

Abstract
The proximal C terminus of the cardiac L-type calcium channel (CaV1.2) contains structural elements important for the binding of calmodulin (CaM) and calcium-dependent inactivation, and exhibits extensive sequence conservation with the corresponding region of the skeletal L-type channel (CaV1.1). However, there are several CaV1.1 residues that are both identical in six species and are non-conservatively changed from the corresponding CaV1.2 residues, including three of the “IQ motif.” To investigate the functional significance of these residue differences, we used native gel electrophoresis and expression in intact myotubes to compare the binding of CaM to extended regions (up to 300 residues) of the C termini of CaV1.1 and CaV1.2. We found that in the presence of Ca2+ (either millimolar or that in resting myotubes), CaM bound strongly to C termini of CaV1.2 but not of CaV1.1. Furthermore, replacement of two residues (Tyr1657 and Lys1662) within the IQ motif of a C-terminal CaV1.2 construct with the divergent residues of CaV1.1 (His1532 and Met1537) led to a weakening of CaM binding (native gels), whereas the reciprocal substitution in CaV1.1 caused a gain of CaM binding. In full-length CaV1.2, substitution of these same two divergent residues with those of CaV1.1 (Y1657H, K1662M) eliminated calcium-dependent inactivation of the heterologously expressed channel. Thus, our results reveal that a conserved difference between the IQ motifs of CaV1.2 and CaV1.1 has a profound effect on both CaM binding and calcium-dependent inactivation.
Calcium ion influx into the cytoplasm via voltage-gated calcium channels not only plays an important role in electrical excitability but also serves as a second messenger controlling cellular functions including muscle contraction, transmitter/hormone release, and transcriptional regulation. It is not surprising, therefore, that voltage-gated calcium channels are subject to diverse regulatory influences that modify both the size and time course of the Ca2+ currents that they generate. One important regulatory mechanism is that of calcium-dependent inactivation, whereby calcium influx via the channel acts as a signal to initiate inactivation of the channel (1–3). Calcium-dependent inactivation has been well documented (4–6) for calcium channels in which the principal subunit is either CaV1.2 (α1C, expressed in cardiac and smooth muscle) or CaV2.1 (α1A, P/Q-type channels expressed in the nervous system). Calcium-dependent inactivation, together with voltage-dependent inactivation, limits the amount of Ca2+ entering the cell. Unlike voltage-dependent inactivation, calcium-dependent inactivation displays a U-shaped dependence on test potential that mirrors the voltage dependence of peak calcium current. Additionally, calcium-dependent inactivation is thought to be reduced or eliminated when Ba2+ is substituted for Ca2+ as the charge carrier (7, 8).
Functional studies of heterologously expressed channels indicate that multiple regions of the channel complex can influence calcium-dependent inactivation. In particular, the involvement of the I-II loop, II-III loop, and C-terminal of CaV1.2 has been suggested on the basis of substitution of these regions by the corresponding segments of CaV1.1 (9). Effects of the accessory β-subunit on calcium-dependent inactivation have also been reported (10). However, a primary role in calcium-dependent inactivation appears to be played by C-terminal domains of CaV1 (4–6). Furthermore, calmodulin (CaM)4 binds to C-terminal segments of CaV1.2 and CaV2.1 in vitro (6, 11–14), and constitutively bound CaM appears to serve as the Ca2+ sensor within intact cells because calcium-dependent inactivation is suppressed by the expression of CaM constructs mutated to lack Ca2+ binding (15, 16). Based on studies with peptides, a number of different regions within the CaV1.2 C terminus have been implicated as important for the binding of the N- and/or C-lobes of Ca2+-free or Ca2+-bound CaM. These include regions “A” (residues 1588–1607), “C” (residues 1615–1636) (12, 17), and the IQ motif (residues 1654–1665). The IQ motif was initially identified as a CaM binding sequence by Rhodes and Friedberg (18) and it was subsequently shown that the Ile → Glu mutation of this motif within CaV1.2 eliminates calcium-dependent inactivation and binding of Ca2+-CaM to a corresponding peptide (19).
Overall, there is a high degree of sequence similarity within the C-terminal region implicated for CaM binding between CaV1.2 and CaV1.1 (α1S), the predominant isoform in skeletal muscle. This similarity is particularly high within the A and C regions (each harboring only a single conservative change of residue). Moreover, there are no differences in the canonical residues (“IQXXXRGXXXRX”) of the IQ motifs. Thus, it has been tacitly assumed that CaM should bind similarly to CaV1.1 and CaV1.2 (20, 21). However, a more detailed comparison of the IQ motifs reveals three non-conservative changes of the “X” residues between CaV1.2 (Tyr1657, Lys1662, and Lys1665) and CaV1.1 (His1532, Met1537, and Gln1540). An indication that these three residues may be functionally significant is that the changes between CaV1.2 and CaV1.1 are identical for six vertebrate species (from zebrafish to humans). Moreover, all three CaV1.2 residues make close contact with the C-lobe of CaM in the crystal structure presented by Van Petegem et al. (21). Thus, a major goal of the studies described here was to determine the significance of these three residues for both the binding of CaM and calcium-dependent inactivation.
For our analysis of binding, we used C-terminal regions of CaV1.1 and CaV1.2, which were up to ∼300 residues in length rather than the relatively short peptides (<60 residues, typically ∼20) used in prior studies. These extended peptides have the advantage that they encompass all of the regions (A, C, and IQ) previously implicated in CaM binding and may preserve a more native structure, a point of importance because dissimilar crystal structures were obtained for two different IQ motif-containing peptides complexed with CaM (21, 22). To overcome the limited solubility of the extended C-terminal peptides (23), we expressed and purified them as fusions to the bacterial NusA protein (24), incubated them with CaM, cleaved the link between NusA and the C-terminal peptide with TEV protease, and then analyzed the resultant protein mixtures by means of non-denaturing gel electrophoresis and mass spectroscopy. With this assay system, we found that Ca2+-CaM binds strongly to C-terminal segments of CaV1.2 but not to the corresponding segments of CaV1.1. Moreover, this difference in CaM binding was also present under resting cellular conditions, based on expression in dysgenic myotubes of fluorescently tagged CaM and tagged C-terminal segments of CaV1.2 or CaV1.1. CaM binding was weakened when the divergent IQ motif residues in CaV1.2 C-terminal segments were converted to those of CaV1.1, and CaM binding was gained in CaV1.1 C-terminal segments by converting the divergent residues to those of CaV1.2. Within full-length CaV1.2, conversion of two of the three divergent IQ motif residues to those of CaV1.1 was sufficient to abolish calcium-dependent inactivation. Thus, our results underscore that, although previously unappreciated, the “X-type residues” of the IQ motifs of CaV1.1 and CaV1.2 are important both for CaM binding and channel inactivation. Furthermore, because the IQ motif differences between CaV1.1 and CaV1.2 are evolutionarily conserved, they are likely to be important for the different mechanisms of calcium handling in skeletal and cardiac muscle.
EXPERIMENTAL PROCEDURES
Construction of Expression Vectors, Bacterial Expression Vectors—Fig. 1 illustrates the C-terminal (CT) segments of α1S (CaV1.1; GenBank™ number M23919) or α1C (CaV1.2; GenBank number X15539), which were expressed as bacterial fusion proteins. The sequences encoding the CT segments were produced by applying conventional PCR to expression plasmids for CaV1.1 (25) or CaV1.2 (26) in combination with primers carrying restriction sites for NcoI (5′) and SalI (3′). The primer pairs used for CaV1.1 were forward (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and reverse (rev) 5′-CTT GTC GAC TAG GGC CGA TAC CCA TAA TAT TCC TC-3′ for CT1 (amino acids 1453–1548), (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and (rev) 5′-CTT GTC GAC TAT CCC GTC CTC CGG AAG ATC CTC TC-3′ for CT2 (amino acids 1453–1602), (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and (rev) 5′-CTT GTC GAC TAT CCC GGG AAC TCC CTT TCA CAG TG-3′ for CT3 (amino acids 1381–1692). The primer pairs used for CaV1.2 were (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and (rev) 5′-CTT GTC GAC TAG GGC TTG CCC ACA AGC CCT TG-3′ for CT1 (amino acids 1578–1673), (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and (rev) 5′-CTT GTC GAC TAG GCC CTC CTG AAG ATG TCA TCT TC-3′ for CT2 (amino acids 1578–1729), and (fw) 5′-CAC CAT GGA CAA CTT TGA CTA CCT GAC AAG-3′ and (rev) 5′-CTT GTC GAC TAG CTG GGG TAG CCG GCG GGG CG-3′ for CT3 (amino acids 1506–1816). The amplified segments were subcloned into the pETM60 vector (24) to allow expression of NusA-CT fusion proteins. In this vector, sequences coding for a hexahistidine tag and for a TEV protease recognition site separate the NusA protein (55 kDa) and the CT segment to allow for metal affinity purification and cleavage of the fusion proteins, respectively (Fig. 2A).
FIGURE 1.

Top, schematic representation of the full-length C-terminal and the CT segments tested for complex formation with CaM. Residue numbers refer to the rabbit isoforms of CaV1.2 (top number) and CaV1.1 (bottom number). The EF-hand (4), A and C (12, 17), and IQ (15) motifs have been implicated as important for calcium-dependent inactivation and/or CaM binding. Bottom, alignment of CaV1.2 residues 1637–1673 and CaV1.1 residues 1512–1548 (numbering according to rabbit sequences). Arrows and bold-face indicate residues that were subjected to mutational exchange between the two isoforms. The species and accession numbers for the illustrated sequences are indicated. The sequence for chicken CaV1.2 and dog CaV1.1/1.2 are predicted (p) only.
FIGURE 2.

A, scheme used to generate reaction mixtures of CaM and C-terminal (CT) fragments of CaV1.1 (Sk) or CaV1.2 (Card). B, Coomassie-stained SDS-PAGE of the reaction mixtures confirms that both skeletal and cardiac CT fragments (see Fig. 1) were expressed and released from the NusA fusion by the TEV-protease treatment. CT1 is shown here as an example. Mass spectrometric analysis (MS) of the low molecular mass bands identified CaM (16.7 kDa) and either the skeletal or cardiac CT1 (both 11.3 kDa). Note that digestion with TEV-protease was not run to completeness in this example, as is indicated by the presence of two bands in the 60–80 kDa range.
An expression plasmid carrying the cDNA encoding rat CaM (GenBank number M19312; plasmid kindly provided by K. Stroffekova, Utah State University, Logan, UT), was subcloned into the bacterial expression vector pRSFDuet-1 (Novagen) using standard PCR with primers adding recognition sequences for NdeI (5′) and XhoI (3′). The primers used were (fw) 5′-GAC GAC AAG CAT ATG GCT GAC CAA CTG ACT GAA GAG CAG GC-3′ and (rev) 5′-GAC TCG AGT CAC TTC GCT GTC ATC ATT TGT ACA AAC TC-3′.
C Terminus Mutations of CaV1.1 or CaV1.2—To replace cardiac-type residues by those present in the skeletal muscle isoform and vice versa, site-directed mutagenesis (Stratagene, La Jolla, CA) was performed using the respective bacterial CT expression plasmids described above as templates. The following primers were used: (fw) 5′-GTC ATC CCT CCC GCA GGA GAT GAC GAG G-3′ and (rev) 5′-CCT CGT CAT CTC CTG CGG GAG GGA TGA C-3′ for CaV1.1 I1513A, (fw) 5′-CCA CAT TCC TCA TCC AGG AGT ACT TCC GGA AGT TCA TGA AGC-3′ and (rev) 5′-GCT TCA TGA ACT TCC GGA AGT ACT CCT GGA TGA GGA ATG TGG-3′ for CaV1.1 H1532Y, (fw) 5′-GCA CTT CCG GAA GTT CAA GAA GCG CCA GGA GG-3′ and (rev) 5′-CCT CCT GGC GCT TCT TGA ACT TCC GGA AGT GC-3′ for CaV1.1 M1537K, (fw) 5′-GGA GTA CTT CCG GAA AGT TCA AGA AGC GCC AGG AGG-3′ and (rev) 5′-C CTC CTG GCG CTT CTT GAA CTT CCG GAA GTA C-3′ for CaV1.1 H1532Y/M1537K, (fw) 5′-CGG AAG TTC AAG AAG CGC AAG GAG GAA TAT TAT GG-3′ and (rev) 5′-CC ATA ATA TTC CTC CTT GCG CTT CTT GAA CTT CC-3′ for CaV1.1 H1532Y/M1537K/Q1540K, (fw) 5′-CCA AGT GGT GCC CCC TAT AGG CGA TGA TGA GGT C-3′ and (rev) 5′-GAC CTC ATC ATC GCC TAT AGG GGG CAC CAC TTG G-3′ for CaV1.2 A1638I, (fw) 5′-CCT TCC TGA TCC AAG AGC ACT TCC GGA AAT TC-3′ and (rev) 5′-GAA TTT CCG GAA GTG CTC TTG GAT CAG GAA GG-3′ for CaV1.2 Y1657H, (fw) 5′-CTT CCG GAA ATT CAT GAA GCG CAA AGA GC-3′ and (rev) 5′-GCT CTT TGC GCT TCA TGA ATT TCC GGA AG-3′ for CaV1.2 K1662M, (fw) 5′-CCA AGA GCA CTT CCG GAA ATT CAT GAA GCG CA-3′ and (rev) 5′-TGC GCT TCA TGA ATT TCC GGA AGT GCT CTT GG-3′ for CaV1.2 Y1657H/K1662M, (fw) 5′-CGG AAA TTC ATG AAG CGC CAA GAG CAA GG-3′ and (rev) 5′-CCC TTG CTC TTG GCG CTT CAT GAA TTT CC-3′ and CaV1.2 Y1657H/K1662M/K1665Q. CTs bearing the mutation(s) were then excised from the amplified template by cutting with NcoI (5′) and SalI (3′) and were subcloned into the original pETM60 vector as described above. Full-length CaV1.2 harboring mutations Y1657H/K1662M was constructed by site-directed mutagenesis using the mammalian expression plasmid GFP-α1C (26) as the template and the same primers described above to introduce this mutation into the bacterial expression vector.
Expression Vectors for YPet-CaM and CyPet-CT1—Mammalian expression vectors encoding the fluorescent proteins CyPet and YPet (27) served as templates and were kindly provided by Dr. Patrick S. Daugherty, University of California, Santa Barbara. First, flanking recognition sites for AgeI (5′) and BglII (3′) were added to the coding sequences of CyPet and YPet by standard PCR using the following primers. The same forward primer was used for both CyPet and YPet, (fw) 5′-CTG CAG TCG ACG GTA CC-3′. For CyPet, the primer (rev) was 5′-CGA GAT CTT TTG TAC AGT TCG TCC ATG C-3′ and for YPet the primer (rev) was 5′-CGA GAT CTC TTA TAG AGC TCG TTC ATG C-3′. The AgeI-BglII fragment was placed into the corresponding restriction sites of pECFP-C1 (Clontech, Palo Alto, CA) to create the vectors pCyPet-C1 and pYPet-C1.
For insertion of CT1 into pCyPet-C1, PCR primers used were (fw) 5′-GGA ATT CCA TGG CCC TGA ACA GTG ACG GCA CG-3′ and (rev) 5′-CTT GTC GAC TAG GGC CGA TAC CCA TAA TAT TCC TC-3′ for CaV1.1, and (fw) 5′-GGA ATT CCA TGG CCC TGA ACA GTG ACG GCA CG-3′ and (rev) 5′-CTT GTC GAC TAG GGC TTG CCC ACA AGC CCT TG-3′ for CaV1.2; subsequently the EcoRI-SalI cleavage fragment was inserted into the corresponding restriction sites of pCyPet-C1 to produce the expression plasmids for skeletal or cardiac CyPet-CT1.
To construct YPet-CaM, XhoI restriction sites flanking the CaM sequence were introduced into the rat CaM expression plasmid, using primers (fw) 5′-GAG CTC GAG ATA TGG CTG ACC AAC TGA CTG AAG AGC-3′ and (rev) 5′-GAC TCG AGC ACT TCG CTG TCA TCA TTT GTA CAA ACT C-3′ and followed by excision of the XhoI-XhoI fragment and its insertion into pYPet-C1 that had been opened by XhoI. Accuracy of all vector sequences described above was confirmed by sequencing (MWG Biotech, Ebersberg, Germany).
Protein Expression and Purification—Escherichia coli (BL21 Rosetta 2 (DE3)pLysS; EMD Biosciences) transformed with the CaM expression vector (described above) were grown to mid-log phase and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 37 °C. The bacteria were lysed by sonication in a lysis buffer containing in mm: 20 Tris (pH 7.4), 300 NaCl, 5 MgCl2, and 0.1% Triton X-100. The lysate was heated to 70 °C for 3 min, immediately cooled on ice, and centrifuged at 100,000 × g for 30 min at 4 °C. The supernatant was subjected to ion exchange chromatography (Vivapure, Sartorius, Germany) following the manufacturer's instructions and then dialyzed overnight against lysis buffer. The solution was then diluted to 400 μg/ml CaM and aliquots were stored at -85 °C.
E. coli (BL21), transformed with the TEV protease expression vector pRK793 (generously provided by Dr. David S. Waugh, NCI-Frederick, MD, National Institutes of Health), were grown to A600 of 0.6 and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 30 °C. The cells were lysed by sonication in a lysis buffer containing in mm: 50 Tris (pH 8.0), 1 EDTA, 1 dithiothreitol, and 0.1% Triton X-100. Soluble TEV protease was prepared using metal affinity chromatography (Protino, Macherey-Nagel, Germany) following the manufacturer's instructions. The solution was dialyzed against lysis buffer, concentrated (Millipore, Centricon-Centrifugal Filter YM-30) to 200 μg/ml, divided into aliquots, and stored at -85 °C.
Transformation and expression of the NusA-CT1, -CT2, and -CT3 fusions were as described for CaM, except that, after induction, bacteria were grown overnight at room temperature. The bacterial lysate was centrifuged at 8,000 × g for 30 min and the His-tagged fusion proteins contained in the supernatant were purified and concentrated using metal affinity chromatography (Protino).
Analysis of CT-CaM Interaction—Protein concentrations of CaM, TEV Protease, and the NuSA-CT fusion proteins were determined by the method of Bradford (28) and a constant molar ratio of CaM:TEV Protease:NuSA-CT fusion protein was used in the CaM binding assays. For the binding assay, 75 μl (corresponding to 22.5 μg) of NusA-CT fusion protein was mixed with 50 μl of CaM protein solution (corresponding to 20 μg CaM), 1.65 μl of 0.1 m dithiothreitol, 1.65 μl of Triton X-100, 3.0 μl of TEV protease, 12.7 μl of 20 mm Tris (pH 8.0), and either 6 μl of 50 mm CaCl2 or 6 μl of 50 mm EDTA to set the nominal concentrations for Ca2+ or EDTA to 2.0 mm. The mixture was incubated overnight at 6 °C. After analysis of aliquots by SDS-PAGE (29) to confirm the presence of TEV protease, CaM, NuS-CT fusion protein, and fragments resulting from exposure to TEV protease (Fig. 2B), complex formation between CaM and CT was assayed by means of nondenaturing PAGE (15%). The gel running buffer contained 20 mm Tris/HCl (pH 8.6), 125 mm glycine, 0.1% Triton X-100, and either 2 mm CaCl2 or EDTA. For densitometry, Coomassie Blue-stained gels were scanned and 8 bit-digitized (Epson Perfection 2400 PHOTO, Epson). Areas under the bands of interest, corrected for background (as defined by the intensity integral of an equal area within an empty lane next to the band) were then measured with the FluoView10 software package that is included in the FV1000 confocal scanning system (Olympus). Bands were defined by zooming into the gel and drawing a region of interest, and integrating the intensity values inside. The intensity of a band corresponding to a given CT·CaM complex was referenced to the intensity of the band corresponding to free CaM within the same lane (Table 1). For mass spectrometric identification of proteins, gel bands were cut out, dissolved, subjected to cleavage by trypsin, and analyzed in-house by matrix-assisted laser desorption ionization time-of-flight/time-of-flight (Ultra flex, Bruker Daltonik).
TABLE 1.
Densitometric analysis of the binding of CT constructs to CaM in the presence of 2 mm Ca2+ Values give the ratio between the band intensity of the CT-CaM complex (ICT:CaM) and the intensity of the corresponding band (in the same lane) of free CaM (ICaM), as determined by densitometry. In the left column indicating mutational exchanges within the CT constructs, native cardiac residues are underlined. The values listed under the skeletal column for CT1 (WT), CT1 (I → A), and CT2 (WT) likely represent nonspecific smearing rather than presence of a CT·CaM complex since comparable values were obtained by densitometric integration of regions above or below the region of expected complex formation.

Significantly different (p < 0.05) from CT1 (WT)
Not significantly different (p = 0.12)
Analyis of cDNAs Expressed in Mammalian Cells—Primary cultures of dysgenic myotubes (30) were prepared from newborn mice as described previously (31). Briefly, myoblasts were plated into 35-mm culture dishes with glass coverslip bottoms (MatTek Corp., Ashland, MA) and cultivated in a humidified 37 °C incubator with 5% CO2. 2 days after replacing the culture medium by differentiation medium, the nuclei of myotubes were microinjected with a small amount of plasmid DNA, which was a mixture (1:1 molar ratio) of YPet-CaM and either skeletal muscle-type or cardiac-type CyPet-CT1. The DNA concentration of the injection solution was 20 ng/μl water. Myotubes were examined 24 h after microinjection using a FW1000 confocal laser-scanning microscope (Olympus). CyPet and YPet were excited using the 458 and 514 nm argon lines, respectively, directed to the cells via a 458/514 nm dual dichroic mirror. CyPet and YPet emissions were photometrically detected using Olympus filters BA465-495 and BA535-565, respectively. FRET efficiency was measured by means of enhanced donor emission after acceptor bleaching (25). In brief, the intensity of the CyPet-CT emission (BA465-495) upon excitation at 458 nm was measured before and after completely bleaching YPet-CaM.
Modified HEK-293 cells (tsA-201; ECACC, Salisbury, UK) were maintained in high glucose (4.5 g/liter) Dulbecco's modified Eagle's medium (Biochrom, Germany) with 10% fetal bovine serum in a humidified incubator with 5% CO2 (32). Cells were plated at a density of 2 × 105 cells in 35-mm dishes and transfected by Lipofectamine 2000 (Invitrogen) hours later with equal concentrations (1 μg/μl) of either GFP-CaV1.2 wt (26) or GFP-CaV1.2[Y1657H/K1662M] (see above), YFP-β2a, and α2-δ1 subunit cDNA. Twenty-four hours following transfection, cells were removed from the dish using Trypsin EDTA 1X (Mediatech, Manassas VA) and re-plated at ∼1 × 104 cells per 35-mm dish to obtain isolated cells for electrophysiological recording. Forty-eight hours following transfection, positively transfected cells were identified by green fluorescence.
Measurement of L-type Ca2+ Current—Whole cell voltage clamping (33) was used to record macroscopic currents. Borosilicate glass pipettes were polished to a final resistance of ∼2.0–3.0 MΩ when filled with internal solution containing in mm: 140 CsAsp, 5 MgCl2, 10 Cs2EGTA, 10 HEPES, pH to 7.4 with CsOH. The External solution contained (mm) 10 CaCl2 or 10 BaCl2, 145 tetraethylammonium-Cl, 10 HEPES, pH to 7.4 with triethanolamine-OH. For solution changes, 20 ml of solution was perfused at a rate of 6 ml min-1. Test currents were elicited by rectangular test potentials applied from a -80 mV holding potential. Test currents were corrected for linear components of leak and capacitive current by digitally scaling and subtracting the average of 8 control currents elicited by a hyperpolarizing step to -100 mV. This same control current was used to calculate linear cell capacitance. Calcium currents were filtered at 0.5–1 kHz and sampled at 1 kHz. Peak currents as a function of test potential were fitted according to,
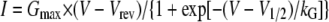 |
(Eq. 1) |
where I is the peak inward Ca2+ current measured at the potential V, Vrev is the reversal potential, and kG is a slope factor. Fractional inactivation of current in 50 ms (R50) was determined by dividing the peak current into the current 50 ms after the peak. All data reported were obtained from cells in which both Ca2+ and Ba2+ currents were recorded.
RESULTS
Non-denaturing Gels Reveal That Homologous C-terminal Regions of CaV1.1 and CaV1.2 Differ in Their Ability to Bind CaM—Fig. 1 illustrates the CT segments of CaV1.1 (α1S, skeletal) and CaV1.2 (α1C, cardiac), which we tested for CaM binding ability. The CT3 segments begin immediately distal to the IVS6 trans-membrane segment and end close to the site of proteolytic cleavage that naturally occurs in CaV1.1 and CaV1.2 expressed in native muscle (34). Compared with CT3, the CT2 segments lack the 72 residues that are immediately distal to IVS6 and that are highly conserved between CaV1.1 and CaV1.2 (differing at only 3 residues). These 72 residues are critical for membrane expression and contain an EF-hand motif whose presence, but not the ability to bind Ca2+, is essential for calcium-dependent inactivation of CaV1.2 (35, 36). CT2 also lacks the ∼90 residues that are present at the distal end of CT3 and are poorly conserved between CaV1.1 and CaV1.2. Finally, CT1 lacks ∼55 residues that are present in the distal portion of CT2 and only partially conserved between CaV1.1 and CaV1.2. Overall, the CT1 segments are well conserved between CaV1.1 and CaV1.2. Alignment of the regions showing the greatest differences is illustrated in Fig. 1 (upstream in CT1, there are two conservative and two non-conservative residue differences between CaV1.1 and CaV1.2). Within the aligned regions of each isoform for six different species, there are four residues (boldface, arrows) that are (i) identical in all of the CaV1.2 isoforms, (ii) identical in all of the CaV1.1 isoforms, and (iii) differ between CaV1.2 and CaV1.1.
For analysis of interactions with CaM, we initially attempted to express hexahistidine-tagged CT segments in bacteria, but this yielded protein (as indicated by SDS-PAGE) that could not be isolated from the bacteria in a soluble form under non-denaturing conditions (data not shown). We then turned to using the bacterial vector pETM60 (24) to produce CTs linked to the C terminus of the bacterial protein NusA via His6 and TEV-protease recognition sequences (Fig. 2A). These fusion proteins were purified by metal ion chromatography and then cleaved by adding TEV protease in the presence of CaM and either 2 mm EDTA or 2 mm Ca2+. Using SDS-PAGE on aliquots of these reaction mixtures, we established that both cardiac and skeletal NusA-CTs were expressed and were released from the fusion by TEV protease (Fig. 2B). Furthermore, mass spectrometric analysis identified the low molecular weight proteins as CaM and the predicted CT regions of CaV1.2 and CaV1.1. This direct confirmation was important because a previous study found that it was not possible to obtain soluble protein from bacterial expression of a His-tagged CT construct similar to our CT1 unless the preceding EF-hand region was also included (23). Evidently, fusion to NusA overcomes this difficulty.
To assay binding, mixtures of NusA-CT, TEV protease, and CaM were incubated overnight and subjected to non-denaturing PAGE. With non-denaturing PAGE it was possible to detect bands corresponding to NusA, TEV protease, CaM, and CT·CaM complex (see below), but not to free CT (Fig. 3). The inability to detect the free C-terminal segments (cardiac or skeletal) is consistent with previous work on C-terminal segments of CaV1.2 (20) and likely is because these unbound CT segments have properties (limited solubility as well a predicted pI ≈ 10 for our cardiac CT1), which interfere with entry into non-denaturing gels.
FIGURE 3.

Nondenaturing PAGE to check for complex formation between CaM and the CaV1.1 (Sk) or CaV1.2 (Card) C-terminal regions CT1, CT2, and CT3 (see Fig. 1 for orientation). In the presence of 2 mm Ca2+, all CTs of the cardiac type were able to form a complex with CaM (underlined labels; confirmed for CT1 by mass spectrometry), whereas the skeletal type CT constructs lacked the additional band. In the absence of Ca2+ and presence of 2 mm EDTA, CaM failed to form a complex with cardiac (or skeletal) CT1 (bottom, right).
In the presence of 2 mm Ca2+, all three cardiac CTs bound CaM as indicated by the presence of four bands: NusA, TEV protease, CaM, and one additional band representing the CT·CaM complex (Fig. 3). For cardiac CT1, mass spectroscopic analysis provided direct confirmation of both its presence and that of CaM in the additional band. By contrast to the cardiac CTs, none of the skeletal CTs formed a detectable complex with CaM (Fig. 3).
Because there was no obvious difference in the ability of the three cardiac CTs to form a complex with CaM, the majority of our experiments focused on CT1 because it harbors the regions often referred to as A, C, and IQ, which have been implicated in CaM binding and calcium-dependent inactivation of CaV1.2 (12, 17, 37, 38). The complex formed between CaM and cardiac CT1 in 2 mm Ca2+ was not observed when Ca2+ was omitted and divalent ions were chelated by the addition of 2 mm EDTA (Fig. 3). However, the CaM·CT1 complex still occurred when Ca2+ was omitted and EDTA was not added (data not shown), indicating that contaminant Ca2+ was sufficient to support formation of this complex. These results are consistent with previous work (15) showing that CaM formed a complex with a cardiac-type 20-amino acid IQ-peptide at 100 nm Ca2+ but not in the presence of EGTA.
Expression in Dysgenic Myotubes Also Reveals Differences in the Ability of Skeletal and Cardiac CT1 to Bind CaM—As described above, we were unable to detect formation of a complex of CaM with any of the skeletal CT fragments using non-denaturing gel-shift analysis. The result for skeletal CT1 (residues 1453–1548) was surprising given its sequence similarity to cardiac CT1. Furthermore, one previous study using a gel shift assay did find an interaction between CaM and CaV1.1 residues 1522–1542 (12), although this interaction was weaker than that of CaM with the corresponding CaV1.2 residues. Another study demonstrated a change of native tryptophan fluorescence that was indicative of an interaction of CaM with CaV1.1 residues 1393–1527 (39). Thus, we sought an alternative approach that would allow comparison of the ability of CaM to interact with skeletal and cardiac CT1 isoforms under conditions that would occur in resting muscle cells. Specifically we constructed cDNAs encoding CyPet fluorescent protein fused to either skeletal or cardiac CT1 and expressed these cDNAs by injection into single nuclei of dysgenic myotubes (which are null for endogenous CaV1.1). We then examined the subcellular distribution of the CyPet-CT1 fusion proteins when expressed either alone or together with YPet-CaM. When expressed without CyPET-CT1 constructs, YPet-CaM displayed a diffuse cytoplasmic distribution (not shown). In the absence of exogenously expressed CaM, both skeletal and cardiac CyPet-CT1 were present only in the vicinity of the injected nucleus (Fig. 4A). We do not know the identity of the cellular structure that corresponded to this perinuclear localization, but an aggresome (40) is one possibility.
FIGURE 4.

Expression of skeletal (Sk) or cardiac (Card) CyPet-CT1 in dysgenic mouse myotubes, either alone (A) or in combination with YPet-CaM (B). In the absence of YPet-CaM, both cardiac and skeletal CT1 were present only around the nucleus injected with cDNA (A). Upon co-expression with YPet-CaM, the skeletal CT1 was still restricted around the injected nucleusm whereas the cardiac CT1 was distributed throughout the cell, with only occasional accumulation around the injected nucleus (not shown). Bars, 10 μm.
The co-expression of YPet-CaM caused an isoform-specific alteration in the subcellular distribution of CyPet-CT1. Specifically, YPet-CaM expression did not alter the perinuclear localization of skeletal CyPet-CT1 (Fig. 4B, upper right) but caused cardiac CyPet-CT1 to become diffusely distributed throughout the cytoplasm (Fig. 4B, lower right). In addition to this diffuse, cytoplasmic distribution, a variable amount of perinuclear aggregation of cardiac CyPet-CT1 was still sometimes observed after co-expression with YPet-CaM (data not shown). As a test of whether the similar cytoplasmic distribution of cyan and yellow fluorescence represented a direct interaction of YPet-CaM and cardiac CyPet-CT1, we assayed for FRET by means of enhanced donor (CyPet) emission after acceptor (YPet) photobleaching (25). The measured FRET efficiency was 11.1 ± 5.7% (mean ± S.D., n = 4), consistent with a close association between CaM and cardiac CT1. In summary, the analysis of expression in dysgenic myotubes, like the gel shift assays described above, indicates that CaM associates readily with the C terminus of CaV1.2 but not with that of CaV1.1.
Sequence Differences between the IQ Motifs of CaV1.1 and CaV1.2 Are Important for CaM Binding—To identify the sequence determinants responsible for differential binding of CaM to the CT1 segments of CaV1.2 and CaV1.1, we focused on the region that includes the IQ motif (Fig. 5A), because outside of this region there are only four residues of CT1 that differ (Met1587-Thr1462, Arg1601-Lys1476, Leu1608-Phe1483, and Val1635-Ile1510, underlining used to indicate CaV1.2 residues). Moreover, the IQ region contains residue differences between CaV1.2 and CaV1.1 that are highly conserved (Fig. 1), including three residues that, in CaV1.2, are revealed by the crystal structure to lie within a region of intimate contact (Fig. 5A, highlighted in gray) with the Ca2+-loaded C-lobe of CaM (21). Thus, we carried out mutational exchange of these three residues (Tyr1657, Lys1662, and Lys1665 of CaV1.2 and His1532, Met1537, and Gln1540 of CaV1.1, respectively), as well as of one residue outside the region of contact with the CaM C-lobe (cardiac Ala1638 and skeletal Ile1513). The Ala1638 Δ Ile1513 exchange did not cause skeletal CT1 to gain the ability to bind CaM or cardiac CT1 to lose this ability (lane 2 of Fig. 5, B and C, respectively; Table 1). By contrast, the Tyr1657 Δ His1532 exchange resulted in detectable CaM binding to skeletal CT1 and a decrease in CaM binding to cardiac CT1 (lane 3, Fig. 5, B and C, respectively; Table 1). Interestingly, the Lys1662 Δ Met1537 also caused a gain of CaM binding by skeletal CT1 (Fig. 5B, lane 4) without causing a decrease in CaM binding to cardiac CT1 (Fig. 5C, lane 4). In addition to single mutational exchanges, we also examined both double Tyr1657 Δ His1532/Lys1662 Δ Met1537 (Fig. 5, B and C, lane 5) and triple Tyr1657 Δ His1532/Lys1662 Δ Met1537/Lys1665 Δ Gln1540 exchanges. The double exchange Tyr1657 Δ His1532/Lys1662 Δ Met1537 appeared to cause a slightly larger decrease in CaM binding to cardiac CT1 (Fig. 5C, lane 5; Table 1) than did the single exchange Tyr1657 Δ His1532. However, the triple exchange Tyr1657 Δ His1532/Lys1662 Δ Met1537/Lys1665 Δ Gln1540 appeared to have no greater effect than the double exchange Tyr1657 Δ His1532/Lys1662 Δ Met1537 (Table 1).
FIGURE 5.

Residues important for CT1-CaM interaction. A, alignment of cardiac and skeletal CT1 in the region analyzed by mutagenesis, with gray shading indicating residues that contact the CaM C-lobe in the crystal structure (21). B and C, non-denaturing PAGE analysis for complex formation between CaM and wild-type (wt) or mutant skeletal or cardiac CT1 in the presence of 2 mm Ca2+.
Calcium-dependent Inactivation of CaV1.2 Is Eliminated by Replacement of the IQ Motif Residues with the Corresponding Residues of CaV1.1—As just described, the gel shift assays (Fig. 5) revealed that the binding of CaM to the cardiac CT1 fragment was greatly reduced by replacement of two of the cardiac residues (Tyr1657 and Lys1662) with the corresponding skeletal residues of CaV1.1 (His1532 and Met1537). To determine the functional consequences of this residue exchange, whole cell currents were measured in tsA-201 cells after expression of wild-type CaV1.2 or CaV1.2[Y1657H/K1662M], together with auxiliary subunits β2a and α2-δ1. Expression in non-muscle cells was chosen for these experiments because previous work has shown that, for unknown reasons, calcium-dependent inactivation does not occur when CaV1.2 is expressed in dysgenic myotubes (41). Fig. 6A compares Ca2+ and Ba2+ currents in single tsA-201 cells expressing either wild-type CaV1.2 (upper panel) or CaV1.2[Y1657H/K1662M] (lower panel). For each construct, the Ca2+ current was scaled up to match the peak amplitude of the corresponding Ba2+ current to facilitate the comparison of decay kinetics. In accord with a large body of previous work (2, 15, 42, 43), substitution of Ba2+ for Ca2+ slowed the decay of current for wild-type CaV1.2, as expected if the entry of Ca2+, but not of Ba2+, accelerates this decay. However, this calcium-dependent inactivation appeared to be absent for CaV1.2[Y1657H/K1662M] because the decay of the scaled Ca2+ and Ba2+ currents was nearly identical. As a means of comparing the decay of currents quantitatively, we determined the fraction of current remaining 50 ms after the peak (R50) and plotted it as a function of test potential. For the wild-type CaV1.2 with Ca2+ as the charge carrier, R50 displayed a characteristic “U” shape (Fig. 6C, filled circles) with a minimum near the potential that elicited the maximal inward current (Fig. 6B). By contrast, R50 was a monotonically decreasing function of voltage for wild-type CaV1.2 with Ba2+ as charge carrier. Moreover, R50 for CaV1.2[Y1657H/K1662M] with either Ca2+ or Ba2+ as charge carrier was very similar at all potentials to R50 for wild-type CaV1.2 in Ba2+. Thus, substitution of the conserved His1532 and Met1537 residues of CaV1.1 for the Tyr1657 and Lys1662 residues within the IQ motif of CaV1.2 both interfered with CaM binding and abolished calcium-dependent inactivation.
FIGURE 6.

The mutations Y1657H/K1662M in the CaV1.2 IQ motif eliminate calcium-dependent inactivation. A, representative whole cell currents in ts-A201 cells expressing either wild-type CaV1.2 or CaV1.2[H1657Y/K1662M], elicited by depolarization to +20 mV in either 10 mm Ca2+ or 10 mm Ba2+. For both wild-type and mutant, the Ca2+ current has been scaled ∼2.5 times. The scale bar, which applies to the Ba2+ currents, represents 1000 or 500 pA (CaV1.2 and CaV1.2 mutant, respectively). B, normalized peak I-V relationships demonstrate that Ba2+ caused a similar shift of activation for wild-type and mutant channels. Average peak calcium and barium current densities (mean ± S.E.) for CaV1.2 were 32.9 ± 8.2 and 78.5 ± 16.0 pA/pF, respectively, and for CaV1.2[Y1657H/K1662M] were 53.2 ± 11.3 and 76.9 ± 13.7 pA/pF. C, ratio of current-50-ms-after-the-peak-inward-current to the peak-inward-current (R50) plotted as a function of test potential. For B and C, the symbols are as defined in A and the error bars represent ±S.E.
DISCUSSION
In this study, we compared the ability of CaM to bind to the proximal C termini of two L-type Ca2+ channels, CaV1.1 (skeletal isoform) and CaV1.2 (cardiac isoform) expressed as fusion proteins of up to >300 residues. Analyzed by means of non-denaturing gel electrophoresis, Ca2+-CaM bound strongly to the proximal CaV1.2 C terminus but not to that of CaV1.1. The differential ability to bind Ca2+-CaM was unaffected by deletion of ∼70 highly conserved N-terminal and ∼140 partially conserved C-terminal residues (Figs. 1 and 3). The remaining ∼100 residues (“CT1”) also displayed a differential ability to bind CaM in resting myotubes, as assayed by expression of fluorescently tagged constructs (Fig. 4). Alignment demonstrates that within CT1, three X residues of the IQ-motif (IQXXXRGXXXRX) displayed consistent, evolutionarily conserved differences between the skeletal (CaV1.1) and cardiac (CaV1.2) isoforms. Within cardiac CT1, converting the first two divergent residues to their skeletal counterparts resulted in a decrease in Ca2+-CaM binding, whereas Ca2+-CaM binding was gained in skeletal CT1 by converting the corresponding two skeletal residues to cardiac (Fig. 5). In full-length CaV1.2 heterologously expressed in ts-A201 cells, conversion of the first two divergent residues from cardiac to skeletal caused a complete loss of calcium-dependent inactivation.
For each of the cardiac C-terminal segments, the formation of a tight complex with Ca2+-CaM resulted in the presence of a distinct band in native gels. Under the same conditions, no distinct band was evident for any of the skeletal C-terminal constructs, which indicates either that there was little binding of Ca2+-CaM or that the binding was sufficiently weak that the complex dissociated during electrophoresis. As an example of the latter, some cardiac C-terminal peptides have been reported to bind CaM weakly enough that no distinct band could be detected in native gels; however, the binding could be inferred from a reduced intensity of the band corresponding to free CaM (20). Because our experiments were carried out with a severalfold molar excess of CaM, we cannot say whether there was weak binding or no binding to the skeletal C-terminal because we would have been unable to detect a small depletion of free CaM.
Injection of single nuclei of dysgenic myotubes with cDNA encoding CyPet-CT1 either alone or together with YPet-CaM was used to probe CaM binding at resting myoplasmic Ca2+ levels. Both skeletal and cardiac CyPet-CT1 aggregated around the injected nucleus if YPet-CaM cDNA was not also expressed. The co-expression of YPet-CaM caused cardiac CyPet-CT1 to assume a diffuse cytoplasmic distribution but did not alter the aggregates of skeletal CyPet-CT1 (Fig. 4). In addition to indicating that CaM binding to cardiac CT1 at resting myoplasmic Ca2+ is much stronger than to skeletal CT1, these results also raise the question as to why endogenous CaM was insufficient to cause cardiac CyPet-CT1 to be diffusely distributed. However, it has been suggested that the total concentration of endogenous CaM within cells is significantly lower than that of its target sites (44). Thus, the concentration of endogenous CaM in myotubes might have been too low to have caused measurable mobilization of cardiac CT1, particularly because the cardiac C-terminal fragments appeared in the non-denaturing gel analysis to have limited solubility when not bound to CaM.
Determinants of CaM Binding—Previously, Pate et al. (12) examined CaM binding to synthetic, 21-residue peptides representing IQ regions of different voltage-gated calcium channels. Consistent with our results, they found that, when a Ca2+ chelator (1 mm EGTA) was present, CaM failed to form a complex with any of the peptides that could be detected with native gels. One apparent difference is that they observed the formation of a complex between CaM and the skeletal IQ-peptide in the presence of 0.2 mm Ca2+, whereas we failed to detect such a complex even in the presence of 2 mm Ca2+. However, CaM interactions appear to be governed by multiple, noncontiguous regions of the C-terminal (45). Thus, it may well be that CaM binding to small peptides differs from that to more extended constructs that we used. Whether or not this explanation is correct, it is important to note that the results of Pate et al. (12) are in overall agreement with ours. In particular, they found that skeletal IQ-peptide had a lower affinity for Ca2+-CaM than the cardiac peptide. Two additional results of Pate et al. (12) also support our view that the cardiac and skeletal isoforms differ significantly in affinity for CaM. First, CaM mutants in which both lobes lack Ca2+ binding form a stable complex with the cardiac IQ-peptide but fail to bind to the skeletal peptide. Second, peptides representing regions proximal to IQ (termed CB in Ref. 12) are able to competitively strip CaM from the skeletal IQ-peptide but not from the cardiac IQ-peptide.
In the crystal structure presented by Van Petegem et al. (21) for the CaM complex with the cardiac IQ-region, Tyr1657, Lys1662, and Lys1665 are part of the IQ domain α-helix interacting with CaM. All three residues intimately contact (distances <4 Å) the C-lobe of Ca2+-CaM, with Tyr1657 deeply buried in the C-lobe hydrophobic pockets. In the crystal structure presented by Fallon et al. (22), the polar interactions with CaM are somewhat (Lys1663) or greatly (Lys1665) reduced compared with those deduced by Van Petegem et al. (21). In both studies, however, Tyr1657 is part of the aromatic-hydrophobic interaction domain. Therefore, it may not be surprising that replacement in the cardiac CT1 of the aromatic Tyr1657 with the weakly basic histidine had the biggest effect on CaM binding of all the single amino exchanges that we examined. With respect to the discrepancy between Van Petegem et al. (21) and Fallon et al. (22) as to the involvement of Lys1662 and Lys1665 in the complex with CaM, perhaps the different target peptide lengths used in the two studies is responsible: the IQ-peptide used by Van Petegem et al. (21) extending 10 residues more toward the C terminus than that used by Fallon et al. (22).
Although our studies indicate that the IQ region plays a major role in the differential binding of CaM to the cardiac and skeletal C-terminals, studies from other labs have indicated roles in CaM binding for proximal and distal regions as well. For example, Lee et al. (46) identified a segment (termed CBD) distal to the IQ region of P/Q-type (Cav2.1) channels that binds CaM in a Ca2+-dependent manner. Our results suggest that there is no comparable region distal to the IQ motif in CaV1.1 because the absence of Ca2+-CaM binding was observed not just for CT1, but also for CT2 and CT3, which contain ∼50 and ∼150 residues, respectively, C-terminal to the IQ motif. Moreover, because cardiac CT2 and CT3 share the ability with cardiac CT1 to bind Ca2+-CaM strongly, it seems that regions distal to the cardiac IQ motif have no strong inhibitory effect on binding. Regions proximal to the IQ motif have also been implicated as contributors to CaM binding, including the regions designated A and C (12, 17), which are both present in CT1. Potentially, these regions may contribute to the residual CaM binding (Table 1) that we observed for cardiac CT1 in which the three divergent IQ motif residues were converted to skeletal (CT1card[Y1657H/K1662M/K1665Q]). However, the region proximal to the IQ motif of cardiac CT1 differs little from that of skeletal CT1 (Met1587-Thr1462, Arg1601-Lys1476, Leu1608-Phe1483, and Val1635-Ile1510), which did not detectably bind CaM. Thus, an alternative possibility is that the residual CaM binding to CT1card[Y1657H/K1662M/K1665Q] is because these three mutations weaken, but do not abolish, binding to the IQ motif. By essentially the same argument (the strong conservation of sequence proximal to the IQ motif), it seems likely that the reduced CaM binding to the reciprocally mutated skeletal CT1 construct, CT1skel[H1532Y/M1537K/Q1540K], compared with cardiac CT1 can be attributed to the significant differences in residues immediately distal to the IQ motif (Fig. 1). Structural studies of CaM binding to CT1skel[H1532Y/M1537K/Q1540K] and CT1card[Y1657H/K1662M/K1665Q] would help in better understanding the role of the regions preceding and following the IQ motif.
Calcium-dependent Inactivation—We found that the mutation Y1657H/K1662M completely eliminated calcium-dependent inactivation of full-length CaV1.2 (Fig. 6), but reduced CaM binding to cardiac CT1 by <60% (Table 1). One way to account for the much more profound effect of the mutation on calcium-dependent inactivation than on CaM binding is to suppose that CaM binding to intact CaV1.2 is reduced to a much greater extent than indicated by the experiments on the isolated C-terminal peptides. However, it is also important to note that CaM appears able to bind, with different affinities and various structural arrangements, to several target sequences within the CaV1.2 C-terminal (45). Furthermore, reorientations of pre-bound CaM upon binding of Ca2+ have been postulated in models for the coupling of the Ca2+ sensor to channel inactivation (23, 47, 48). Thus, the residual binding seen for CT1card[Y1657H/K1662M] may represent Ca2+-CaM complexed at sites that do not directly promote inactivation. Alternatively, the Y1657H/K1662M mutation may interfere with reorientations important for triggering calcium-dependent inactivation.
Recently, Stroffekova (49) reported the presence of calcium-dependent inactivation of the L-type Ca2+ current in normal myotubes, which endogenously express CaV1.1. However, it is important to note that the analysis of inactivation in normal myotubes is complicated by the very slow activation of CaV1.1, which would prevent the detection of “cardiac” calcium-dependent inactivation with kinetics like those of CaV1.2. In fact, the inactivation process described for normal myotubes is more than 10-fold slower than calcium-dependent inactivation of CaV1.2, making it uncertain how the two relate to one another. The inactivation in myotubes is suppressed by overexpression of mutant CaM lacking Ca2+ binding, and FRET measurements suggest an association between CaM and CaV1.1 (49). However, our results provide evidence against the idea that this association involves binding of CaM to the CaV1.1 C terminus. Indeed, the FRET measurements of Stroffekova (49) suggest that the N-lobe of CaM lies close (<10 nm) to the N terminus of CaV1.1, whereas FRET measurements of Erickson et al. (50) indicate that for CaV1.2, the N-lobe of CaM is positioned close to a portion of the C terminus just distal (residue 1671) to the IQ motif. Thus, the available data seem to indicate that rapid, cardiac-like calcium-dependent inactivation may not occur for CaV1.1.
Functional Implications—Our studies raise the obvious question: what functional adaptations that are important for skeletal muscle depend upon weak or absent CaM binding to the CaV1.1 C-terminal? One possibility is that cardiac-like, calcium-dependent inactivation of the skeletal L-type Ca2+ current would negatively affect muscle function. At least for brief stimuli, however, Ca2+ entry is not required for excitation-contraction coupling in skeletal muscle (51, 52). Furthermore, selective pressure on Ca2+ current does not appear to be a tenable idea because the residues within the CaV1.1 IQ motif that weaken CaM binding are present in the zebrafish isoform, despite the fact that this isoform conducts essentially no Ca2+ current (53).
A second possibility for why the CaV1.1 C terminus might lack strong CaM binding is that such binding could interfere with the interaction of this region with other proteins and these interactions could be important for excitation-contraction coupling. For example, Sencer et al. (39) have shown that CaV1.1 C-terminal regions that bind CaM are also able to bind directly to the type 1 ryanodine receptor (RyR1) and suggest that this interaction may stabilize contacts between CaV1.1 and RyR1. One could imagine that strong binding of CaM would interfere with this stabilizing interaction. Moreover, measurements of FRET (25) and streptavidin accessibility (54, 55) suggest that RyR1 may be closely apposed to the CaV1.1 C terminus. However, strong, skeletal-type excitation-contraction coupling is mediated by the chimeric construct “CSk3” (56, 57) in which the entire C terminus has the CaV1.2 sequence. Thus, the ability of the C-terminal of this construct to bind CaM strongly did not interfere with its ability to carry out excitation-contraction coupling.
A third possibility that could explain why it is important that the CaV1.1 C terminus lacks strong CaM binding is that calcium-dependent inactivation might cause CaV1.1 to enter a state refractory for excitation-contraction coupling, much as occurs for voltage-dependent inactivation. In particular, it is known that during prolonged depolarization CaV1.1 loses the ability to generate L-type Ca2+ current (58) and to elicit Ca2+ release via RyR1 (59), which is to say that both processes inactivate. Thus, it seems reasonable to propose that calcium-dependent inactivation could also inactivate not only L-type current but also excitation-contraction coupling. In principle, one could test this hypothesis by the functional analysis of dysgenic myotubes expressing a chimera in which the C terminus of CaV1.1 was replaced by that of CaV1.2. However, it appears that the absence of CaM binding is not the only adaptation in skeletal muscle to prevent rapid calcium-dependent inactivation. In particular, calcium-dependent inactivation is not present for the L-type calcium currents that result when CaV1.2 is expressed in dysgenic myotubes (41),5 raising the possibility that yet to be identified factors in skeletal muscle actively inhibit calcium-dependent inactivation in a channel that is otherwise capable of this process. An important goal for future research will be to identify these factors and to test the hypothesis that if calcium-dependent inactivation occurred in skeletal muscle, it would inhibit excitation-contraction coupling.
Acknowledgments
We greatly appreciate the help provided by Ulrike Fuhrmann.
This work was supported, in whole or in part, by National Institutes of Health Grant NS24444 (to K. G. B.). This work was also supported by grants from the Deutsche Forschungsgemeinschaft and Deutscher Akademischer Austauschdienst (DAAD) (to S. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: CaM, calmodulin; CT, C-terminal; FRET, fluorescence resonance energy transfer; RyR1, ryanodine receptor type 1.
J. Ohrtman and K. G. Beam, unpublished data.
References
- 1.Dascal, N., Snutch, T. P., Lubbert, H., Davidson, N., and Lester, H. A. (1986) Science 231 1147-1150 [DOI] [PubMed] [Google Scholar]
- 2.Imredy, J. P., and Yue, D. T. (1994) Neuron 12 1301-1318 [DOI] [PubMed] [Google Scholar]
- 3.Budde, T., Meuth, S., and Pape, H. C. (2002) Nat. Rev. Neurosci. 3 873-883 [DOI] [PubMed] [Google Scholar]
- 4.de Leon, M., Wang, Y., Jones, L., Perez-Reyes, E., Wei, X., Soong, T. W., Snutch, T. P., and Yue, D. T. (1995) Science 270 1502-1506 [DOI] [PubMed] [Google Scholar]
- 5.Soldatov, N. M., Zuhlke, R. D., Bouron, A., and Reuter, H. (1997) J. Biol. Chem. 272 3560-3566 [DOI] [PubMed] [Google Scholar]
- 6.Zuhlke, R. D., and Reuter, H. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 3287-3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brehm, P., and Eckert, R. (1978) Science 202 1203-1206 [DOI] [PubMed] [Google Scholar]
- 8.Kass, R. S., and Sanguinetti, M. C. (1984) J. Gen. Physiol. 84 705-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams, B., and Tanabe, T. (1997) J. Gen. Physiol. 110 379-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cens, T., Restituito, S., Rousset, M., and Charnet, P. (2005) in Voltagegated Calcium Channels (Zamponi, G. W., ed) pp. 95-112 Landes Bioscience, Georgetown, TX
- 11.Peterson, B. Z., DeMaria, C. D., Adelman, J. P., and Yue, D. T. (1999) Neuron 22 549-558 [DOI] [PubMed] [Google Scholar]
- 12.Pate, P., Mochca-Morales, J., Wu, Y., Zhang, J. Z., Rodney, G. G., Serysheva, I. I., Williams, B. Y., Anderson, M. E., and Hamilton, S. L. (2000) J. Biol. Chem. 275 39786-39792 [DOI] [PubMed] [Google Scholar]
- 13.Mouton, J., Feltz, A., and Maulet, Y. (2001) J. Biol. Chem. 276 22359-22367 [DOI] [PubMed] [Google Scholar]
- 14.Lee, A., Zhou, H., Scheuer, T., and Catterall, W. A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 16059-16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuhlke, R. D., Pitt, G. S., Deisseroth, K., Tsien, R. W., and Reuter, H. (1999) Nature 399 159-162 [DOI] [PubMed] [Google Scholar]
- 16.Alseikhan, B. A., DeMaria, C. D., Colecraft, H. M., and Yue, D. T. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 17185-17190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pitt, G. S., Zuhlke, R. D., Hudmon, A., Schulman, H., Reuter, H., and Tsien, R. W. (2001) J. Biol. Chem. 276 30794-30802 [DOI] [PubMed] [Google Scholar]
- 18.Rhoads, A. R., and Friedberg, F. (1997) FASEB J. 11 331-340 [DOI] [PubMed] [Google Scholar]
- 19.Zuhlke, R. D., Pitt, G. S., Tsien, R. W., and Reuter, H. (2000) J. Biol. Chem. 275 21121-21129 [DOI] [PubMed] [Google Scholar]
- 20.Tang, W., Halling, D. B., Black, D. J., Pate, P., Zhang, J. Z., Pedersen, S., Altschuld, R. A., and Hamilton, S. L. (2003) Biophys. J. 85 1538-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Petegem, F., Chatelain, F. C., and Minor, D. L., Jr. (2005) Nat. Struct. Mol. Biol. 12 1108-1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fallon, J. L., Halling, D. B., Hamilton, S. L., and Quiocho, F. A. (2005) Structure 13 1881-1886 [DOI] [PubMed] [Google Scholar]
- 23.Kim, J., Ghosh, S., Nunziato, D. A., and Pitt, G. S. (2004) Neuron 41 745-754 [DOI] [PubMed] [Google Scholar]
- 24.De Marco, V., Stier, G., Blandin, S., and de Marco, A. (2004) Biochem. Biophys. Res. Commun. 322 766-771 [DOI] [PubMed] [Google Scholar]
- 25.Papadopoulos, S., Leuranguer, V., Bannister, R. A., and Beam, K. G. (2004) J. Biol. Chem. 279 44046-44056 [DOI] [PubMed] [Google Scholar]
- 26.Grabner, M., Dirksen, R. T., and Beam, K. G. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 1903-1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen, A. W., and Daugherty, P. S. (2005) Nat. Biotechnol. 23 355-360 [DOI] [PubMed] [Google Scholar]
- 28.Bradford, M. M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 29.Laemmli, U. K. (1970) Nature 227 680-685 [DOI] [PubMed] [Google Scholar]
- 30.Knudson, C. M., Chaudhari, N., Sharp, A. H., Powell, J. A., Beam, K. G., and Campbell, K. P. (1989) J. Biol. Chem. 264 1345-1348 [PubMed] [Google Scholar]
- 31.Beam, K. G., and Franzini-Armstrong, C. (1997) Methods Cell Biol. 52 283-306 [DOI] [PubMed] [Google Scholar]
- 32.Wilkens, C. M., and Beam, K. G. (2003) J. Muscle Res. Cell. Motil. 24 99-109 [DOI] [PubMed] [Google Scholar]
- 33.Hamill, O. P., Marty, A., Neher, E., Sakmann, B., and Sigworth, F. J. (1981) Pflugers Arch. 391 85-100 [DOI] [PubMed] [Google Scholar]
- 34.Hulme, J. T., Konoki, K., Lin, T. W., Gritsenko, M. A., Camp, D. G., 2nd, Bigelow, D. J., and Catterall, W. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 5274-5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou, J., Olcese, R., Qin, N., Noceti, F., Birnbaumer, L., and Stefani, E. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 2301-2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernatchez, G., Talwar, D., and Parent, L. (1998) Biophys. J. 75 1727-1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romanin, C., Gamsjaeger, R., Kahr, H., Schaufler, D., Carlson, O., Abernethy, D. R., and Soldatov, N. M. (2000) FEBS Lett. 487 301-306 [DOI] [PubMed] [Google Scholar]
- 38.Erickson, M. G., Liang, H., Mori, M. X., and Yue, D. T. (2003) Neuron 39 97-107 [DOI] [PubMed] [Google Scholar]
- 39.Sencer, S., Papineni, R. V., Halling, D. B., Pate, P., Krol, J., Zhang, J. Z., and Hamilton, S. L. (2001) J. Biol. Chem. 276 38237-38241 [DOI] [PubMed] [Google Scholar]
- 40.Johnston, J. A., Ward, C. L., and Kopito, R. R. (1998) J. Cell Biol. 143 1883-1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mikami, A., Imoto, K., Tanabe, T., Niidome, T., Mori, Y., Takeshima, H., Narumiya, S., and Numa, S. (1989) Nature 340 230-233 [DOI] [PubMed] [Google Scholar]
- 42.Cavalié, A., Ochi, R., Pelzer, D., and Trautwein, W. (1983) Pflugers Arch. 398 284-297 [DOI] [PubMed] [Google Scholar]
- 43.Romanin, C., Karlsson, J. O., and Schindler, H. (1992) Pflugers Arch. 421 516-518 [DOI] [PubMed] [Google Scholar]
- 44.Persechini, A., and Stemmer, P. M. (2002) Trends Cardiovasc. Med. 12 32-37 [DOI] [PubMed] [Google Scholar]
- 45.Halling, D. B., Aracena-Parks, P., and Hamilton, S. L. (2006) Sci. STKE 318 er1. [DOI] [PubMed] [Google Scholar]
- 46.Lee, A., Wong, S. T., Gallagher, D., Li, B., Storm, D. R., Scheuer, T., and Catterall, W. A. (1999) Nature 399 155-159 [DOI] [PubMed] [Google Scholar]
- 47.Soldatov, N. M. (2003) Trends Pharmacol. Sci. 24 167-171 [DOI] [PubMed] [Google Scholar]
- 48.Cens, T., Rousset, M., Leyris, J. P., Fesquet, P., and Charnet, P. (2006) Prog. Biophys. Mol. Biol. 90 104-117 [DOI] [PubMed] [Google Scholar]
- 49.Stroffekova, K. (2008) Pflugers Arch. 455 873-884 [DOI] [PubMed] [Google Scholar]
- 50.Erickson, M. G., Alseikhan, B. A., Peterson, B. Z., and Yue, D. T. (2001) Neuron 31 973-985 [DOI] [PubMed] [Google Scholar]
- 51.Armstrong, C. M., Bezanilla, F. M., and Horowicz, P. (1972) Biochim. Biophys. Acta 267 605-608 [DOI] [PubMed] [Google Scholar]
- 52.Chiarandini, D. J., Sanchez, J. A., and Stefani, E. (1980) J. Physiol. 303 153-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schredelseker, J., Di Biase, V., Obermair, G. J., Felder, E. T., Flucher, B. E., Franzini-Armstrong, C., and Grabner, M. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 17219-17224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorenzon, N. M., Haarmann, C. S., Norris, E. E., Papadopoulos, S., and Beam, K. G. (2004) J. Biol. Chem. 279 44057-44064 [DOI] [PubMed] [Google Scholar]
- 55.Lorenzon, N. M., and Beam, K. G. (2007) J. Gen. Physiol. 130 379-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanabe, T., Beam, K. G., Adams, B. A., Niidome, T., and Numa, S. (1990) Nature 346 567-569 [DOI] [PubMed] [Google Scholar]
- 57.Nakai, J., Tanabe, T., Konno, T., Adams, B., and Beam, K. G. (1998) J. Biol. Chem. 273 24983-24986 [DOI] [PubMed] [Google Scholar]
- 58.Beam, K. G., and Knudson, C. M. (1988) J. Gen. Physiol. 91 781-798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beam, K. G., and Horowicz, P. (2004) in Myology (Engel, A. G., and Franzini-Armstrong, C., eds) 3rd Ed., pp. 257-280, McGraw-Hill, New York