-FRET) structure.
It is a limitation of the present study that we cannot fully evaluate the structural transition that gives rise to the observed decrease in FRETmax with phosphomimetic mutations. Future high resolution studies will be helpful in this regard. However, co-crystals of PLB-SERCA suitable for diffraction studies have been elusive, perhaps because of the rapid subunit exchange kinetics of the regulatory complex (25). The lack of high resolution structure information has motivated a variety of alternative theoretical and experimental approaches, including the present FRET imaging study.
The observed shifts in PLB binding equilibria are also expected to be functionally significant. Fig. 2C shows that phosphorylation-mimicking mutations of PLB result in a 2.1-fold increase in the oligomerization affinity of PLB in the biological membranes of live cells. This effect was matched by a shift in the PLB-SERCA binding equilibrium that was of approximately equal magnitude (1.9-fold lower apparent affinity of PLB for SERCA), leading to the conclusion that there is not a direct effect of phosphorylation on the intrinsic PLB affinity for SERCA (Fig. 4C). It is remarkable that there is so little direct effect of phosphorylation on the thermodynamics of the PLB-SERCA complex, particularly since the pseudo-phosphorylated PLB-SERCA complex quaternary conformation is different by more than 4 Å (Fig. 4D and Table 1). It may be that the putative conformational change does not involve aspects of the interface that contribute significant binding energy. Nevertheless, the seemingly modest effect of phosphorylation on the monomer-pentamer equilibrium (and indirectly on the PLB-SERCA equilibrium) compares favorably with the known functional effect of PLB phosphorylation. SERCA activity increases about 2-fold in submicromolar calcium after phosphorylation of PLB (40). By comparison, the shifts in the coupled equilibria with pseudophosphorylation are approximately half the magnitude of those observed for the superinhibitory I40A mutation (10) and of opposite direction, consistent with the respective functional effects of these mutations (9, 18).
In the heart, the relative contributions of structure changes and equilibria shifts to functional regulation must be determined by the absolute concentration of PLB and SERCA in the native sarcoplasmic reticulum. The effect of altered binding equilibria would be most pronounced at low protein concentrations. At very high concentrations PLB could saturate SERCA binding sites regardless of phosphorylation, and the regulatory effects of structural changes would dominate. Because transgenic studies indicate the PLB-pump interaction is not saturated (41), it is likely that both mechanisms play complimentary roles in vivo: Phosphorylation directly increases PLB5 at the expense of PLB1 (Figs. 2 and 4B), indirectly causing unbinding of PLB1 from SERCA (Figs. 3 and 4C); it also stabilizes a regulatory complex structure with a high calcium affinity (Figs. 3 and 4D). Together, these mechanisms promote faster calcium uptake and result in positive lusitropy (faster cardiac relaxation). Increased calcium transport also supports a larger sarcoplasmic reticulum calcium load and results in positive inotropy (more forceful contractions). Thus, the regulation of SERCA by PLB plays an important role in the heart's inotropic and lusitropic responses to adrenaline.
Supplementary Material
Acknowledgments
We thank Jose Puglisi and Kenneth S. Campbell for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Mentored Research Scientist Development Award EB006061. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental information.
Footnotes
The abbreviations used are: PLB, phospholamban; FRET, fluorescence resonance energy transfer; PKA, cAMP-dependent protein kinase; CFP, cyan fluorescent protein; YFP, yellow fluorescent protein; WT, wild type; AU, arbitrary units; SERCA, sarco(endo)plasmic reticulum calcium ATPase.
References
- 1.Traaseth, N. J., Verardi, R., Torgersen, K. D., Karim, C. B., Thomas, D. D., and Veglia, G. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14676–14681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamoon, J., Mascioni, A., Thomas, D. D., and Veglia, G. (2003) Biophys. J. 85 2589–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacLennan, D. H., and Kranias, E. G. (2003) Nat. Rev. Mol. Cell. Biol. 4 566–577 [DOI] [PubMed] [Google Scholar]
- 4.Tada, M., Kirchberger, M. A., and Katz, A. M. (1975) J. Biol. Chem. 250 2640–2647 [PubMed] [Google Scholar]
- 5.Wegener, A. D., Simmerman, H. K., Lindemann, J. P., and Jones, L. R. (1989) J. Biol. Chem. 264 11468–11474 [PubMed] [Google Scholar]
- 6.Kranias, E. G., and Solaro, R. J. (1982) Nature 298 182–184 [DOI] [PubMed] [Google Scholar]
- 7.Wegener, A. D., and Jones, L. R. (1984) J. Biol. Chem. 259 1834–1841 [PubMed] [Google Scholar]
- 8.Autry, J. M., and Jones, L. R. (1997) J. Biol. Chem. 272 15872–15880 [DOI] [PubMed] [Google Scholar]
- 9.Kimura, Y., Kurzydlowski, K., Tada, M., and MacLennan, D. H. (1997) J. Biol. Chem. 272 15061–15064 [DOI] [PubMed] [Google Scholar]
- 10.Kelly, E. M., Hou, Z., Bossuyt, J., Bers, D. M., and Robia, S. L. (2008) J. Biol. Chem. 283 12202–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robia, S. L., Flohr, N. C., and Thomas, D. D. (2005) Biochemistry 44 4302–4311 [DOI] [PubMed] [Google Scholar]
- 12.Oxenoid, K., and Chou, J. J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 10870–10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith, S. O., Kawakami, T., Liu, W., Ziliox, M., and Aimoto, S. (2001) J. Mol. Biol. 313 1139–1148 [DOI] [PubMed] [Google Scholar]
- 14.Tatulian, S. A., Jones, L. R., Reddy, L. G., Stokes, D. L., and Tamm, L. K. (1995) Biochemistry 34 4448–4456 [DOI] [PubMed] [Google Scholar]
- 15.Simmerman, H. K., and Jones, L. R. (1998) Physiol. Rev. 78 921–947 [DOI] [PubMed] [Google Scholar]
- 16.Abu-Baker, S., and Lorigan, G. A. (2006) Biochemistry 45 13312–13322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karim, C. B., Zhang, Z., Howard, E. C., Torgersen, K. D., and Thomas, D. D. (2006) J. Mol. Biol. 358 1032–1040 [DOI] [PubMed] [Google Scholar]
- 18.Hoshijima, M., Ikeda, Y., Iwanaga, Y., Minamisawa, S., Date, M. O., Gu, Y., Iwatate, M., Li, M., Wang, L., Wilson, J. M., Wang, Y., Ross, J., Jr., and Chien, K. R. (2002) Nat. Med. 8 864–871 [DOI] [PubMed] [Google Scholar]
- 19.Oxenoid, K., Rice, A. J., and Chou, J. J. (2007) Protein Sci. 16 1977–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen, Z., Akin, B. L., and Jones, L. R. (2007) J. Biol. Chem. 282 20968–20976 [DOI] [PubMed] [Google Scholar]
- 21.Asahi, M., McKenna, E., Kurzydlowski, K., Tada, M., and MacLennan, D. H. (2000) J. Biol. Chem. 275 15034–15038 [DOI] [PubMed] [Google Scholar]
- 22.Li, J., Bigelow, D. J., and Squier, T. C. (2003) Biochemistry 42 10674–10682 [DOI] [PubMed] [Google Scholar]
- 23.Negash, S., Yao, Q., Sun, H., Li, J., Bigelow, D. J., and Squier, T. C. (2000) Biochem. J. 351(Pt 1), 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cornea, R. L., Jones, L. R., Autry, J. M., and Thomas, D. D. (1997) Biochemistry 36 2960–2967 [DOI] [PubMed] [Google Scholar]
- 25.Robia, S. L., Campbell, K. S., Kelly, E. M., Hou, Z., Winters, D. L., and Thomas, D. D. (2007) Circ. Res. 101 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winters, D. L., Autry, J. M., Svensson, B., and Thomas, D. D. (2008) Biochemistry 47 4246–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Förster, T. (1948) Ann. Phys. (Leipzig) 2 55–75 [Google Scholar]
- 28.Patterson, G. H., Piston, D. W., and Barisas, B. G. (2000) Anal. Biochem. 284 438–440 [DOI] [PubMed] [Google Scholar]
- 29.Li, M., Reddy, L. G., Bennett, R., Silva, N. D., Jr., Jones, L. R., and Thomas, D. D. (1999) Biophys. J. 76 2587–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004) Nucleic Acids Res. 32(Web Server issue), W665–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, H., Robertson, A. D., and Jensen, J. H. (2005) Proteins 61 704–721 [DOI] [PubMed] [Google Scholar]
- 32.Karim, C. B., Kirby, T. L., Zhang, Z., Nesmelov, Y., and Thomas, D. D. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 14437–14442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, J., Bigelow, D. J., and Squier, T. C. (2004) Biochemistry 43 3870–3879 [DOI] [PubMed] [Google Scholar]
- 34.Traaseth, N. J., Thomas, D. D., and Veglia, G. (2006) J. Mol. Biol. 358 1041–1050 [DOI] [PubMed] [Google Scholar]
- 35.Clayton, J. C., Hughes, E., and Middleton, D. A. (2005) Biochemistry 44 17016–17026 [DOI] [PubMed] [Google Scholar]
- 36.Hutter, M. C., Krebs, J., Meiler, J., Griesinger, C., Carafoli, E., and Helms, V. (2002) Chembiochem. 3 1200–1208 [DOI] [PubMed] [Google Scholar]
- 37.Toyoshima, C., Asahi, M., Sugita, Y., Khanna, R., Tsuda, T., and MacLennan, D. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahaney, J. E., Albers, R. W., Waggoner, J. R., Kutchai, H. C., and Froehlich, J. P. (2005) Biochemistry 44 7713–7724 [DOI] [PubMed] [Google Scholar]
- 39.Traaseth, N. J., Ha, K. N., Verardi, R., Shi, L., Buffy, J. J., Masterson, L. R., and Veglia, G. (2008) Biochemistry 47 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antipenko, A. Y., Spielman, A. I., Sassaroli, M., and Kirchberger, M. A. (1997) Biochemistry 36 12903–12910 [DOI] [PubMed] [Google Scholar]
- 41.Brittsan, A. G., Carr, A. N., Schmidt, A. G., and Kranias, E. G. (2000) J. Biol. Chem. 275 12129–12135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.