

Abstract
Redox control in the mitochondrion is essential for the proper functioning of this organelle. Disruption of mitochondrial redox processes contributes to a host of human disorders, including cancer, neurodegenerative diseases, and aging. To better characterize redox control pathways in this organelle, we have targeted a green fluorescent protein-based redox sensor to the intermembrane space (IMS) and matrix of yeast mitochondria. This approach allows us to separately monitor the redox state of the matrix and the IMS, providing a more detailed picture of redox processes in these two compartments. To verify that the sensors respond to localized glutathione (GSH) redox changes, we have genetically manipulated the subcellular redox state using oxidized GSH (GSSG) reductase localization mutants. These studies indicate that redox control in the cytosol and matrix are maintained separately by cytosolic and mitochondrial isoforms of GSSG reductase. Our studies also demonstrate that the mitochondrial IMS is considerably more oxidizing than the cytosol and mitochondrial matrix and is not directly influenced by endogenous GSSG reductase activity. These redox measurements are used to predict the oxidation state of thiol-containing proteins that are imported into the IMS.
Maintenance of the thiol-disulfide balance in cells is critical for the proper functioning of numerous enzymes and proteins with functionally important cysteine residues. The cellular redox balance can be disrupted by unregulated production of reactive oxygen species (ROS)2 that interfere in redox signaling pathways and oxidatively damage DNA, proteins, and lipids (1). To control the cellular redox environment, cells contain two primary redox regulatory systems that utilize thiol-disulfide redox chemistry: the glutathione (GSH)/glutathione disulfide (GSSG) redox couple and the reduced/oxidized thioredoxin redox couple (1, 2). The tripeptide glutathione (γ-glutamylcysteinylglycine) and the small protein thioredoxin can serve as reductants themselves or as cofactors for anti-oxidant enzymes (3). Glutathione is considered the primary determinant of the cellular redox environment, because it has a relatively low redox potential (-240 mV at pH 7.0) and a high intracellular abundance (1–13 mm) (4).
Measurements of GSH:GSSG levels in subcellular compartments demonstrate that individual organelles have different redox requirements. The endoplasmic reticulum maintains a relatively oxidizing environment (-170 to -185 mV at pH 7.0, or a GSH:GSSG ratio of 1:1 to 3:1) (5), whereas the cytosol is quite reducing in comparison (-290 mV at pH 7.0, or a GSH: GSSG ratio of 3300:1) (6). GSH:GSSG measurements in isolated mitochondria indicate a redox potential of -250 mV to -280 mV at pH 7.8 or GSH:GSSG ratios of 20:1 to 40:1 (7–10). However, measuring the GSH:GSSG redox state in isolated mitochondria has several drawbacks. First, GSH:GSSG levels in the matrix and the intermembrane space (IMS) cannot be measured separately, because the IMS is quite small (≤5% of the total mitochondria volume), making it difficult to effectively isolate IMS GSH:GSSG from matrix pools. Second, GSH may be oxidized during cell lysis and fractionation steps creating an artificially low GSH:GSSG ratio. Finally, metabolites may be lost or exchanged during the mitochondrial isolation procedure thereby altering the physiology and redox state of the organelle.
Nevertheless, defining redox control in the IMS is critical given the various redox-dependent pathways in this compartment, including apoptotic signaling (11, 12), assembly of respiratory chain components (13), anti-oxidant activation (14), and protein import (15). It is not known if the redox state of this compartment is relatively oxidizing or reducing in comparison to the mitochondrial matrix and cytosol. On the one hand, this compartment is phylogenetically linked to the oxidizing periplasm of bacteria (16). Furthermore, a substantial number of IMS proteins have functionally essential disulfide bonds (17, 18). On the other hand, porin channels in the mitochondrial outer membrane presumably allow free exchange of GSH and GSSG between the IMS and cytosol (15, 19), suggesting that the GSH:GSSG redox state in the IMS is similar to the reducing cytosol.
An in vivo method for measuring the subcellular redox state of GSH:GSSG is an effective approach to address redox control in individual compartments. Østergaard and coworkers (6) have developed a genetically encoded, cytosolic redox sensor based on the yellow variant of green fluorescent protein (GFP) called redox-sensitive YFP (rxYFP). GFP and its derivatives provide ideal scaffolds for creating in vivo sensors due to their protease resistance and high stability in a broad range of pH and buffer conditions (20). The rxYFP protein in particular can be used to measure the redox potential in live cells via formation of an engineered disulfide bond that perturbs the local chromophore environment without significantly altering the overall β-can fold (21). The relative ratio of oxidized to reduced rxYFP can also be assessed via non-reducing SDS-PAGE in which the two forms have different electrophoretic mobilities. Østergaard and coworkers (6) have shown both in vivo and in vitro that the cysteines in rxYFP specifically equilibrate with GSH and GSSG via rapid disulfide exchange reactions with the cytosolic glutaredoxins (GRXs). In contrast, the two cytosolic thioredoxins are unable to undergo thiol-disulfide exchange with this sensor. The ratio of oxidized to reduced rxYFP measured in the cell can be used to generate an in vivo readout of the GSH:GSSG redox state. Their measurements indicated that the ratio of GSH: GSSG in the eukaryotic cytosol was 3300:1, which corresponds to a redox potential of -289 mV (6). This value is considerably more reducing than whole cell redox measurements (-221 to -236 mV) (5), highlighting the importance of examining subcellular compartments separately.
In this study we have modified rxYFP for expression in the mitochondrial IMS and matrix of the yeast Saccharomyces cerevisiae to compare redox differences between these compartments and the cytosol. We have demonstrated exclusive targeting of IMS- and matrix-rxYFP to these compartments and have confirmed their dynamic response to an exogenous oxidant and reductant. We have measured the IMS redox potential, demonstrating that this compartment is considerably more oxidizing than the cytosol or matrix. Furthermore, we have manipulated the subcellular redox potential using GSSG reductase mutants to verify that the sensors respond to specific intracellular redox changes that are localized to subcellular compartments. Overall, our data suggest that redox control is independently regulated within these individual compartments in the cell.
EXPERIMENTAL PROCEDURES
Yeast Strains, Media, and Growth Conditions—S. cerevisiae strains used in this study were BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and BY4741 glr1Δ::kanMX4 obtained from Research Genetics. Yeast transformations were performed by the lithium acetate procedure (22). Strains were maintained at 30 °C on either enriched yeast extract-peptone-based medium supplemented with 2% glucose (YPD) or synthetic complete medium (SC) supplemented with 2% glucose or 2% galactose and the appropriate amino acids.
Plasmids—The plasmid pJH208 expressing cytosol-rxYFP was constructed by digesting the rxYFP yeast expression plasmid pHOJ150 (6) with NotI and SacII. The NotI-SacII-digested fragment containing the PGK1 promoter, the coding sequence of rxYFP, and the TDH3 terminator was then inserted into the URA3-CEN vector pRS316 (23).
The IMS-rxYFP plasmid pJH200 was constructed as follows. An NdeI site downstream of the rxYFP coding sequence in pHOJ150 was removed by site-directed mutagenesis forming pJH300, which leaves one NdeI site at the start codon for rxYFP. The mitochondrial targeting sequence of S. cerevisiae cytochrome b2 (Cyb2) (codons for amino acids 1–88) was amplified by PCR introducing an NheI site and an NdeI site at the N and C terminus, respectively. The PCR fragment was inserted in-frame at the N terminus of rxYFP at the SpeI and NdeI sites of pHOJ150, forming pJH200.
The matrix-rxYFP plasmid pLD207 was constructed by amplifying the promoter region and mitochondrial targeting signal (codons for amino acids 1–25) of cytochrome oxidase subunit IV (COX4) with primers that introduced NotI site and NdeI sites at the 5′ and 3′ ends, respectively. The PCR fragment was inserted in-frame at the N terminus of rxYFP at the NotI and NdeI sites of pJH300. The COX4 promoter was later replaced with the manganese superoxide dismutase (SOD2) promoter by creating a SpeI site just upstream of the Cox4 mitochondrial targeting signal. The COX4 promoter was removed by digestion with NotI and SpeI and a PCR fragment containing SOD2 promoter was inserted in its place. This plasmid was then digested with NotI and SacII, and the fragment containing the SOD2 promoter, the coding sequence of COX4-rxYFP, and the TDH3 terminator was inserted into the URA3-CEN vector pRS316, yielding pLD207.
The GLR1-expressing plasmids pJH201 (WT Glr1), pJH202 (M17L Glr1), and pJH203 (M1L Glr1) were created by inserting the GLR1 gene sequence from the plasmids pCO113 (WT), pCO114 (M17L), or pCO116 (M1L) (9) into the SacI and XhoI sites of the HIS3-CEN vector pRS413 (23). The sequence integrity of all plasmids was verified by double-stranded DNA sequencing (USC Environmental Genomics Core Facility).
Subcellular Fractionation—Yeast cells were grown aerobically to mid-log phase in selecting SC medium with 2% galactose. Mitochondrial and post-mitochondrial supernatant (PMS) fractions were obtained as previously described by converting cells to spheroplasts followed by gentle lysis by Dounce homogenization and differential centrifugation (24). Mitochondrial intermembrane and mitoplast fractions were prepared by osmotic shock as previously described (25). Mitoplasts and IMS fractions were subsequently treated with 50 μg/ml proteinase K in the absence or presence of 0.2% Triton X-100.
Immunoblotting Techniques—Yeast extracts were subjected to electrophoresis on Tris-glycine gels (Invitrogen) and analyzed by Western blotting using an anti-rxYFP antibody (kind gift of J. Winther) or an anti-GFP antibody (Invitrogen) and a secondary anti-rabbit IgG (IRDye, LI-COR Lincoln, NE). PMS fractions were monitored by anti-3-phosphoglycerate kinase (PGK1) antibodies (Invitrogen). Mitochondrial fractions were monitored by using antibodies directed against Cyb2 in the IMS, mitochondrial processing protease Mas2 in the matrix (kind gifts of R. Jensen), or mitochondrial NADH kinase Pos5 in the matrix (26). Proteins were analyzed by Western blot using an Odyssey Infrared Imaging System (LI-COR). Protein concentrations were determined using the Bradford method (Bio-Rad) with bovine serum albumin as the calibration standard.
Fluorescence Microscopy—The BY4741 parental strain transformed with pJH208 (cytosol-rxYFP), pJH200 (IMS-rxYFP), or pLD207 (matrix-rxYFP) was grown aerobically to mid-log phase in selecting SC medium containing 2% galactose. Cells were incubated with 2.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) for 30 min to stain mitochondrial DNA. Live cells were examined with a Zeiss LSM 510 META Confocal Scanning Laser Microscope (Instrumentation Resource Facility at the University of South Carolina School of Medicine).
Redox Western—Redox Western blot analysis of rxYFP was adapted from previous methods (6). Briefly, cells were grown in selecting SC medium to mid-log phase. In some experiments, cells were treated with 180 μm 4,4′-dithiodipyridine (4-DPS) (Sigma) or 50 mm dithiothreitol (DTT) (Sigma) and incubated at 30 °C for an additional 20 min. 4-DPS and DTT are both membrane-permeable. Cell cultures were acid-quenched with trichloroacetic acid (Sigma) (15% (w/v) final concentration) at 4 °C for 20 min. Five A600 units of cells were harvested by centrifugation and resuspended in 1 ml of 10% trichloroacetic acid. Following glass bead lysis, the lysed cells were transferred to a new tube and pelleted by centrifugation. The pellet was resuspended in 500 μl of 1X non-reducing SDS sample buffer containing 40 mm N-ethylmaleimide (Sigma). Following a 10-min incubation at room temperature, the proteins were separated on a 16% Tris-glycine gel (Invitrogen). Reduced and oxidized forms of rxYFP were analyzed by quantitative immunoblot using an Odyssey Infrared Imaging System (LI-COR).
Redox Potential Calculations—The rxYFP sensor equilibrates with GSH:GSSG pools according to the following reaction (Scheme 1) (6).
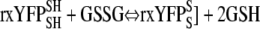 |
SCHEME 1 |
The ratio of oxidized to reduced rxYFP and the standard reduction potentials of rxYFP and GSH were inserted into the Nernst equation (Equation 1) to estimate the oxidation state of GSH: GSSG.
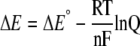 |
(Eq. 1) |
In this equation, R is the gas constant (8.314 J K-1 mol-1), T is the temperature in K, n is the number of electrons, and F is Faraday's constant (96,485 C mol-1). At 30 °C, for the reaction shown in Scheme 1, Equation 1 can be rewritten as Equation 2,
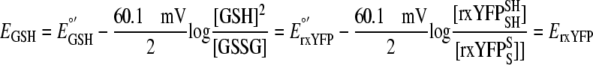 |
(Eq. 2) |
According to Equation 2,
ErxYFP is dependent on the ratio of reduced:oxidized rxYFP
but not the absolute concentration, [rxYFP]. However, estimation of the
GSH:GSSG ratio from ErxYFP requires the absolute
concentrations [GSH] or [GSSG]. At pH 7.0, the standard reduction potential of
rxYFP (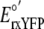 ) is -265 mV
(6) and the standard reduction
potential of GSH (
) is -265 mV
(6) and the standard reduction
potential of GSH (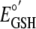 )
is -240 mV (4). The reduction
potentials of rxYFP and GSH at different pH values were calculated using the
expression shown in Equation 3
(4),
)
is -240 mV (4). The reduction
potentials of rxYFP and GSH at different pH values were calculated using the
expression shown in Equation 3
(4),
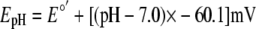 |
(Eq. 3) |
In this expression, E°′ is the standard reduction potential of GSH or rxYFP at pH 7.0.
GSH/GSSG Assays—Total glutathione (GSH + GSSG) and oxidized glutathione (GSSG) in PMS and mitochondrial extracts were measured by the 5,5′-dithiobis(2-nitrobenzoic acid)-GSSG reductase cycling assay as previously described (9).
RESULTS
Targeting rxYFP to the Mitochondrial Matrix and Intermembrane Space—Mitochondria-targeted versions of rxYFP were created by fusing the mitochondrial targeting signals from native mitochondrial proteins to the N terminus of rxYFP (Fig. 1A). The mitochondrial targeting signal of Cox4 was used to generate matrix-rxYFP, whereas the mitochondrial targeting signal of Cyb2 was used to make IMS-rxYFP. Both sequences have been successfully employed in the past for targeting non-native proteins to the matrix or intermembrane space (27–29). The matrix targeting sequence encodes an amphipathic helix that is recognized by the mitochondrial import machinery and subsequently removed by matrix proteases (Fig. 1B) (30). The IMS targeting sequence includes an additional hydrophobic sorting domain, which is cleaved by an inner membrane protease (Fig. 1C) (31).
FIGURE 1.

Mitochondria-targeted constructs of rxYFP. A, the gray box represents the rxYFP protein, the black coil represents the amphipathic helix required for matrix targeting, and the black box represents the hydrophobic sorting domain required for IMS targeting. The N termini of native mitochondrial proteins (Cox4 and Cyb2) were fused in-frame to rxYFP. B and C, schematics depicting import into the mitochondrial matrix (B) and IMS (C) via N-terminal targeting sequences. The targeting signals are cleaved during import by matrix and/or inner membrane (IM) proteases and the protein folds into its native conformation.
To confirm the correct localization of the rxYFP constructs, we conducted a Western blot analysis of crude mitochondria and largely cytosolic (PMS) fractions from WT strains transformed with cytosol-, matrix-, or IMS-rxYFP. As shown in Fig. 2A, cytosol-rxYFP is localized to the cytosol as previously determined (6). In contrast, matrix-rxYFP and IMS-rxYFP are localized to mitochondria (Fig. 2, B and C, left panels). The molecular weights of the rxYFP bands in Fig. 2 (B and C) are consistent with the mature, processed forms of both sensors. Mitochondria were further fractionated into intermembrane space and mitoplasts, revealing that matrix-rxYFP is correctly localized to the matrix (Fig. 2B, right panel), whereas IMS-rxYFP is located in the soluble IMS fraction (Fig. 2C, right panel, lane 5). However, a portion of IMS-rxYFP is also found associated with mitoplasts (Fig. 2C, lane 2). To determine whether IMS-rxYFP in this fraction is located on the outside (IMS side) or inside (matrix side) of the mitoplast membrane, proteinase K was added to intact mitoplasts to digest exposed (IMS side) proteins. IMS-rxYFP was largely degraded by this protease (Fig. 2C, lane 3) in a similar manner to soluble IMS-rxYFP (Fig. 2C, lane 6), whereas the control matrix protein Pos5 (26) was unaffected. The Pos5 matrix control is only digested (indicated by a change in molecular weight) upon addition of Triton X-100, which disrupts the mitoplast membrane allowing access to matrix proteins (Fig. 2C, lane 4). In contrast, proteolysis of mitoplast-associated IMS-rxYFP is similar with and without the addition of Triton X-100 (compare Fig. 2C, lanes 3 and 4), indicating that it is located on the outside of the mitochondrial inner membrane in the IMS compartment. We note that, unlike the Cyb2 control, rxYFP in the soluble IMS fraction is only partially digested (Fig. 2C, lane 6), even though all proteins in this fraction are soluble and therefore accessible to proteinase K. This observation may be explained by the fact that GFP-based proteins have an unusually stable core structure that is resistant to complete digestion by proteases (20).
FIGURE 2.

Subcellular localization of cytosol- (A), matrix- (B), and IMS-rxYFP (C). WT yeast cells (BY4741) expressing cytosol-, matrix-, or IMS-rxYFP were grown to mid-log phase in SC galactose media. Cells were lysed and fractionated, and fractions were analyzed by SDS-PAGE and immunoblotting using antibodies directed against rxYFP, PGK1 (cytosol marker), Pos5 or Mas2 (mitochondrial matrix markers), or Cyb2 (mitochondrial IMS marker). In the left panel of A–C, 75 μg of total cell protein (Total) was fractionated into post-mitochondrial supernatant (PMS) and mitochondria (Mito), and the entire amount of each fraction was analyzed. In the right panel of B, mitochondria (15 μg of protein) from cells expressing matrix-rxYFP were further fractionated into IMS and mitoplast components, and the entire amount of each fraction was analyzed. In the right panel of C, mitochondria (15 μg of protein) from cells expressing IMS-rxYFP were fractionated as in B. Mitoplast and IMS fractions were further treated with proteinase K and/or Triton X-100 as indicated. rxYFP-expressing plasmids utilized include: pJH208 (cytosol), pLD207 (matrix), and pJH200 (IMS).
Localization and fluorescence signal of the rxYFP sensors were further verified by fluorescence microscopy. Live yeast cells expressing cytosol-, matrix-, or IMS-rxYFP were stained with DAPI for co-localization experiments. DAPI staining for DNA highlights both mitochondria (string-like structures) and the nucleus (large, rounded structure) as shown in Fig. 3. Matrix- and IMS-rxYFP specifically co-localized with DAPI staining of the mitochondrial, but not nuclear DNA, indicating that these sensors are exclusively localized to the mitochondria and are correctly folded with the expected fluorescence properties.
FIGURE 3.

Differential interference contrast (DIC) and fluorescence microscopy of WT yeast cells (BY4741) expressing cytosol-, matrix, or IMS-rxYFP. Cells were grown to mid-log phase in SC galactose media and incubated with DAPI for 30 min to stain DNA. Live cells were examined with Zeiss LSM 510 META confocal scanning laser microscope at a magnification of 605×. Merge = merged images of DAPI and YFP fluorescence with white areas indicating overlap. Plasmids and strains are the same as in Fig. 2.
Matrix- and IMS-rxYFP Register Dynamic Subcellular Redox Changes—To test the reactivity and response of the targeted rxYFP sensors, a strong oxidant or reductant was added to cells expressing cytosol-, matrix-, or IMS-rxYFP. The redox state of rxYFP can be monitored by redox Western blot or fluorescence spectroscopy as described for cytosol-rxYFP (6). Both of these techniques avoid the artificial oxidation problems and lengthy subcellular fractionation steps encountered by conventional methods for measuring subcellular redox potential.
The redox Western blots shown in Fig. 4 indicate that the mitochondrial rxYFP constructs are responsive to redox changes generated by exogenous reagents, as previously demonstrated for cytosol-rxYFP (6). Addition of the thiol-oxidant 4-DPS or the disulfide reductant DTT shifts all three sensors toward the oxidized state (Fig. 4A, lane 2) or reduced state (Fig. 4A, lane 3), respectively. Fluorescence measurements were also performed using live cells expressing cytosol-, matrix-, and IMS-rxYFP. The fluorescence response of cells expressing cytosol-rxYFP upon addition of DTT or 4-DPS was similar to published reports (6) (data not shown). However, fluorometer measurements of whole cells expressing matrix- or IMS-rxYFP were more difficult to obtain over the background autofluorescence. This difficulty may be explained by the relatively small mitochondrial volume of these cells, which constitutes only ∼3% of the total cell volume under these growth conditions (32).
FIGURE 4.

rxYFP redox response to an exogenous oxidant or reductant. WT yeast cells expressing cytosol-, matrix-, or IMS-rxYFP were grown to mid-log phase in SC glucose media. Cells were treated with 4-DPS or DTT as described under “Experimental Procedures.” A, redox Western blot of the samples separated by non-reducing SDS-PAGE and immunoblotted with anti-GFP antibodies. B, reduced (red) and oxidized (ox) forms of rxYFP were quantified using an Odyssey Infrared Imaging System. The reported values are the mean of three to four independent experiments. Error bars are the means ± S.D. Plasmids and strains are the same as in Fig. 2.
The IMS Redox State Is More Oxidizing Than the Matrix or Cytosol—Using the differently targeted versions of rxYFP, we compared the redox state in the cytosol, mitochondrial matrix, and mitochondrial IMS. As shown in Fig. 5 and Table 1, cytosol-rxYFP is ∼16% oxidized under steady-state conditions, which is similar to published results (6), whereas matrix-rxYFP is ∼38% oxidized. Inserting these values into Equation 2 and assuming an intracellular [GSH] of 13 mm (6), we estimate that the GSH: GSSG ratio in the cytosol and matrix are ∼3000:1 and ∼900:1, respectively (Table 1). Interestingly, IMS-rxYFP is significantly more oxidized (∼68%) under steady-state conditions compared with both cytosol- and matrix-rxYFP. Assuming that the GSH concentration in this compartment is similar to the cytosol (i.e. 13 mm) (6), this value corresponds to a GSH:GSSG ratio of ∼250:1 (see “Discussion”).
FIGURE 5.

rxYFP redox response in WT and glr1Δ cells. WT (BY4741) and glr1Δ (BY4741 glr1Δ) yeast cells expressing cytosol-, matrix-, or IMS-rxYFP were grown to mid-log phase in SC glucose media. Redox Western blot was performed as described in Fig. 4.
TABLE 1.
GSH:GSSG ratios and redox potential measurements in the cytosol, mitochondrial matrix, and mitochondrial IMS of yeast cells
|
Strains
|
Cytosol
|
Matrix
|
IMS
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Oxidized rxYFPa | GSH:GSSGb | Ec | Oxidized rxYFP | GSH:GSSG | Ed | Oxidized rxYFP | GSH:GSSG | Ec | |
| % | mV | % | mV | % | mV | ||||
| WT | 16 ± 5 | ∼3000:1 | –286 ± 5 | 38 ± 9 | ∼900:1 | –296 ± 5 | 68 ± 4 | ∼250:1 | –255 ± 3 |
| glr1Δ | 74 ± 2 | ∼200:1 | –252 ± 2 | 84 ± 3 | ∼100:1 | –267 ± 3 | 62 ± 10 | ∼300:1 | –259 ± 6 |
The reported values of percent oxidized rxYFP are the mean ± S.D. for 3–8 independent experiments
GSH:GSSG ratios were calculated using intracellular [GSH] = 13 mm (6)
Cytosolic and IMS redox potentials were calculated from Equation 2 using ErxYFP° = –265 mV at pH 7.0 (6)
Matrix redox potentials were calculated from Equation 2 using ErxYFP° = –289 mV at pH 7.4
The in vivo redox state of rxYFP was also used to generate redox
potentials for each subcellular compartment using published cytosol and matrix
pH values (Table 1). Yeast
cytosolic pH measurements for glucose-grown cells vary from 6.7 to 7.3
(33–36),
so 7.0 was used for our calculations. The redox potential of rxYFP at pH 7.0
(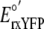 = -265 mV)
(6) was inserted into
equation 2 along with the ratio
of reduced-to-oxidized sensor to calculate the cytosolic redox potential.
Similarly, the redox potential of rxYFP at pH 7.4 (E7.4 =
= -265 mV)
(6) was inserted into
equation 2 along with the ratio
of reduced-to-oxidized sensor to calculate the cytosolic redox potential.
Similarly, the redox potential of rxYFP at pH 7.4 (E7.4 =
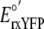 -24 mV according
to Equation 3) was used to
calculate the matrix redox potential, because this matrix pH value was
reported for yeast cells grown in glucose
(36). Using these values, the
cytosolic redox potential corresponds to -286 mV, whereas the matrix redox
potential is -296 mV (Table 1).
Studies in mammalian cells suggest that the IMS pH is typically 0–0.7 pH
units lower than cytosolic pH
(37,
38). By using pH 7.0 as a
conservative estimate, the IMS redox potential calculated with IMS-rxYFP is
-255 mV (Table 1). If the IMS
pH is lower than 7.0, the calculated E value will increase by 6.0 mV
for every 0.1 unit of decrease in pH (see
Equation 3). Therefore it is
possible that the IMS redox state is even more oxidizing than our estimate at
pH 7.0. In any case, these values are considerably more oxidizing than both
the cytosol and mitochondrial matrix.
-24 mV according
to Equation 3) was used to
calculate the matrix redox potential, because this matrix pH value was
reported for yeast cells grown in glucose
(36). Using these values, the
cytosolic redox potential corresponds to -286 mV, whereas the matrix redox
potential is -296 mV (Table 1).
Studies in mammalian cells suggest that the IMS pH is typically 0–0.7 pH
units lower than cytosolic pH
(37,
38). By using pH 7.0 as a
conservative estimate, the IMS redox potential calculated with IMS-rxYFP is
-255 mV (Table 1). If the IMS
pH is lower than 7.0, the calculated E value will increase by 6.0 mV
for every 0.1 unit of decrease in pH (see
Equation 3). Therefore it is
possible that the IMS redox state is even more oxidizing than our estimate at
pH 7.0. In any case, these values are considerably more oxidizing than both
the cytosol and mitochondrial matrix.
Deletion of GLR1 Alters Redox Status in the Cytosol and Matrix but Not the IMS—To examine how the GSH:GSSG ratio influences redox status within submitochondrial compartments, we tested the sensors in glr1Δ mutant cells that lack the GSSG reductase gene (GLR1). Because this enzyme is responsible for regenerating GSH from GSSG, deletion of this gene creates a higher than normal intracellular GSSG:GSH ratio (6, 9, 39). As shown in Fig. 5 and Table 1, cytosol-rxYFP undergoes dramatic redox changes (∼74% oxidized) in glr1Δ mutant cells compared with WT cells (∼16% oxidized), as previously demonstrated (6). Likewise, the matrix-rxYFP sensor shifts from ∼38% oxidized to ∼84% oxidized in WT versus glr1Δ strains. Glr1 is localized to the cytosol and mitochondrial matrix (9), therefore deletion of this gene should have direct consequences for both of these compartments. In contrast, there is very little change in the percent oxidized IMS-rxYFP upon deletion of the GLR1 gene (Fig. 5 and Table 1). We have previously shown that Glr1 is not localized to the IMS (9), and these results support that conclusion. Taken together, these experiments suggest that redox control in the IMS is maintained separately from the cytosol and mitochondrial matrix.
Localized rxYFP Sensors Are Responsive to Redox Changes in Distinct Subcellular Compartments—We next determined whether the targeted rxYFP sensors are responsive to Glr1-induced redox changes that specifically alter either cytosolic or mitochondrial matrix redox status. The GLR1 gene encodes both a cytosolic and mitochondrial form of Glr1 via two different start codons in the mRNA sequence. Translation from the first start codon (encoding M1) generates the long mitochondrial form with a mitochondrial targeting signal, while translation from the second start codon (encoding M17) generates a shorter cytosolic form that lacks this signal peptide (9). Mutation of the first Met to a Leu (M1L) results in exclusive expression of cytosolic Glr1, while mutation of Met-17 to a Leu (M17L) primarily generates the mitochondrial form. However, mitochondrial Glr1 is somewhat overexpressed in the M17L mutant compared with WT, resulting in incomplete mitochondrial import. Consequently, some residual Glr1 is present in the cytosol in M17L Glr1 strains (9).
To test whether rxYFP is responsive to redox changes within distinct subcellular compartments, matrix-rxYFP, IMS-rxYFP, or cytosol-rxYFP was expressed in glr1Δ yeast strains harboring plasmid-encoded WT Glr1, no Glr1 (vector), cytosolic Glr1 (M1L), or mitochondrial Glr1 (M17L). When Glr1 is exclusively localized to the cytosol (M1L), the oxidation state of the cytosolic redox sensor is similar to cells expressing WT Glr1 (Fig. 6A, compare lanes 1 and 3), whereas the matrix sensor measurements are similar to glr1Δ strains (compare lanes 2 and 3). This result confirms that the matrix and cytosolic sensors are registering redox changes in distinct compartments. Accordingly, expression of mitochondrial Glr1 (M17L) shifts the matrix sensor equilibrium back toward the reduced form (lane 4), which is similar to cells expressing WT Glr1 (lane 1). However, the cytosolic sensor is also more reduced in this strain (lane 4) compared with glr1Δ with the vector control (lane 2). This observation can be explained by the fact that the M17L Glr1 mutant exhibits some cytosolic localization due to inefficient mitochondrial import (9). This small cytosolic amount may be sufficient to maintain cytosolic GSH:GSSG in a more reduced form compared with the strain with no Glr1 expressed.
FIGURE 6.

Cytosolic and mitochondrial GSH:GSSG pools are maintained separately. The glr1Δ strain was doubly transformed with an rxYFP expression plasmid (pJH208 (cytosol-rxYFP), pLD207 (matrix-rxYFP), or pJH200 (IMS-rxYFP)) and a Glr1 expression plasmid (pJH201 (WT Glr1), pRS413 (vector control), pJH203 (M1L Glr1), or pJH202 (M17L Glr1)). Cells were grown to mid-log phase in selecting SC glucose media. A, redox Western blot and quantification (B) were performed as described in Fig. 4. C, the percent of oxidized glutathione (percent GSS/(GSH + GSS)) was calculated from total GSH and GSSG levels in each extract. GSS = 2XGSSG. For B and C, the reported values are the mean of three independent experiments. Error bars are the means ± S.D.
To confirm the rxYFP sensor responses, we also measured GSH and GSSG levels in cytosolic and whole mitochondrial extracts using conventional subfractionation methods. As pointed out in the introduction, this technique has several drawbacks, but it provides a rough estimation for comparison to the sensor measurements. The same yeast strains shown in Fig. 6 (A and B) were subjected to lysis and subcellular fractionation to generate cytosolic and whole mitochondrial extracts. These extracts were assayed for GSH and GSSG by enzyme cycling and spectrophotometric analysis (9). We cannot assess matrix and IMS GSH:GSSG separately with this method, so the data shown represent whole mitochondria. However, because the matrix volume is much larger than the IMS, these values are primarily a reflection of the matrix redox state. As shown in Fig. 6C, direct measurement of GSH:GSSG showed a close correlation with the cytosol and matrix sensor readouts. The percent oxidized GSSG was high in both the cytosol and mitochondria in glr1Δ strains with no Glr1 expressed, whereas expression of cytosolic Glr1 exclusively lowered cytosolic GSSG, but not mitochondrial matrix GSSG. Expression of primarily mitochondrial matrix Glr1 (which also has some cytosolic Glr1) reduced mitochondrial matrix GSSG as well as cytosolic GSSG to some extent. However, the percent oxidized GSSG in the cytosol was not quite at WT levels in this M17L strain. Likewise, our cytosolic sensor was slightly more oxidized in the M17L strain (Fig. 6B), indicating that it is sensitive to this small cytosolic redox potential difference between WT and M17L Glr1 strains. Overall, these results confirm that cytosol- and matrix-rxYFP are functional and provide localized readouts of the subcellular redox potential.
The IMS sensor readout in glr1Δ mutants was quite different from both the cytosol and matrix. As seen in Figs. 5 and 6, the redox state of IMS-rxYFP did not change dramatically in a glr1Δ strain. Accordingly, expression of WT or cytosolic Glr1 (M1L) in a glr1Δ strain did not significantly alter the redox state of IMS-rxYFP. However, we observed a slight decrease in the percent oxidized IMS-rxYFP upon overexpression of matrix Glr1 (M17L) compared with cytosolic Glr1. We postulated that overexpression of matrix Glr1 could cause some Glr1 to mislocalize to the IMS. However, using subcellular fractionation and Western blotting techniques we determined that M17L Glr1 was not found in IMS extracts (data not shown). Overall these results indicate that endogenous Glr1 expression does not strongly influence the redox state of the mitochondrial IMS. Additionally, our data suggest that the IMS redox state is largely insulated from redox shifts that occur in both the matrix and cytosol.
DISCUSSION
Developing Sensors for Mitochondria Redox Measurements—Numerous studies have established that the mitochondrial redox state and the GSH:GSSG pool in particular play important roles in the cell (1, 40, 41). To measure GSH:GSSG in the matrix versus the IMS, we created GFP-based redox sensors that are separately targeted to these compartments. We tested the redox functionality of matrix- and IMS-rxYFP by verifying their in vivo response to addition of an oxidant (4-DPS) or reductant (DTT). Furthermore, we manipulated the subcellular GSH:GSSG ratio genetically via mutations in Glr1 that favor cytosolic versus mitochondrial expression of this reductase. These results demonstrate that the mitochondrial and cytosolic rxYFP sensors are selectively registering redox variations within subcellular compartments.
Equilibration of IMS- and Matrix-rxYFP with Mitochondrial GSH:GSSG Pools—To obtain an accurate in vivo redox measurement, a targeted redox sensor must meet several requirements: 1) the redox couple with which the sensor equilibrates must be clearly identified, 2) the sensor must rapidly reach equilibrium with this couple, 3) the reduced and oxidized forms of the sensor must have equivalent stabilities at different pH values, and 4) the sensor's redox potential must be tuned to accommodate the subcellular redox environment (6, 42). Østergaard and coworkers have demonstrated that rxYFP largely fulfills these requirements. It specifically registers the intracellular GSH:GSSG redox state via disulfide exchange reactions with cytosolic GRXs. This reaction has a similar equilibrium constant between pH 6.7 and 7.9 and occurs rapidly in the cytosol (within 20 min) (6). However, pulse-chase analysis revealed that steady-state rxYFP redox measurements slightly underestimated the redox potential (by ∼6–10 mV) in rapidly growing cells, because rxYFP is synthesized in the reduced form. If the oxidation rate is comparable to the protein synthesis rate, steady-state pools may be slightly skewed toward reduced rxYFP (6).
Similarly, in the mitochondria, rxYFP is imported in the reduced form and must interact with the local redox environment to become oxidized. Are GRXs available in the mitochondrial matrix and IMS to catalyze the rapid equilibration of rxYFP? Grx1 and Grx2 are primarily localized in the cytosol, however, a portion of Grx2 is also located in the mitochondrial matrix (43), where it is available to catalyze equilibration of rxYFP with matrix GSH:GSSG pools. In fact, matrix-rxYFP and GSH:GSSG measurements in Glr1 localization mutants indicate a close correlation between the mitochondrial GSH:GSSG redox state and the matrix-rxYFP redox state (see Fig. 6, B and C). Grx2 is also localized to the mitochondrial outer membrane, however the active site of this form is proposed to face the cytosolic side (43). The presence of yeast Grx1 in the IMS has not been addressed, but a recent publication suggests that a fraction of human Grx1 is found in the IMS (44). Therefore, it is possible that yeast Grx1 is available in the IMS to catalyze equilibration of IMS-rxYFP with the local GSH:GSSG pool. The fact that IMS-rxYFP is predominantly in the oxidized form argues that the equilibration rate is on the same order as the rate of protein synthesis and import. Future studies will focus on the role of Grx1 and Grx2 in IMS redox control and the kinetics of IMS-rxYFP oxidation.
Comparing the Redox State of the Cytosol and Matrix—In WT cells, cytosol- and matrix-rxYFP were 16 and 38% oxidized, respectively, reflecting substantial differences in the thiol-disulfide equilibrium in these individual compartments. In the mitochondrial matrix, the balance is shifted more toward disulfide formation than in the cytosol. This shift may reflect higher levels of ROS in the matrix versus the cytosol, which can oxidize free thiols to disulfides. Using published estimates of pH in the yeast cytosol (∼7.0) (33–36) and matrix (∼7.4) (36), the cytosolic and matrix reduction potentials measured with rxYFP are -286 mV and -296 mV. Because reduction potential is strongly dependent on pH (4), the matrix value is actually more reducing that the cytosol despite the higher percentage of oxidized sensor in this compartment. A similar trend was observed with in vivo redox measurements taken in mammalian cells. The redox sensor roGFP1 was 16% oxidized in the cytosol (45) and 33% oxidized in the matrix (46), yet the calculated matrix redox potential (-360 mV at pH 7.9) was more reducing than the cytosol (-315 mV at pH 7.0) due to the large pH difference between these compartments.
The IMS Redox State and Implications for IMS Thiol-disulfide Equilibrium—The critical role that thiol-disulfide balance plays in the IMS has been highlighted in some recent work on IMS protein import (for recent reviews see Refs. 15, 17, and 47). Several IMS proteins with highly conserved cysteines are imported through the outer membrane in a vectorial fashion via the Erv1-Mia40 disulfide relay system (18, 48–50). This system catalyzes disulfide bond formation in the imported proteins, which facilitates their folding and retention in the IMS. How does the IMS redox state influence this process? Because porin channels in the outer mitochondrial membrane are thought to allow free exchange of small molecules like GSH and GSSG between the cytosol and IMS (51), a logical assumption is that the redox state of these two compartments is similar. Therefore, it has been proposed that, once oxidized, these disulfide-containing IMS proteins must be resistant to reduction by GSH (52). The data presented herein provide an alternate explanation. Our redox measurements suggest that the IMS redox state in WT cells is more oxidizing (-255 mV) compared with both the cytosol (-286 mV) and matrix (-296 mV). This redox environment, in turn, may support the oxidative folding of IMS imported proteins, rather than opposing it. The redox potentials of structural disulfides in several proteins imported via the Erv1-Mia40 system have been determined, thus allowing us to test this argument by predicting the redox state of these cysteine pairs in the cytosol versus the IMS, assuming that they are in equilibrium with local GSH:GSSG pools. These predictions, shown in Table 2, indicate that the structural disulfide of Erv1-Mia40 substrates are all >90% oxidized in the IMS, but their putative oxidation state in the cytosol ranges from 46 to 92% oxidized. Whereas several structural studies on disulfide-containing IMS proteins have proposed this type of redox differential between the cytosol and IMS (53, 54), we provide here direct evidence for a more oxidizing IMS environment.
TABLE 2.
Predicted oxidation states of cysteine pairs in IMS-imported proteins in the cytosol versus the IMS
| Protein | Published E | Ref. | E°′ at pH 7.0a | Oxidation in cytosolb | Oxidation in IMSb | |
|---|---|---|---|---|---|---|
| mV | mV | % | ||||
| Erv1 (C30-C33) | –320 at pH 7.0 | (57) | –320 | 93 | 99 | |
| Tim10 (twin CX3C) | –320 at pH 7.4 | (58) | –296 | 68 | 96 | |
| Tim9 (twin CX3C) | –310 at pH 7.4 | (52) | –286 | 49 | 91 | |
| Cox17 (twin CX9C) | –340 at pH 7.6 | (54) | –304 | 79 | 98 | |
E°′ values at pH 7.0 were calculated using Equation 3
The percent oxidized protein in the cytosol and IMS was calculated from Equation 2 using Ecyto = –286 mV and EIMS = –255 mV from Table 1
According to Equation 2, the IMS redox measurements reflect the [GSH]2/[GSSG] ratio, but do not provide the absolute concentration of GSH or the GSH:GSSG ratio. Therefore the relatively oxidizing IMS environment may be due to a lower GSH:GSSG ratio or lower overall GSH levels in this compartment compared with the cytosol. The latter possibility implies that the IMS has a lower redox buffering capacity than the cytosol. Our measurements also assume that the IMS-rxYFP redox state is primarily influenced by GSH:GSSG pools. Alternatively, rxYFP may interact with another redox pathway, such as the Mia40-Erv1 disulfide relay system, which may play a more dominant role in this compartment than GSH:GSSG. However, previous studies have shown that this relay system selectively interacts with substrates that possess a twin CX9Cor CX3C motif (18), which is absent in rxYFP. In any case, the factors that influence the IMS redox state are apparently more complex that simple diffusion of GSH:GSSG from the cytosol to the IMS. By using IMS-rxYFP as a localized gauge of GSH: GSSG, as well as other markers of redox status, these factors will be identified in future studies.
The question still remains: what is oxidizing rxYFP and presumably GSH:GSSG in the IMS? One potential clue may be the lack of GSSG reductase (Glr1) in this compartment (9). Without this antioxidant factor, the IMS may be prone to ROS-induced oxidation of reactive thiols. As demonstrated by redox proteomic studies in yeast (55), molecular oxygen is the primary source of oxidizing equivalents for cellular thiols, including GSH. In the cytosol and matrix, Glr1 counteracts the oxidizing effects of aerobic metabolism by continuously catalyzing reduction of GSH to maintain a high GSH:GSSG ratio. Without Glr1 in the IMS, GSSG produced via ROS oxidation may accumulate, leading to a lower GSH:GSSG ratio, which in turn influences the rxYFP redox state. In support of this argument, it is interesting to note that, in a glr1Δ mutant, the redox states of the cytosol and matrix are very similar to the IMS (see Table 1). Future studies will determine whether factors that influence ROS production, such as oxygen tension (anaerobic versus aerobic growth), carbon source (i.e. fermentative versus non-fermentative), and growth phase, alter the subcellular redox state.
Independent Redox Control in Subcellular Compartments—On a broader scale, our redox measurements provide some interesting insights into redox control in subcellular compartments. Our redox measurements in Glr1 localization mutants (Fig. 6) suggest that redox homeostasis is primarily maintained independently in the cytosol, matrix, and IMS, because the thiol-disulfide balance in each of these compartments is quite different. Furthermore, alteration of the matrix redox state by abrogating Glr1 mitochondrial import has little influence on the cytosolic and IMS redox state. We were unable to test the reciprocal experiment (selective expression of matrix Glr1) because our matrix Glr1 construct (M17L) exhibits some cytosolic localization due to inefficient mitochondrial import (see Ref. 9). However, we did observe that matrix Glr1 completely restores GSH:GSSG balance in the matrix but not the cytosol. Interestingly, we also noted a subtle effect of matrix Glr1 overexpression on the IMS redox state, because our IMS-rxYFP was slightly less oxidized in these strains compared with cytosol Glr1 expression. This results suggests that the matrix GSH: GSSG pool has more influence over the IMS redox state than cytosol GSH:GSSG pools. In fact, exchange of GSH between the matrix and IMS has been suggested to occur in purified rat liver mitochondria (56). The specific role that matrix versus cytosolic GSH:GSSG pools play in influencing the IMS redox state will be addressed in future studies.
Overall, the existence of independent GSH:GSSG pools in subcellular compartments parallels previous studies on the separate nature of NADPH pools in the cytosol and mitochondrial matrix (26). Taken together, these experiments suggest that the redox status of the mitochondrial matrix, IMS, and cytosol are influenced separately by factors that are specifically targeted to these compartments.
Acknowledgments
We thank Jakob Winther (University of Copenhagen) for providing rxYFP antibodies and technical advice, Jakob Winther and the Carlsberg Laboratory (Copenhagen Valby, Denmark) for providing the rxYFP expression vector pHOJ150, and Robert Jensen (Johns Hopkins University School of Medicine) for providing Cyb2 and Mas2 antibodies. We also thank F. Wayne Outten for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant ES 13780 (to C. E. O.). This work was also supported by the University of South Carolina Center for Colon Cancer Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; GSH, reduced glutathione; GSSG, oxidized glutathione; IMS, intermembrane space; GFP, green fluorescent protein; rxYFP, redox-sensitive yellow fluorescent protein; GRX, glutaredoxin; PMS, post-mitochondrial supernatant; DAPI, 4′,6-diamidino-2-phenylindole; 4-DPS, 4,4′-dithiodipyridine; DTT, dithiothreitol; Cyb2, cytochrome b2; WT, wild type.
References
- 1.Jones, D. P. (2006) Chem. Biol. Interact. 163 38-53 [DOI] [PubMed] [Google Scholar]
- 2.Holmgren, A., Johansson, C., Berndt, C., Lonn, M. E., Hudemann, C., and Lillig, C. H. (2005) Biochem. Soc. Trans. 33 1375-1377 [DOI] [PubMed] [Google Scholar]
- 3.Carmel-Harel, O., and Storz, G. (2000) Annu. Rev. Microbiol. 54 439-461 [DOI] [PubMed] [Google Scholar]
- 4.Schafer, F. Q., and Buettner, G. R. (2001) Free Radic. Biol. Med. 30 1191-1212 [DOI] [PubMed] [Google Scholar]
- 5.Hwang, C., Sinskey, A. J., and Lodish, H. F. (1992) Science 257 1496-1502 [DOI] [PubMed] [Google Scholar]
- 6.Østergaard, H., Tachibana, C., and Winther, J. R. (2004) J. Cell Biol. 166 337-345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen, D., Dalton, T. P., Nebert, D. W., and Shertzer, H. G. (2005) J. Biol. Chem. 280 25305-25312 [DOI] [PubMed] [Google Scholar]
- 8.Rebrin, I., Zicker, S., Wedekind, K. J., Paetau-Robinson, I., Packer, L., and Sohal, R. S. (2005) Free Radic. Biol. Med. 39 549-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Outten, C. E., and Culotta, V. C. (2004) J. Biol. Chem. 279 7785-7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteiro, G., Kowaltowski, A. J., Barros, M. H., and Netto, L. E. (2004) Arch. Biochem. Biophys. 425 14-24 [DOI] [PubMed] [Google Scholar]
- 11.Aon, M. A., Cortassa, S., Maack, C., and O'Rourke, B. (2007) J. Biol. Chem. 282 21889-21900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda, S., Masutani, H., Nakamura, H., Tanaka, T., Ueno, M., and Yodoi, J. (2002) Antioxid. Redox Signal. 4 405-414 [DOI] [PubMed] [Google Scholar]
- 13.Khalimonchuk, O., and Winge, D. R. (2007) Biochim. Biophys. Acta 1783 618-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldsteins, G., Keksa-Goldsteine, V., Ahtoniemi, T., Jaronen, M., Arens, E., Akerman, K., Chan, P. H., and Koistinaho, J. (2008) J. Biol. Chem. 283 8446-8452 [DOI] [PubMed] [Google Scholar]
- 15.Herrmann, J. M., and Kohl, R. (2007) J. Cell Biol. 176 559-563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamoto, H., and Bardwell, J. C. (2004) Biochim. Biophys. Acta 1694 111-119 [DOI] [PubMed] [Google Scholar]
- 17.Hell, K. (2008) Biochim. Biophys. Acta 1783 601-609 [DOI] [PubMed] [Google Scholar]
- 18.Gabriel, K., Milenkovic, D., Chacinska, A., Muller, J., Guiard, B., Pfanner, N., and Meisinger, C. (2007) J. Mol. Biol. 365 612-620 [DOI] [PubMed] [Google Scholar]
- 19.Koehler, C. M., Beverly, K. N., and Leverich, E. P. (2006) Antioxid. Redox Signal. 8 813-822 [DOI] [PubMed] [Google Scholar]
- 20.Cubitt, A. B., Heim, R., Adams, S. R., Boyd, A. E., Gross, L. A., and Tsien, R. Y. (1995) Trends Biochem. Sci. 20 448-455 [DOI] [PubMed] [Google Scholar]
- 21.Østergaard, H., Henriksen, A., Hansen, F. G., and Winther, J. R. (2001) EMBO J. 20 5853-5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gietz, R. D., and Schiestl, R. H. (1991) Yeast 7 253-263 [DOI] [PubMed] [Google Scholar]
- 23.Sikorski, R. S., and Hieter, P. (1989) Genetics 122 19-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daum, G., Bohni, P. C., and Schatz, G. (1982) J. Biol. Chem. 257 13028-13033 [PubMed] [Google Scholar]
- 25.Diekert, K., de Kroon, A. I., Kispal, G., and Lill, R. (2001) Methods Cell Biol. 65 37-51 [DOI] [PubMed] [Google Scholar]
- 26.Outten, C. E., and Culotta, V. C. (2003) EMBO J. 22 2015-2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sesaki, H., and Jensen, R. E. (1999) J. Cell Biol. 147 699-706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sturtz, L. A., Diekert, K., Jensen, L. T., Lill, R., and Culotta, V. C. (2001) J. Biol. Chem. 276 38084-38089 [DOI] [PubMed] [Google Scholar]
- 29.Beasley, E. M., Muller, S., and Schatz, G. (1993) EMBO J. 12 2303-2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Heijne, G. (1986) EMBO J. 5 1335-1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herrmann, J. M., and Hell, K. (2005) Trends Biochem. Sci. 30 205-211 [DOI] [PubMed] [Google Scholar]
- 32.Stevens, B. J. (1977) Biol. Cellulaire 28 37-56 [Google Scholar]
- 33.Imai, T., and Ohno, T. (1995) J. Biotechnol. 38 165-172 [DOI] [PubMed] [Google Scholar]
- 34.Melvin, B. K., and Shanks, J. V. (1996) Biotechnol. Prog. 12 257-265 [DOI] [PubMed] [Google Scholar]
- 35.Breeuwer, P., and Abee, T. (2000) J. Microbiol. Methods 39 253-264 [DOI] [PubMed] [Google Scholar]
- 36.Matsuyama, S., Llopis, J., Deveraux, Q. L., Tsien, R. Y., and Reed, J. C. (2000) Nat. Cell Biol. 2 318-325 [DOI] [PubMed] [Google Scholar]
- 37.Cortese, J. D., Voglino, A. L., and Hackenbrock, C. R. (1992) Biochim. Biophys. Acta 1100 189-197 [DOI] [PubMed] [Google Scholar]
- 38.Porcelli, A. M., Ghelli, A., Zanna, C., Pinton, P., Rizzuto, R., and Rugolo, M. (2005) Biochem. Biophys. Res. Commun. 326 799-804 [DOI] [PubMed] [Google Scholar]
- 39.Muller, E. G. (1996) Mol. Biol. Cell 7 1805-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hancock, J. T., Desikan, R., and Neill, S. J. (2003) Ann. N. Y. Acad. Sci. 1010 446-448 [DOI] [PubMed] [Google Scholar]
- 41.Hurd, T. R., Costa, N. J., Dahm, C. C., Beer, S. M., Brown, S. E., Filipovska, A., and Murphy, M. P. (2005) Antioxid. Redox Signal. 7 999-1010 [DOI] [PubMed] [Google Scholar]
- 42.Bjornberg, O., Østergaard, H., and Winther, J. R. (2006) Antioxid. Redox Signal. 8 354-361 [DOI] [PubMed] [Google Scholar]
- 43.Porras, P., Padilla, C. A., Krayl, M., Voos, W., and Barcena, J. A. (2006) J. Biol. Chem. 281 16551-16562 [DOI] [PubMed] [Google Scholar]
- 44.Pai, H. V., Starke, D. W., Lesnefsky, E. J., Hoppel, C. L., and Mieyal, J. J. (2007) Antioxid. Redox Signal. 9 2027-2033 [DOI] [PubMed] [Google Scholar]
- 45.Dooley, C. T., Dore, T. M., Hanson, G. T., Jackson, W. C., Remington, S. J., and Tsien, R. Y. (2004) J. Biol. Chem. 279 22284-22293 [DOI] [PubMed] [Google Scholar]
- 46.Hanson, G. T., Aggeler, R., Oglesbee, D., Cannon, M., Capaldi, R. A., Tsien, R. Y., and Remington, S. J. (2004) J. Biol. Chem. 279 13044-13053 [DOI] [PubMed] [Google Scholar]
- 47.Stojanovski, D., Muller, J. M., Milenkovic, D., Guiard, B., Pfanner, N., and Chacinska, A. (2008) Biochim. Biophys. Acta 1783 610-617 [DOI] [PubMed] [Google Scholar]
- 48.Allen, S., Balabanidou, V., Sideris, D. P., Lisowsky, T., and Tokatlidis, K. (2005) J. Mol. Biol. 353 937-944 [DOI] [PubMed] [Google Scholar]
- 49.Mesecke, N., Terziyska, N., Kozany, C., Baumann, F., Neupert, W., Hell, K., and Herrmann, J. M. (2005) Cell 121 1059-1069 [DOI] [PubMed] [Google Scholar]
- 50.Müller, J. M., Milenkovic, D., Guiard, B., Pfanner, N., and Chacinska, A. (2007) Mol. Biol. Cell 19 226-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benz, R. (1994) Biochim. Biophys. Acta 1197 167-196 [DOI] [PubMed] [Google Scholar]
- 52.Morgan, B., and Lu, H. (2008) Biochem. J. 411 115-122 [DOI] [PubMed] [Google Scholar]
- 53.Banci, L., Bertini, I., Ciofi-Baffoni, S., Janicka, A., Martinelli, M., Kozlowski, H., and Palumaa, P. (2008) J. Biol. Chem. 283 7912-7920 [DOI] [PubMed] [Google Scholar]
- 54.Voronova, A., Meyer-Klaucke, W., Meyer, T., Rompel, A., Krebs, B., Kazantseva, J., Sillard, R., and Palumaa, P. (2007) Biochem. J. 408 139-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le Moan, N., Clement, G., Le Maout, S., Tacnet, F., and Toledano, M. B. (2006) J. Biol. Chem. 281 10420-10430 [DOI] [PubMed] [Google Scholar]
- 56.Mårtensson, J., Lai, J. C., and Meister, A. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 7185-7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dabir, D. V., Leverich, E. P., Kim, S. K., Tsai, F. D., Hirasawa, M., Knaff, D. B., and Koehler, C. M. (2007) EMBO J. 26 4801-4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu, H., and Woodburn, J. (2005) J. Mol. Biol. 353 897-910 [DOI] [PubMed] [Google Scholar]