

Abstract
Experiments were performed to test the hypothesis that tyrosine kinase activity contributes to renal arteriolar contractile responses to angiotensin II (AngII). Rats were subjected to acute enalaprilat treatment to decrease endogenous AngII formation prior to harvesting tissue for experiments using the in vitro blood-perfused juxtamedullary nephron technique. Acute surgical papillectomy was employed to avoid the indirect afferent arteriolar effect of AngII that arises via increased tubuloglomerular feedback sensitivity. Arteriolar lumen diameter responses to 1 and 10 nmol/L AngII were monitored using videomicroscopic methods before and during treatment with various tyrphostin compounds: 100 μmol/L AG18 (broad-spectrum tyrosine kinase inhibitor), 100 nmol/L AG1478 (selective EGF-receptor tyrosine kinase inhibitor) or 100 μmol/L AG9 (inactive analog). Baseline afferent arteriolar lumen diameter averaged 23.5 ± 1.2 μm and was not influenced by any tyrphostin. AngII (10 nM) decreased afferent diameter by 11.1 ± 1.0 μm under untreated conditions, a response that was not altered by AG9 but significantly blunted by AG18 (34 ± 9% inhibition) or AG1478 (52 ± 8% inhibition). AG18 did not suppress afferent arteriolar contractile responses to membrane depolarization (20−55 mmol/L bath K+). Efferent arteriolar baseline diameter averaged 24.1 ± 0.8 μm and was unaltered by AG18 or AG1478; however, efferent diameter responses to 10 nmol/L AngII were diminished 52 ± 10% by AG18 and 51 ± 13% by AG1478. These observations indicate that AngII signaling in renal afferent and efferent arteriolar vascular smooth muscle is either mediated or modulated by tyrosine kinase activity, including that of the EGF receptor tyrosine kinase.
Keywords: afferent arteriole, angiotensin II, efferent arteriole, tyrphostins, EGF receptor
Introduction
Although the G-protein-coupled angiotensin II type 1 (AT1) receptor lacks intrinsic tyrosine kinase activity, angiotensin II (AngII) binding to this receptor induces tyrosine phosphorylation of multiple signaling proteins. Tyrosine phosphorylation of phospholipase C (PLC)-γ has been reported to be necessary for AngII-stimulated inositol trisphosphate (IP3) formation, Ca2+ mobilization and contraction of mesangial and aortic smooth muscle cells.1,2 Moreover, transactivation of the epidermal growth factor (EGF) receptor is evident in vascular smooth muscle within 1−2 minute of AngII exposure,3,4 consistent with the possibility that this process may contribute to the vasoconstrictor response to the peptide. Similarly, AngII-induced activation of p38 mitogen-activated protein (MAP) kinase occurs within 5 min, and inhibition of p38 kinase activity reduces the contractile effect of AngII on rat aortic rings.5 Nevertheless, Watts and colleagues6 have demonstrated dissociation of AngII-induced tyrosine kinase activity from the contractile response of rat aortic rings.
Virtually all studies probing the involvement of tyrosine kinase activity in AngII activation of vascular smooth muscle have employed large arteries or cultured myocytes from these vessels. The involvement of tyrosine kinase activity in evoking AngII-induced contraction within the microvasculature, including that of the kidney, remains virtually unexplored. Accordingly, the present experiments addressed the hypothesis that tyrosine phosphorylation participates in the renal arteriolar constrictor response to AngII. Tyrosine kinase activity has also been reported to modulate L-type voltage-gated Ca2+ channels. In particular, tonic phosphorylation by tyrosine kinases has been suggested to maintain the channels in an available state for activation by depolarization.7 These channels are functionally prominent in the afferent arteriole and represent a critical component of the constrictor response to AngII in this vascular segment.8-11 Accordingly, we also assessed the influence of tyrosine kinase activity on the afferent arteriolar contractile response to membrane depolarization.
Methods
Animals
The procedures used in this study were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee and conducted according to the NIH Guide for the Care and Use of Laboratory Animals. Thirty-two male Sprague-Dawley rats (SAS:VAF strain) weighing 250−300 g were purchased from Charles River Laboratories (Wilmington, MA) and provided free access to food and water prior to study.
The in vitro blood-perfused juxtamedullary nephron technique
Arteriolar contractile function was assessed in experiments performed using the in vitro blood-perfused juxtamedullary nephron technique.12 After anesthetization with pentobarbital sodium (50 mg/kg i.p), a cannula was inserted into the left carotid artery and enalaprilat was administered (2 mg in 1 ml isotonic saline, i.a.) to reduce endogenous AngII formation. The left renal artery and vein were ligated and the right renal artery was cannulated via the superior mesenteric artery, thereby initiating renal perfusion with Tyrode's solution containing 52 g/L dialyzed bovine serum albumin and a mixture of L-amino acids.13 Blood was collected via the carotid cannula prior to harvesting the kidney. Renal perfusion was maintained throughout the ensuing dissection procedure needed to reveal the tubules, glomeruli, and related vasculature of juxtamedullary nephrons. Tight ligatures were placed around the most distal accessible segments of the large arterial branches that supply the exposed microvasculature. Acute surgical papillectomy was performed to avoid an indirect, tubuloglomerular feedback-dependent influence of AngII on the vasculature.14 The Tyrode's perfusate was then replaced with reconstituted blood prepared as described previously.13 Renal arterial perfusion pressure was set at 110 mmHg and maintained at that level throughout the experiment. The perfusion chamber was warmed, and the tissue surface was bathed continuously with Tyrode's solution containing 10 g/L bovine serum albumin at 37°C.
The tissue was transilluminated on the fixed stage of a compound microscope equipped with a water-immersion objective (×40). Video images of the microvessels were stored on videotape for later analysis. An afferent or efferent arteriole was selected for study based on visibility and acceptable blood flow. All protocols assessed arteriolar diameter at a single measurement site under several experimental conditions. Afferent arteriolar diameter was monitored at sites ≥100 μm upstream from the glomerulus, whereas efferent arterioles were studied at sites ≤100 μm from the point of emergence from the glomerulus.
Protocols
Effect of tyrosine kinase blockade on renal arteriolar constrictor responses to AngII
Following a 15-min stabilization period, afferent or efferent arteriolar baseline diameter was established during an initial 5-min control period, after which the effects of exogenous AngII were assessed. Because AngII causes similar afferent arteriolar constrictor responses when administered from the bath and lumen of in vitro perfused juxtamedullary nephrons,15 the present studies employed the technically simpler method of administering the peptide (1 and 10 nmol/L; 3 min treatment periods) via the bathing solution. After a 10 min recovery period, 100 μmol/L tyrphostin AG18 (broad-spectrum tyrosine kinase blocker) was added to the perfusate bath. Ten min later, the AngII exposure sequence was repeated. In other experiments, AG18 was replaced with either 100 μmol/L AG9 (inactive analog) or 100 nmol/L AG1478 (EGF receptor tyrosine kinase blocker). Previous studies from our laboratory have documented the stability of juxtamedullary afferent arteriolar lumen diameter in tissue not exposed to exogenous vasoactive agents for the duration of these experiments.16,17
Effect of tyrosine kinase blockade on afferent arteriolar responses to depolarization
Following the stabilization period, baseline afferent arteriolar diameter was established during exposure to the normal bathing solution (Tyrode's solution containing 2.7 mmol/L K+). Subsequently, arteriolar diameter responses to increasing extracellular [K+] were assessed (20, 40, and 55 mmol/L bath K+; substitution for Na+). After recovery from K+-induced depolarization, 100 μmol/L AG18 was added to the bath (10 min) and arteriolar responses to increasing bath [K+] were repeated in the continued presence of AG18.
Data analysis
Microvessel inside diameters were measured from videotaped images utilizing a digital image-shearing monitor (model 908, IPM, San Diego, CA, USA) calibrated using a stage micrometer, a system that allows diameter measurements reproducible to within <1 μm. Microvessel diameter was measured at 12-sec intervals from a single site along the vessel length. The average diameter during the final minute of each treatment period was utilized for statistical analysis (analysis of variance for repeated measures and Newman-Keuls tests). P values <0.05 were considered significant. All data are reported as means ± SEM (n = number of arterioles).
Results
Effect of tyrosine kinase blockade on renal arteriolar constrictor responses to AngII
Figure 1A illustrates the impact of tyrosine kinase blockade on afferent arteriolar lumen diameter responses to AngII. Baseline afferent arteriolar diameter averaged 23.1 ± 2.0 μm (n = 5) and decreased by 3.4 ± 1.5 and 12.7 ± 2.1 μm upon exposure to 1 and 10 nmol/L AngII, respectively. Removal of AngII from the bath allowed restoration of afferent diameter to 24.0 ± 2.9 μm (P>0.05 vs baseline). Although AG18 (100 μmol/L) alone did not significantly alter baseline afferent diameter (23.9 ± 2.6 μm), 1 and 10 nmol/L AngII decreased lumen diameter by only 1.9 ± 0.7 and 8.2 ± 1.5 μm (P<0.05 vs untreated), respectively, during AG18 treatment. Thus, exposure to the broad-spectrum tyrosine kinase blocker reduced afferent arteriolar responsiveness to 10 nmol/L AngII by 34 ± 9%. However, the time required to develop 50% of the peak response to 10 nmol/L AngII (t50) was unaffected by AG18, averaging 39 ± 7 sec before and 47 ± 4 sec during AG18 treatment (P=0.32 by paired t-test).
Figure 1.
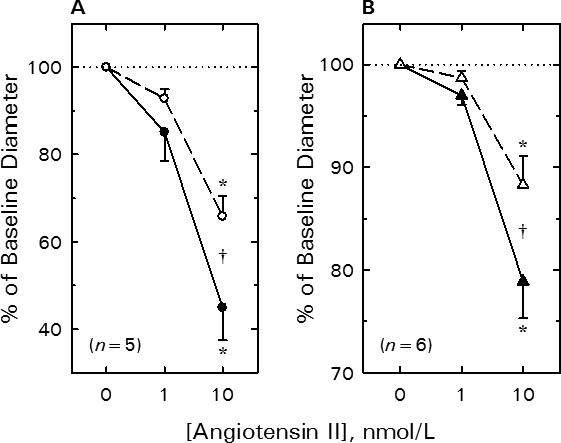
Effect of tyrosine kinase blockade on renal arteriolar constrictor responses to AngII. A) Afferent arteriole. B) Efferent arteriole. See text for baseline values. Solid symbols, untreated; Open symbols, 100 μmol/L AG18. Data are means ± SEM. *P < 0.05 vs baseline (0 nmol/L AngII), †P < 0.05 Untreated vs AG18.
AG18 also attenuated efferent arteriolar AngII responsiveness (Figure 1B). Efferent arteriolar lumen diameter averaged 23.6 ± 1.1 μm (n = 6) during the untreated baseline period. AngII evoked reductions in efferent diameter, with 1 nmol/L AngII decreasing diameter by 0.7 ± 0.2 μm and 10 nmol/L AngII decreasing diameter by 5.0 ± 0.9 μm. Recovery from the AngII exposure restored efferent diameter to 22.5 ± 1.1 μm (P>0.05 vs baseline). AG18 did not significantly alter efferent arteriolar baseline diameter (22.4 ± 1.1 μm); however, responses to AngII were attenuated such that 10 nmol/L AngII only reduced efferent diameter by 2.6 ± 0.6 μm (P<0.005 vs untreated). Thus, tyrosine kinase blockade suppressed the efferent constrictor response to 10 nmol/L AngII by 52 ± 10%. Values for t50 averaged 38 ± 8 sec before and 43 ± 17 sec during AG18 treatment in efferent arterioles (P=0.822 by paired t-test), indicating no change in the time course of the response to 10 nmol/L AngII.
Figure 2A depicts the impact of EGF receptor tyrosine kinase blockade on AngII-induced afferent arteriolar constriction. Afferent lumen diameter averaged 22.3 ± 1.5 μm (n = 7) under baseline conditions, and 1 and 10 nmol/L AngII reduced afferent diameter by 2.4 ± 0.5 and 9.9 ± 1.0 μm, respectively. Removal of AngII from the bath allowed restoration of arteriolar diameter to 22.5 ± 1.5 μm. AG1478 (100 nmol/L) did not alter baseline afferent arteriolar diameter (22.6 ± 1.6 μm), but suppressed AngII-induced vasoconstriction. During exposure to AG1478, 1 and 10 nmol/L AngII reduced afferent diameter by 0.8 ± 0.7 and 4.5 ± 0.7 μm (P<0.001 vs untreated), respectively. Thus, blockade of the EGF receptor tyrosine kinase inhibited the afferent arteriolar response to 10 nmol/L AngII by 52 ± 8%.
Figure 2.
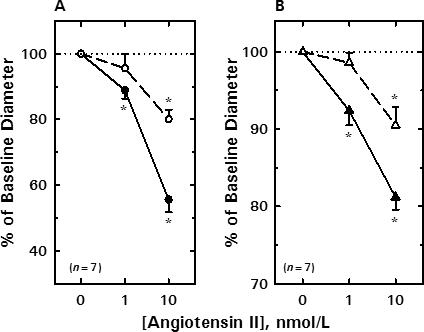
Effect of EGF receptor tyrosine kinase blockade on renal arteriolar constrictor responses to AngII. A) Afferent arteriole. B) Efferent arteriole. See text for baseline values. Solid symbols, untreated; Open symbols, 100 nmol/L AG1478. Data are means ± SEM. *P < 0.05 vs baseline (0 nmol/L AngII), †P < 0.05 Untreated vs AG1478.
The effect of EGF receptor blockade on efferent arteriolar responsiveness to AngII is summarized in Figure 2B. Efferent arteriolar diameter averaged 24.6 ± 1.2 μm (n = 7) under baseline conditions and decreased significantly by 1.9 ± 0.5 and 4.6 ± 0.4 μm in response to 1 and 10 nmol/L AngII, respectively. During recovery from AngII treatment, efferent arteriolar diameter was restored to 23.8 ± 1.3 μm (P>0.05 vs baseline). AG1478 (100 nmol/L) did not significantly alter baseline efferent diameter (23.3 ± 1.5 μm); however, during continued exposure to AG1478, 1 nmol/L AngII failed to alter lumen diameter (Δ= −0.3 ± 0.3 μm) and 10 nmol/L AngII only reduced diameter by 2.1 ± 0.5 μm (P<0.05 vs untreated). Thus, the EGF receptor tyrosine kinase blocker reduced efferent arteriolar responsiveness to 10 nmol/L AngII by 51 ± 13%.
The effect of the inactive tyrphostin (AG9) on afferent arteriolar diameter responses to exogenous AngII is depicted in Figure 3A. Afferent arteriolar diameter averaged 22.7 ± 1.8 μm (n = 4) under baseline conditions, and decreased by 3.9 ± 1.4 and 11.2 ± 2.5 μm during exposure to 1 and 10 nmol/L AngII, respectively. During the recovery period, afferent lumen diameter returned to 21.8 ± 2.2 μm (P > 0.05 vs untreated baseline). AG9 (100 μmol/L) did not alter baseline diameter (22.7 ± 2.2 μm) or responsiveness to AngII (1 nM, Δ = −3.3 ± 0.8 μm; 10 nM, Δ = −9.9 ± 2.3 μm).
Figure 3.
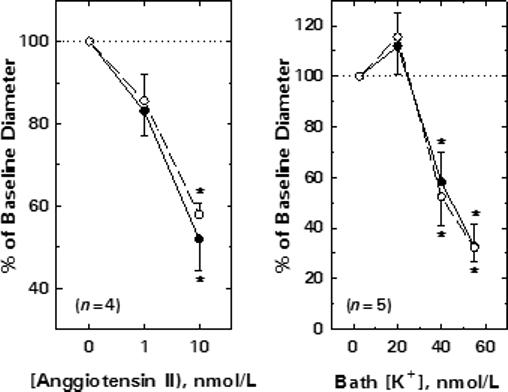
Specificity of tyrphostin effects on afferent arteriolar contractile responses. A) Effect of the inactive tyrphostin analog on afferent arteriolar responses to AngII. B) Effect of tyrosine kinase blockade on afferent arteriolar responses to K+-induced depolarization. Data are expressed as means ± SEM. Solid symbols, untreated; Open symbols, 100 μmol/L AG9 (Panel A) or 100 μmol/L AG18 (Panel B). Data are means ± SEM. *P < 0.05 vs baseline (Panel A, 0 nmol/L AngII; Panel B, 2.7 mmol/L K+).
Effect of tyrosine kinase blockade on afferent arteriolar responses to depolarization
Figure 3B illustrates the impact of increases in bath [K+] on afferent arteriolar diameter before and during tyrosine kinase blockade (100 μmol/L AG18). Afferent diameter averaged 22.5 ± 2.1 μm (n = 5) under untreated baseline conditions (2.7 mmol/L K+), and was significantly reduced by 40 mmol/L K+ (Δ= −10.1 ± 3.3 μm) and 55 mmol/L K+ (Δ= −15.5 ± 2.9 μm). Restoration of bath [K+] to 2.7 mmol/L allowed full recovery of afferent diameter to baseline values (22.5 ± 2.1 μm). AG18 did not significantly alter baseline diameter (22.5 ± 2.1 μm) or responses to K+-induced depolarization (40 mmol/L K+, Δ= −10.4 ± 3.2 μm; 55 mmol/L K+, Δ= −14.2 ± 2.2 μm).
Discussion
In both afferent and efferent arterioles, AngII induces constriction via its interaction with AT1 receptors;18 however, disparate intracellular signaling events are utilized by these vascular segments to generate the increase in intracellular Ca2+ concentration necessary to produce vasoconstriction. Available evidence indicates that activation of AT1 receptors in the afferent arteriole activates PLC to elicit IP3-dependent Ca2+ mobilization, resulting in a rise in intracellular [Ca2+] that increases the open-probability of Ca2+-activated Cl− channels, with the subsequent membrane depolarization prompting Ca2+ influx through voltage-gated channels.8,19,20 In contrast, activation of efferent arteriolar AT1 receptors appears to trigger activation of PLC to provoke Ca2+ mobilization and protein kinase C-dependent Ca2+ influx through a voltage-independent mechanism, possibly via store-operated Ca2+ channels.11,19,20 The present study employed a pharmacological approach to implicate tyrosine phosphorylation in the AngII-induced signaling events of both afferent and efferent arterioles.
The role of tyrosine phosphorylation in AngII-induced constriction was assessed through the use of synthetic tyrphostin compounds with characteristic effects on tyrosine kinase activity. AG18, also known as tyrphostin A23, is a broad-spectrum tyrosine kinase inhibitor that blocks EGF receptor autophosphorylation and platelet-derived growth factor (PDGF) receptor kinase with IC50 values in the range of 25−40 μmol/L.21,22 Because AG18 inhibition of EGF receptor protein kinase is maximally effective at a concentration of 100 μM,23 this concentration was employed in the present study. AG1478 is a potent and specific EGF receptor tyrosine kinase inhibitor (IC50 = 3 nmol/L), affecting PDGF receptor kinase activity only at much higher concentrations (IC50>100 μmol/L).24 At concentrations within the range of 100−250 nM, AG1478 markedly abates EGF- and AngII-stimulated increases in MAP kinase activity in rat aortic smooth muscle.4 AG9, also known as tyrphostin A1, is an inactive compound used as a negative control (IC50>1250 μmol/L for EGF receptor kinase).25
None of the tyrphostin compounds employed in the present study significantly altered baseline diameter of afferent or efferent arterioles; however, AG18 attenuated both afferent and efferent arteriolar constrictor responses to AngII. The failure of the inactive tyrphostin analog to alter afferent arteriolar AngII responsiveness is consistent with the contention that AG18 suppressed AngII responsiveness via its ability to inhibit tyrosine kinase activity, rather than via a nonspecific effect of tyrphostin compounds. These observations are in accord with reports that tyrosine kinase blockade attenuates AngII-induced pHi, [Ca2+]i and/or contractile responses in vascular smooth muscle from aorta or mesenteric artery.26-28
Tyrphostin compounds have been reported to inhibit L-type voltage-gated Ca2+ channels, either secondary to tyrosine kinase blockade7,29 or via a direct nonspecific (tyrosine kinase-independent) effect on channel activity.30 Because these channels are prominent in the renal afferent arteriole9,10 and in evoking the vasoconstrictor response of this vessel to AngII,8 we assessed the impact of AG18 on K+-induced vasoconstriction. Increases in extracellular [K+] contract vascular smooth muscle via the effect of membrane depolarization to increase the open probability of L-type Ca2+ channels. We have shown previously that the afferent arteriolar intracellular [Ca2+] and constrictor responses to this maneuver are blocked by nifedipine and diltiazem.9,31 In contrast with the behavior of non-renal vascular beds,6,32-34 tyrosine kinase blockade did not alter K+-induced afferent arteriolar constriction in the present study. This observation has several mechanistic implications. First, the effect of AG18 on AngII responsiveness cannot be attributed to a direct (tyrosine kinase-independent) effect of AG18 on the L-type Ca2+ channel. Second, it is also unlikely that a direct effect of AG18 on the contractile apparatus or on the Ca2+-sensitivity of the contractile proteins35,36 underlies the impact of this compound on AngII responsiveness. Finally, it unlikely that either membrane depolarization or the resulting Ca2+ influx initiates the tyrosine kinase activation involved in the afferent arteriolar AngII signaling. However, we cannot rule out the possibility the relatively large transient increase in intracellular [Ca2+] resulting from AngII-induced Ca2+ mobilization triggers tyrosine kinase activation.
In recent years, it has become clear that activation of various G-protein-coupled receptors (such as the AT1 receptor) rapidly induces transactivation of the EGF receptor, and that inhibition of EGF receptor tyrosine kinase activity prevents the subsequent events that lead to MAP kinase activation and transmission of mitogenic signals to the nucleus.37 AT1-EGF receptor cross-talk has been studied primarily in the context of the mitogenic effect of AngII on aortic myocytes. Although the present study does not address the sequence of events linking AT1 receptor activation and tyrosine kinase activity to evoke renal arteriolar constriction, involvement of the EGF receptor tyrosine kinase is indicated by the ability of AG1478 to attenuate renal arteriolar constrictor responses to AngII. These observations provide evidence of cross-talk between the AT1 receptor and the EGF receptor in both afferent and efferent arterioles, despite differences in other aspects of AngII-induced signaling at these sites. For example, although AngII has been suggested to constrict afferent arterioles via Gq and efferent arterioles via Gi,19 both of these G proteins are capable of utilizing Src to phosphorylate the EGF receptor for recruitment of adaptor proteins and MAP kinase activation.38 Some studies employing cultured rat vascular smooth muscle suggest that Src family tyrosine kinases mediate AngII-induced phosphorylation of PLC-γ, resulting in IP3 formation and Ca2+ mobilization.2,39 Alternatively, AngII may trigger Gq-mediated PLC-β activation, with the resulting Ca2+ mobilization triggering EGF receptor transactivation and phosphorylation by Src.4,40 The involvement of PLC-β or PLC-γ may represent a critical distinction between these two scenarios; however, although PLC is involved in AngII-induced signaling in both afferent and efferent arterioles, the role of specific PLC isoforms has not been evaluated. Hence, further investigation is necessary to elucidate the mechanism(s) linking the AT1 receptor to tyrosine kinase activation in the afferent and efferent arteriole, as well as the mechanism through which EGF receptor activation influences contractile tone.
In summary, a broad-spectrum tyrosine kinase inhibitor attenuated AngII responsiveness in both afferent and efferent arterioles, but did not significantly alter afferent arteriolar constrictor responses to KCl-induced depolarization. Afferent and efferent arteriolar constrictor responses to AngII were also diminished by an EGF receptor tyrosine kinase inhibitor. These observations suggest that tyrosine phosphorylation event(s) contribute to AngII-induced vasoconstrictor signaling in the renal microvasculature and that this process involves the EGF receptor tyrosine kinase. Further studies are required to determine if tyrosine phosphorylation is a critical step mediating AngII-induced renal arteriolar constriction or, alternatively, if tyrosine kinase activity exerts a modulatory influence on AngII responsiveness.
Acknowledgments
This work was supported by the National Institutes of Health (DK39202). Merck Sharp and Dohme Research Laboratories (Rahway, NJ, USA) provided enalaprilat for use in these studies.
References
- 1.Marrero MB, Schieffer B, Ma HP, Bernstein KE, Ling BN. ANG II-induced tyrosine phosphorylation stimulates phospholipase C-γ1 and Cl− channels in mesangial cells. Am J Physiol Cell Physiol. 1996;270:C1834–C1842. doi: 10.1152/ajpcell.1996.270.6.C1834. [DOI] [PubMed] [Google Scholar]
- 2.Marrero MB, Paxton WG, Duff JL, Berk BC, Bernstein KE. Angiotensin II stimulates tyrosine phosphorylation of phospholipase C-γ1 in vascular smooth muscle cells. J Biol Chem. 1994;269:10935–10939. [PubMed] [Google Scholar]
- 3.Bokemeyer D, Schmitz U, Kramer HJ. Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor. Kidney Int. 2000;58:549–558. doi: 10.1046/j.1523-1755.2000.t01-1-00201.x. [DOI] [PubMed] [Google Scholar]
- 4.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 5.Meloche S, Landry J, Huot J, Houle F, Marceau F, Giasson E. p38 MAP kinase pathway regulates angiotensin II-induced contraction of rat vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2000;279:H741–H751. doi: 10.1152/ajpheart.2000.279.2.H741. [DOI] [PubMed] [Google Scholar]
- 6.Watts SW, Florian JA, Monroe KM. Dissociation of angiotensin II-stimulated activation of mitogen-activated protein kinase kinase from vascular contraction. J Pharmacol Exp Ther. 1998;286:1431–1438. [PubMed] [Google Scholar]
- 7.Liu H, Sperelakis N. Tyrosine kinases modulate the activity of single L-type calcium channels in vascular smooth muscle cells from rat portal vein. Can J Physiol Pharmacol. 1997;75:1063–1068. [PubMed] [Google Scholar]
- 8.Carmines PK, Navar LG. Disparate effects of Ca channel blockade on afferent and efferent arteriolar responses to ANG II. Am J Physiol Renal Fluid Electrolyte Physiol. 1989;256:F1015–F1020. doi: 10.1152/ajprenal.1989.256.6.F1015. [DOI] [PubMed] [Google Scholar]
- 9.Carmines PK, Fowler BC, Bell PD. Segmentally distinct effects of depolarization on intracellular [Ca2+] in renal arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1993;265:F677–F685. doi: 10.1152/ajprenal.1993.265.5.F677. [DOI] [PubMed] [Google Scholar]
- 10.Loutzenhiser R, Hayashi K, Epstein M. Divergent effects of KCl-induced depolarization on afferent and efferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1989;257:F561–F564. doi: 10.1152/ajprenal.1989.257.4.F561. [DOI] [PubMed] [Google Scholar]
- 11.Loutzenhiser K, Loutzenhiser R. Angiotensin II-induced Ca2+ influx in renal afferent and efferent arterioles: Differing roles of voltage-gated and store-operated Ca2+ entry. Circ Res. 2000;87:551–557. doi: 10.1161/01.res.87.7.551. [DOI] [PubMed] [Google Scholar]
- 12.Casellas D, Navar LG. In vitro perfusion of juxtamedullary nephrons in rats. Am J Physiol Renal Fluid Electrolyte Physiol. 1984;246:F349–F358. doi: 10.1152/ajprenal.1984.246.3.F349. [DOI] [PubMed] [Google Scholar]
- 13.Ohishi K, Carmines PK, Inscho EW, Navar LG. EDRF-angiotensin II interactions in rat juxtamedullary afferent and efferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1992;263:F900–F906. doi: 10.1152/ajprenal.1992.263.5.F900. [DOI] [PubMed] [Google Scholar]
- 14.Ikenaga H, Fallet RW, Carmines PK. Contribution of tubuloglomerular feedback to renal arteriolar angiotensin II responsiveness. Kidney Int. 1996;49:34–39. doi: 10.1038/ki.1996.5. [DOI] [PubMed] [Google Scholar]
- 15.Ito S, Amin J, Ren YL, Arima S, Abe K, Carretero OA. Heterogeneity of angiotensin action in renal circulation. Kidney Int. 1997;52:S128–S131. [PubMed] [Google Scholar]
- 16.Harrison-Bernard LM, Carmines PK. Juxtamedullary microvascular responses to arginine vasopressin in rat kidney. Am J Physiol. 1994;267:F249–F256. doi: 10.1152/ajprenal.1994.267.2.F249. [DOI] [PubMed] [Google Scholar]
- 17.Ikenaga H, Bast JP, Fallet RW, Carmines PK. Exaggerated impact of ATP-sensitive K+ channels on afferent arteriolar diameter in diabetes mellitus. J Am Soc Nephrol. 2000;11:1199–1207. doi: 10.1681/asn.v1171199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loutzenhiser R, Epstein M, Hayashi K, Takenaka T, Forster H. Characterization of the renal microvascular effects of angiotensin II antagonist, DuP 753: Studies in isolated perfused hydronephrotic kidneys. Am J Hypertens. 1991;4:309S–314S. doi: 10.1093/ajh/4.4.309s. [DOI] [PubMed] [Google Scholar]
- 19.Takenaka T, Suzuki H, Fujiwara K, Kanno Y, Ohno Y, Hayashi K, Nagahama T, Saruta T. Cellular mechanisms mediating rat renal microvascular constriction by angiotensin II. J Clin Invest. 1997;100:2107–2114. doi: 10.1172/JCI119745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casellas D, Carmines PK. Control of the renal microvasculature: cellular and integrative perspectives. Current Opin Nephrol Hyperten. 1996;5:57–63. doi: 10.1097/00041552-199601000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Bilder GE, Krawiec JA, McVety K, Gazit A, Gilon C, Lyall R, Zilberstein A, Levitzki A, Perrone MH, Schreiber AB. Tyrphostins inhibit PDGF-induced DNA synthesis and associated early events in smooth muscle cells. Am J Physiol Cell Physiol. 1991;260:C721–C730. doi: 10.1152/ajpcell.1991.260.4.C721. [DOI] [PubMed] [Google Scholar]
- 22.Levitzki A, Gilon S. Tyrphostins as molecular tools and potential antiproliferative drugs. Trends Pharmacol Sci. 1991;12:171–174. doi: 10.1016/0165-6147(91)90538-4. [DOI] [PubMed] [Google Scholar]
- 23.Lyall RM, Zilberstein A, Gazit A, Gilon C, Levitzki A, Schlessinger J. Tyrphostins inhibit epidermal growth factor (EGF)-receptor tyrosine kinase activity in living cells and EGF-stimulated cell proliferation. J Biol Chem. 1989;264:14503–14509. [PubMed] [Google Scholar]
- 24.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 25.Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: synthesis and biological activity of protein tyrosine kinase inhibitors. J Med Chem. 1989;32:2344–2352. doi: 10.1021/jm00130a020. [DOI] [PubMed] [Google Scholar]
- 26.Touyz RM, Schiffrin EL. Angiotensin II regulates vascular smooth muscle cell pH, contraction, and growth via tyrosine kinase-dependent signaling pathways. Hypertension. 1997;30:222–229. doi: 10.1161/01.hyp.30.2.222. [DOI] [PubMed] [Google Scholar]
- 27.Touyz RM, Schiffrin EL. Tyrosine kinase signaling pathways modulate angiotensin II-induced calcium ([Ca2+]i) transients in vascular smooth muscle cells. Hypertension. 1996;27:1097–1103. doi: 10.1161/01.hyp.27.5.1097. [DOI] [PubMed] [Google Scholar]
- 28.Malloy LG, Sauro MD. Tyrosine kinase inhibition suppresses angiotensin contraction in hypertensive and normotensive small resistance arteries. Life Sci. 1996;58:PL317–PL324. doi: 10.1016/0024-3205(96)00147-6. [DOI] [PubMed] [Google Scholar]
- 29.Wijetunge S, Hughes AD. Effect of platelet-derived growth factor on voltage-operated calcium channels in rabbit isolated ear artery cells. Br J Pharmacol. 2000;115:534–538. doi: 10.1111/j.1476-5381.1995.tb16367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wijetunge S, Aalkjaer C, Schachter M, Hughes AD. Tyrosine kinase inhibitors block calcium channel currents in vascular smooth muscle cells. Biochem Biophys Res Commun. 1992;189:1620–1623. doi: 10.1016/0006-291x(92)90262-j. [DOI] [PubMed] [Google Scholar]
- 31.Carmines PK, Ohishi K, Ikenaga H. Functional impairment of renal afferent arteriolar voltage-gated calcium channels in rats with diabetes mellitus. J Clin Invest. 1996;98:2564–2571. doi: 10.1172/JCI119075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masumoto N, Nakayama K, Oyabe A, Uchino M, Ishii K, Obara K, Tanabe Y. Specific attenuation of the pressure-induced contraction of rat cerebral artery by herbimycin A. Eur J Pharmacol. 1997;330:55–63. doi: 10.1016/s0014-2999(97)00166-0. [DOI] [PubMed] [Google Scholar]
- 33.Watts SW, Yeum CH, Campbell G, Webb RC. Serotonin stimulates protein tyrosyl phosphorylation and vascular contraction via tyrosine kinase. J Vasc Res. 1996;33:288–298. doi: 10.1159/000159156. [DOI] [PubMed] [Google Scholar]
- 34.Gould EM, Rembold CM, Murphy RA. Genistein, a tyrosine kinase inhibitor, reduces Ca2+ mobilization in swine carotid media. Am J Physiol Cell Physiol. 1995;268:C1425–C1429. doi: 10.1152/ajpcell.1995.268.6.C1425. [DOI] [PubMed] [Google Scholar]
- 35.Ohanian J, Ohanian V, Shaw L, Bruce C, Heagerty AM. Involvement of tyrosine phosphorylation in endothelin-1-induced calcium-sensitization in rat small mesenteric arteries. Br J Pharmacol. 1997;120:653–661. doi: 10.1038/sj.bjp.0700950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Salvo J, Pfitzer G, Semenchuk LA. Protein tyrosine phosphorylation, cellular Ca2+, and Ca 2+ sensitivity for contraction of smooth muscle. Can J Physiol Pharmacol. 1994;72:1434–1439. doi: 10.1139/y94-207. [DOI] [PubMed] [Google Scholar]
- 37.Harris RC. EGF receptor activation by G-protein coupled receptors. Kidney Int. 2000;58:898–899. doi: 10.1046/j.1523-1755.2000.00240.x. [DOI] [PubMed] [Google Scholar]
- 38.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. The EMBO Journal. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marrero MB, Schieffer B, Paxton WG, Schieffer E, Bernstein KE. Electroporation of pp60c-src antibodies inhibits the angiotensin II activation of phospholipase C-g1 in rat aortic smooth muscle cells. J Biol Chem. 1995;270:15734–15738. doi: 10.1074/jbc.270.26.15734. [DOI] [PubMed] [Google Scholar]
- 40.Inagami T, Eguchi S, Numaguchi K, Motley ED, Tao H, Matsumoto T, Yamakawa T. Cross-talk between angiotensin II receptors and the tyrosine kinases and phosphatases. J Am Soc Nephrol. 1999;10:S57–S61. [PubMed] [Google Scholar]