

Abstract
Experiments were performed to evaluate the hypothesis that opening of Ca2+-activated K+ channels (BKCa channels) promotes juxtamedullary arteriolar dilation and curtails constrictor responses to depolarizing agonists. Under baseline conditions, afferent and efferent arteriolar lumen diameters averaged 23.4 ± 0.9 μm (n = 36) and 22.8 ± 1.1 μm (n = 13), respectively. The synthetic BKCa channel opener, NS1619, evoked concentration-dependent afferent arteriolar dilation. BKCa channel blockade (1 mM tetraethylammonium; TEA) decreased afferent diameter by 15 ± 3% and prevented the dilator response to 30 μM NS1619. Angiotensin II (ANG II; 10 nM) decreased afferent arteriolar diameter by 44 ± 4%, a response that was reduced by 30% during NS1619 treatment; however, TEA failed to alter afferent constrictor responses to either ANG II or arginine vasopressin. Neither NS1619 nor TEA altered agonist-induced constriction of the efferent arteriole. Thus, although the BKCa channel agonist was able to curtail afferent (but not efferent) arteriolar constrictor responses to ANG II, BKCa channel blockade did not allow exaggerated agonist-induced arteriolar constriction. These observations suggest that the BKCa channels evident in afferent arteriolar smooth muscle do not provide a prominent physiologic brake on agonist-induced constriction under our experimental conditions.
Keywords: afferent arteriole, angiotensin II, arginine vasopressin, efferent arteriole, NS1619, tetraethylammonium
INTRODUCTION
Calcium influx into arteriolar myocytes through dihydropyridine-sensitive voltage-gated channels and the resulting increase in intracellular Ca2+ concentration ([Ca2+]i) are important processes for maintaining renal vascular tone and evoking agonist-induced vasoconstriction. These events are especially prominent in the preglomerular afferent arteriole, exerting little impact on efferent arteriolar function (32). Because the open probability of voltage-gated Ca2+ channels is governed primarily by membrane potential, regulation of this parameter is crucial for appropriate control of preglomerular microvascular tone. The membrane potential of vascular smooth muscle is determined largely by sarcolemmal K+ conductance, with opening of K+ channels promoting hyperpolarization and closing of K+ channels favoring depolarization. Patch clamp studies have revealed several types of K+ channel in renal preglomerular vascular smooth muscle cells (15, 16, 18, 37, 44), including the large conductance Ca2+-activated K+ channel (BKCa channel). Expressed in smooth muscle cells from a variety of sources, the open probability of the BKCa channel is elevated exponentially by increases in [Ca2+]i and by membrane depolarization (5).
While involvement of BKCa channels in vascular control can be envisioned in several scenarios, their functional role is most widely viewed as providing negative feedback regulation of intrinsic vascular tone (3). This phenomenon arises by virtue of the effect of vasoconstrictor stimuli to induce membrane depolarization and increase [Ca2+]i, events that promote opening of BKCa channels, thereby preventing excessive depolarization and limiting Ca2+ influx through voltage-gated channels. Patch clamp studies have demonstrated that angiotensin II (ANG II) provokes negative feedback activation of BKCa channels in human mesangial cells (41) and vascular smooth muscle from canine renal artery (16). Conversely, a reduction in BKCa channel activity may contribute to agonist-induced membrane depolarization under some conditions. Such a process is indicated by the ability of ANG II to inhibit BKCa channels from coronary artery smooth muscle (43), a phenomenon proposed to involve channel phosphorylation (5). The ability of depolarizing and Ca2+ mobilizing agonists such as ANG II to variably increase or decrease BKCa channel activity could reflect functional differences between vascular beds (12, 26) or between macrovessels and microvessels (22).
The role of BKCa channels in regulating renal microvascular function has been scrutinized primarily with regard to the ability of P450 metabolites of arachidonic acid to alter channel activity (44, 45). However, little information is available regarding the involvement of BKCa channels in promoting or limiting agonist-induced renal vasoconstriction. Pressurized intrarenal arteries (150−250 μm diameter) isolated from rat kidney and studied under conditions of phenylephrine-induced tone have been shown to constrict in response BKCa channel blockade (37, 44), indicating that negative-feedback opening of BKCa channels tempers the constrictor impact of phenylephrine on the renal vasculature. In addition, electrophysiological studies have shown that endothelin-1 provokes negative-feedback activation of BKCa channels in smooth muscle cells isolated from interlobar and arcuate arteries (18). The impact of BKCa channel activity on afferent and efferent arteriolar responsiveness to peptide vasoconstrictor agonists remains unexplored. Accordingly, experiments were performed to test the hypothesis that opening of BKCa channels limits the afferent arteriolar contractile response to peptide agonists known to provoke depolarization and Ca2+ mobilization. Because efferent arteriolar function is relatively independent of membrane potential, we further postulated that BKCa channel activity would have little impact on contractile responses in this vascular segment.
METHODS
Animals
The procedures used in this study were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee and conducted according to the NIH Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (SAS:VAF strain) were purchased from Charles River Laboratories (Wilmington, MA) and provided ad libitum access to food and water prior to study.
The In Vitro Blood-Perfused Juxtamedullary Nephron Technique
Arteriolar contractile function was assessed in experiments performed using the in vitro blood-perfused juxtamedullary nephron technique (10). Each rat was anesthetized with pentobarbital sodium (50 mg/kg ip). For some experiments, enalaprilat (2 mg ia) was administered to suppress endogenous ANG II formation and its impact on vascular tone. Thirty minutes later, the right renal artery was cannulated via the superior mesenteric artery. This procedure initiated in situ perfusion of the kidney with Tyrode's solution containing 52 g/L dialyzed bovine serum albumin. The rat was then exsanguinated via a carotid arterial cannula into a heparinized syringe and the kidney was harvested for in vitro study. Renal perfusion was maintained throughout the dissection procedure needed to reveal the tubules, glomeruli, and vasculature of juxtamedullary nephrons. Ligatures were placed around the distal segments of the large arterial branches that supplied the exposed microvasculature.
The collected blood was processed to remove leukocytes and platelets, as detailed previously (20). The resulting perfusate was stirred continuously in a closed reservoir that was pressurized under 95% O2–5% CO2, thus providing both oxygenation and the driving force for perfusion of the dissected kidney at a constant renal arterial pressure of 110 mmHg. The renal perfusion chamber was warmed and the tissue surface was superfused with Tyrode's solution containing 10 g/L bovine serum albumin at 37°C. The renal microvasculature was transilluminated on the stage of a compound microscope and a single afferent or efferent arteriole was selected for study based on visibility and blood flow. Video images of each microvessel were generated continuously and stored on videotape for later analysis. In three experiments, two vessels could be visualized clearly within the same field of view, a situation that allowed responses of both vessels to be recorded simultaneously and analyzed separately during videotape playback.
Experimental Protocols
Effects of BKCa channel manipulation on afferent arteriolar diameter
The ability of BKCa channels to elicit afferent arteriolar dilation was assessed in experiments employing NS1619, a synthetic BKCa channel agonist (36). NS1619 was dissolved in ethanol to a concentration of 69 mM, and diluted in Tyrode's bath on the day of the experiment. Afferent arteriolar lumen diameter was monitored under baseline conditions and during sequential exposure to increasing concentrations of NS1619 via the bathing solution (10−300 μM; 5−7 min at each concentration), followed by a recovery period. The extent to which the dilator response to NS1619 could be attributed to opening of BKCa channels was examined based on responses to NS1619 during exposure to the BKCa channel blocker, tetraethylammonium chloride (TEA, 1mM; included in both the perfusate blood and the superfusate bath). Recovery of TEA-treated arterioles from NS1619 was followed by exposure to the organic Ca2+ channel blocker, diltiazem (10 μM), during the continued presence of TEA. Other studies assessed the ability of charybdotoxin (CbTX; 50 and 100 nM) to reverse the afferent response to 30 μM NS1619.
Effect of BKCa channel activation on ANG II-induced arteriolar vasoconstriction
These experiments evaluated the ability of pharmacological BKCa channel activation to suppress agonist-induced arteriolar constriction. Using tissue harvested from enalaprilat-treated rats, afferent and efferent arteriolar diameter responses to increasing concentrations of ANG II were evaluated by exposing the juxtamedullary microvasculature to the following superfusate bathing solutions: 1) Tyrode's solution alone (5−10 min); 2) Tyrode's solution containing 1 and 10 nM ANG II (3 min at each concentration); and 3) Tyrode's solution alone (10 min). Following this recovery period, NS1619 was added to the Tyrode's bathing solution to achieve a final concentration of 30 μM. A 5-min NS1619 treatment period preceded initiation of the second ANG II exposure sequence (1 and 10 nM ANG II; 3 min each) in the continued presence of NS1619. This was followed by a recovery period during which the tissue was exposed to Tyrode's solution containing NS1619 alone. Imposition of two consecutive ANG II exposure sequences according to this protocol evokes indistinguishable juxtamedullary arteriolar diameter responses, even at peptide concentrations 10-fold higher than those employed in the present study (6). The NS1619-containing bathing solutions employed in this protocol contained 0.04% ethanol. We previously found no effect of 0.1% ethanol on afferent arteriolar basal diameter or responsiveness to changes in perfusion pressure (42).
Effect of BKCa channel blockade on agonist-induced contractile responses
To determine the extent to which agonist-induced constriction is blunted by physiological (negative-feedback) activation of BKCa channels, TEA treatment was employed to block BKCa channels during exposure to vasoconstrictor agonists. Using tissue harvested from enalaprilat-treated rats, afferent or efferent arteriolar diameter responses to ANG II (1 and 10 nM) were documented before and during TEA exposure (1 mM in both perfusate blood and superfusate bath) according to the protocol described above. To determine the agonist-specificity of this phenomenon, additional experiments determined the effect of TEA on afferent and efferent arteriolar constrictor responses to 0.1 and 1 nM arginine vasopressin (AVP). AVP responsiveness studies employed tissue harvested from rats not subjected to enalaprilat pretreatment.
Chemicals and Reagents
Enalaprilat was a gift from Merck Research Laboratories (Rahway, NJ). NS1619 was purchased from RBI (Natick, MA) and diltiazem HCl from Marion Merrell Dow (Kansas City, MO). All other reagents were purchased from Sigma Chemical (St. Louis, MO).
Data Analysis
Arteriolar lumen diameter was measured from videotaped images at 12-sec intervals from a single point along the length of the vessel. The average diameter (in μm) during the final minute of each treatment period was utilized for statistical analysis by repeated measures ANOVA or Friedman repeated measures ANOVA on ranks, as appropriate, followed by Newman-Keuls test. Between-group comparisons employed the unpaired t-test. Statistical computations were performed utilizing the SigmaStat 2.03 software package (SPSS Inc, Chicago, IL), with statistical significance defined as P < 0.05. All data are reported as mean ± SE (n = number of arterioles).
RESULTS
Acute enalaprilat treatment was employed to suppress endogenous ANG II formation for experiments examining arteriolar responsiveness to ANG II, while other experiments utilized tissue from rats not receiving enalaprilat. As detailed in Table 1, neither afferent nor efferent arteriolar baseline diameter differed significantly between untreated and enalaprilat-treated groups.
Table 1.
Baseline arteriolar lumen diameters
| Afferent Arteriole | Efferent Arteriole | |
|---|---|---|
| Untreated tissue donors | 22.2 ± 1.1 μm (n = 19) |
21.5 ± 1.6 μm (n = 5) |
| Enalaprilat-treated tissue donors | 24.7 ± 1.4 μm (n = 17) |
23.7 ± 1.5 μm (n = 8) |
Values are means ± SE (n = number of arterioles). Kidneys and blood harvested from untreated or acutely enalaprilat-treated (2 mg, ia) Sprague-Dawley rats were utilized for in vitro studies of renal microvascular function.
Effects of BKCa channel manipulation on afferent arteriolar diameter
Figure 1 illustrates the effect of the BKCa agonist, NS1619, on afferent arteriolar lumen diameter. Baseline afferent diameter averaged 24.3 ± 1.9 μm (n = 5). Addition of NS1619 to the bathing solution produced a concentration-dependent increase in diameter, with 30 μM NS1619 increasing diameter by 2.8 ± 0.8 μm (P < 0.05 vs. baseline) and 300 μM NS1619 further increasing diameter to a value averaging 6.5 ± 0.8 μm greater than baseline (P < 0.05 vs. baseline; P < 0.05 vs 30 μM NS1619). Removal of NS1619 from the bathing solution restored of afferent arteriolar lumen diameter to 25.5 ± 1.9 μm (P > 0.05 vs. baseline). The reversible, concentration-dependent afferent arteriolar dilation evoked by NS1619 is consistent with the previously described actions of this agent to increase the open probability of BKCa channels in vascular smooth muscle (36).
Figure 1.
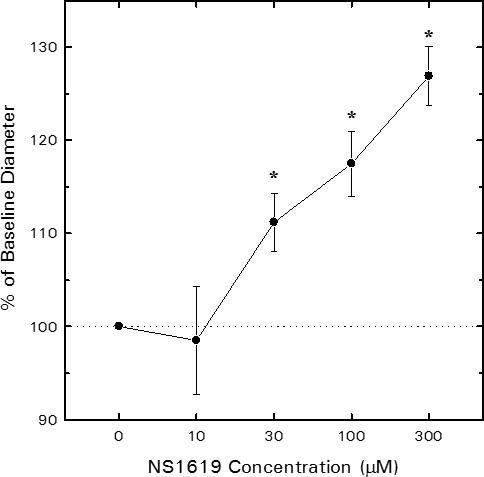
Influence of the synthetic BKCa channel agonist, NS1619, on juxtamedullary afferent arteriolar lumen diameter. Arteriolar diameter was monitored before and during sequential exposure to increasing concentrations of NS1619 via the bathing solution. Concentration-response profiles are shown as % of baseline diameter for tissue harvested from untreated rats (n = 5 arterioles). *P < 0.05 vs. baseline (0 μM NS1619).
Figure 2 summarizes data regarding the extent to which the afferent arteriolar diameter response to NS1619 can be attributed to opening of BKCa channels in our experimental setting. For comparison purposes, the dilator response to 30 μM NS1619 in the absence of any BKCa blocker (Figure 1) is also included in Figure 2. In experiments performed in their entirety with 1 mM TEA present in both the perfusate blood and the superfusate bath, 30 μM NS1619 failed to provoke afferent arteriolar dilation (−0.5 ± 1.1 μm, n = 4; Figure 2). Subsequent exposure to 100 and 300 μM NS1619 increased afferent diameter by 3.0 ± 1.1 and 5.4 ± 1.1 μm, respectively, responses that did not differ significantly from those observed in experiments performed in the absence of TEA (Figure 1). After recovery from NS1619, diltiazem (10 μM) evoked a 6.1 ± 2.5 μm increase in afferent diameter during TEA treatment (n = 4), confirming that this BKCa channel blocker does not provoke a non-specific abolition of vasodilator responsiveness. In other experiments, the afferent arteriolar dilation elicited by 30 μM NS1619 (Δ = 1.3 ± 0.4 μm; n = 4) was reversed upon addition of 50 nM CbTX to the NS1619-containing bath (−0.2 ± 0.6 μm vs baseline; Figure 2) and 100 nM CbTX further reduced arteriolar diameter to a value averaging 1.8 ± 0.7 μm below baseline. Because the CbTX- and TEA-sensitive nature of the afferent dilation provoked by 30 μM NS1619 indicates the involvement of BKCa channels in this response, this concentration was employed in all further experiments involving pharmacological BKCa channel activation.
Figure 2.
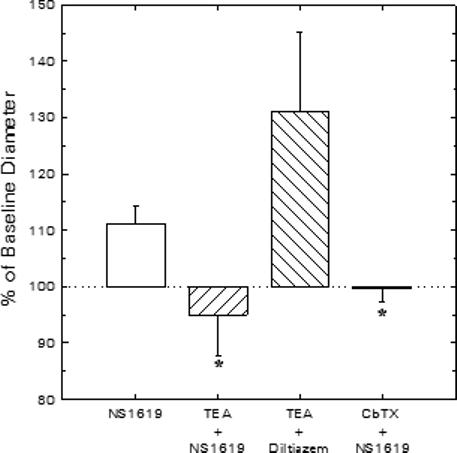
Effects of BKCa blockade on vasodilator responses of juxtamedullary afferent arterioles. Diameter responses to 30 μM NS1619 (BKCa agonist) or 10 μM diltiazem (Ca2+ channel antagonist) were assessed in the absence or presence of the BKCa channel blockers, TEA (1 mM) or CbTX (50 nM). *P < 0.05 vs. NS1619 alone.
Effect of BKCa channel activation on ANG II-induced arteriolar vasoconstriction
Figure 3 summarizes the effects of NS1619 on afferent and efferent arteriolar diameter responses to exogenous ANG II in kidneys harvested from enalaprilat-treated rats. Afferent arteriolar lumen diameter averaged 23.0 ± 1.4 μm (n = 8) under untreated baseline conditions and declined by 3.9 ± 1.2 and 10.5 ± 1.3 μm during exposure to 1 and 10 nM ANG II, respectively (Figure 3A). Upon removal of ANG II from the bathing solution, arteriolar diameter was restored to 102 ± 4% of baseline. Subsequent exposure to 30 μM NS1619 increased afferent diameter to 25.2 ± 1.7 μm (P < 0.05 vs untreated baseline). During continued NS1619 exposure, ANG II reduced afferent diameter to values averaging 2.6 ± 1.8 μm (1 nM ANG II) and 7.3 ± 1.1 μm (10 nM ANG II) below the NS1619 baseline. The response to 10 nM ANG II during NS1619 treatment was diminished significantly compared with to the response observed in the same arterioles prior to NS1619 treatment. On average, pharmacological opening of BKCa channels by NS1619 exposure suppressed afferent arteriolar responsiveness to 10 nM ANG II by 29 ± 7%.
Figure 3.
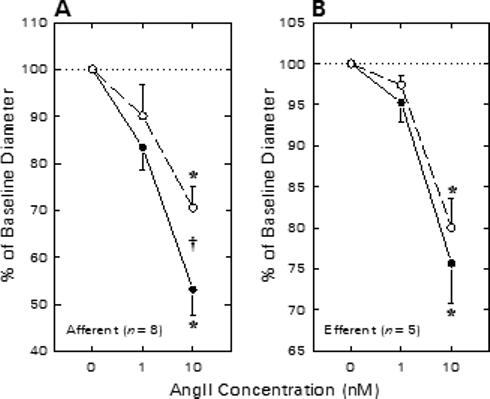
Effect of NS1619 on afferent (A) and efferent (B) arteriolar contractile responses to ANG II in kidneys from enalaprilat-treated rats. Arteriolar diameter responses to ANG II were monitored before (closed symbols) and during (open symbols) exposure to 30 μM NS1619. *P < 0.05 vs. baseline (0 nM ANG II); †P < 0.05 Untreated vs. NS1619.
Figure 3B summarizes the efferent arteriolar vasoconstrictor responses to ANG II in the absence and presence of NS1619. Efferent arteriolar baseline diameter averaged 24.8 ± 2.2 μm (n = 5) under untreated baseline conditions. ANG (1 nM) tended to decrease efferent diameter (Δ = −1.2 ± 0.6 μm) and 10 nM ANG II decreased lumen diameter by 5.7 ± 0.7 μm (P < 0.05 vs baseline). Efferent diameter was restored to 24.1 ± 2.5 μm during recovery from ANG II exposure. Subsequent exposure to 30 μM NS1619 did not significantly alter efferent diameter (23.7 ± 2.7 μm) or constrictor responses to ANG II (1 nM, Δ = −0.7 ± 0.3 μm; 10 nM, Δ = −4.5 ± 0.6 μm; both P > 0.05 vs untreated). Thus, in contrast to responses observed in afferent arterioles, treatment with the BKCa channel opener failed to alter efferent arteriolar constrictor responses to exogenous ANG II.
Effect of BKCa channel blockade on agonist-induced contractile responses
To determine the impact of physiological opening of BKCa channels on agonist-induced arteriolar constriction, ANG II responses were assessed before and during BKCa channel blockade. Afferent arteriolar diameter averaged 25.5 ± 3.3 μm (n = 6) under baseline conditions. As illustrated in Figure 4A, afferent diameter decreased by 4.3 ± 1.6 μm upon exposure to 1 nM ANG II (P < 0.05) and further declined to a value averaging 10.6 ± 2.0 μm below baseline when exposed to 10 nM ANG II (P < 0.05 vs. baseline). Removal of ANG II from the bath restored diameter to 25.4 ± 3.1 μm. Addition of 1 mM TEA to both the perfusate blood and the superfusate bath reduced afferent arteriolar diameter by 3.4 ± 1.0 μm (P < 0.05 vs. untreated baseline). During TEA treatment, ANG II reduced afferent arteriolar lumen diameter by 2.4 ± 0.7 μm (1 nM) and 9.4 ± 1.7 μm (10 nM). These responses to ANG II in the presence of TEA did not differ from those observed in the same arterioles prior to TEA treatment. In additional experiments, responses to 1 and 10 nM ANG II were assessed before and during exposure to either 100 nM iberiotoxin (n=2) or 50 nM charybdotoxin (n=1). ANG II (10 nM) decreased afferent diameter by 11.6 ± 1.5 μm before and 11.4 ± 0.4 μm during exposure to these highly specific BKCa antagonists. As these studies failed to reveal any tendency for BKCa blockade to alter ANG II-induced afferent arteriolar constriction, they were not further pursued because of the virtually prohibitive expense of using the scorpion toxins in our experimental setting.
Figure 4.
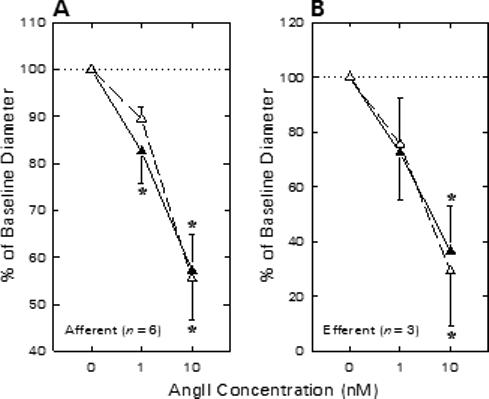
Effect of TEA on afferent (A) and efferent (B) arteriolar contractile responses to ANG II in kidneys from enalaprilat-treated rats. Arteriolar diameter responses to ANG II were monitored before (closed symbols) and during (open symbols) exposure to 1 mM TEA. *P < 0.05 vs. baseline (0 nM ANG II).
The effect of TEA treatment on efferent arteriolar ANG II responsiveness is shown in Figure 4B. Baseline efferent arteriolar diameter averaged 21.9 ± 1.1 μm (n = 3) and 1 and 10 nM ANG II reduced lumen diameter by 6.0 ± 3.6 and 13.8 ± 3.5 μm (P < 0.05 vs. baseline). During subsequent TEA treatment, baseline diameter averaged 20.5 ± 2.1 μm and ANG II reduced efferent diameter by 4.6 ± 2.9 and 13.7 ± 3.1 μm, respectively (both P > 0.05 vs. responses prior to TEA). Thus, although the BKCa channel agonist (NS1619) suppressed afferent arteriolar diameter responses to ANG II, neither the afferent nor the efferent arteriolar ANG II response was exaggerated after BKCa channel blockade.
The impact of BKCa channel blockade (TEA) on AVP-induced renal arteriolar constriction is illustrated in Figure 5. These experiments were performed using tissue harvested from rats not receiving enalaprilat treatment. Afferent arteriolar diameter averaged 21.6 ± 2.3 μm (n = 6) under untreated baseline conditions, and was diminished by 4.6 ± 1.8 and 15.3 ± 2.6 μm, respectively, during exposure to 0.1 and 1.0 nM AVP (Figure 5A). Upon removal of AVP from the bathing solution, afferent diameter was restored to 20.4 ± 2.0 μm (P > 0.05 vs baseline). Subsequent addition of TEA to both the perfusate blood and the superfusate bath reduced afferent diameter to 18.2 ± 1.8 μm. In the continued presence of TEA, 0.1 and 1 nM AVP reduced afferent diameter by 3.9 ± 1.0 and 13.2 ± 1.2 μm, respectively. These responses did not differ significantly from those exhibited by the same arterioles prior to TEA treatment. Hence, as was the case for ANG II, AVP-induced afferent arteriolar constriction was not exaggerated by TEA treatment.
Figure 5.
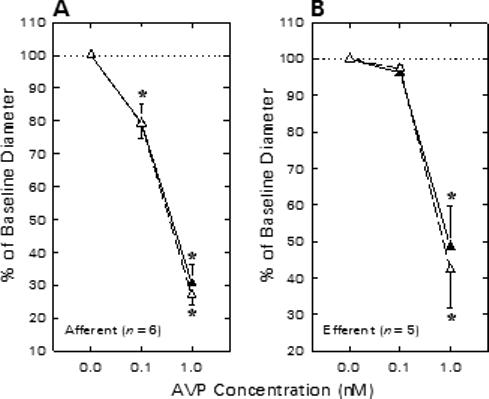
Effect of TEA on afferent (A) and efferent (B) arteriolar contractile responses to AVP. Arteriolar diameter responses to AVP were monitored before (closed symbols) and during (open symbols) exposure to 1 mM TEA. *P < 0.05 vs. baseline (0 nM AVP).
Efferent arteriolar AVP responsiveness in the absence and presence of 1 mM TEA is summarized in Figure 5B. Efferent diameter averaged 21.5 ± 1.6 μm under untreated baseline conditions (n = 5), tended to decline upon exposure to 0.1 nM AVP (Δ = −0.8 ± 0.2 μm) and significantly decreased in response to 1 nM AVP (Δ = −10.9 ± 2.1 μm). TEA treatment failed to alter efferent diameter (20.2 ± 2.1 μm) or the change in diameter elicited by AVP (0.1 nM, Δ = −0.5 ± 0.2 μm; 1 nM, Δ = −10.9 ± 1.4 μm). Thus, the efferent arteriolar constrictor response to AVP was unaffected by TEA treatment.
DISCUSSION
The results of the present study confirm and extend previous reports concerning the role of BKCa channels in regulating renal microvascular function. Afferent arteriolar diameter was reduced by BKCa channel blockade (TEA) and increased by a synthetic BKCa channel agonist (NS1619), indicating the ability of resident BKCa channels to influence preglomerular resistance. Pharmacological opening of BKCa channels attenuated the afferent arteriolar contractile response to AngII; however, BKCa channel blockade did not alter the constrictor response to either ANG II or AVP. Efferent arteriolar function was unaffected by pharmacological manipulation of BKCa channels. These observations suggest that renal arteriolar responses to these peptide vasoconstrictor agonists do not normally involve changes in BKCa channel activity. The validity of this reasoning relies on the efficacy and specificity of the pharmacologic agents employed to manipulate BKCa channel activity in our experimental setting.
Low millimolar extracellular concentrations of TEA are widely employed to achieve BKCa channel blockade. In rat mesenteric arterial myocytes, TEA inhibits mean unitary BKCa current with a dissociation constant (Kd) of 0.2 mM (27). The effect of 1 mM TEA is relatively specific, inhibiting KCa channels by ∼90% but exerting little or no impact on other K+ channels (Kd >7 mM for ATP-sensitive, voltage-dependent, and inward rectifier K+ channels; ref. (34)). Although the present studies relied heavily on the BKCa blocking effect of 1 mM TEA, similar effects of CbTX and IbTX were confirmed in a few experiments. Nanomolar concentrations of these peptidyl scorpion toxins provide potent and specific blockade of BKCa channels in a variety of experimental settings (25).
In recent years, several types of small-molecule compounds have been shown to possess BKCa agonist activity. Although some of these compounds act at intracellular sites and exhibit poor membrane permeability, benzimidazolone analogs (e.g. NS1619) have proven useful in physiological studies because of their ability to reversibly stimulate BKCa channel activity in intact cells (36). NS1619-induced hyperpolarization of basilar artery smooth muscle is rapidly reversed by IbTX, indicating the critical role of BKCa channels in this response, while NS1619-induced relaxation of the basilar artery is only partially blocked by IbTX (19). This phenomenon reflects the ability of NS1619 to inhibit L-type Ca2+ channels under some conditions (14, 19), and introduces the possibility that the NS1619-induced afferent arteriolar dilation observed in the present study might arise not only by direct activation of the BKCa channel (which subsequently decreases voltage-gated Ca2+ influx) but also via direct inhibition of the Ca2+ channel. L-type Ca2+ channels contribute significantly to basal afferent arteriolar tone in our experimental setting (8). Hence, if NS1619 influences the afferent arteriole via direct inhibition of the L-type Ca2+ channel, the vasodilator response to this agent would remain evident in the presence of TEA (which does not block Ca2+ channels). Indeed, the TEA-insensitive nature of the afferent arteriolar dilator response to high concentrations (100 and 300 μM) of NS1619 is consistent with the possibility that that this dilation arises via inhibition of L-type Ca2+ channels. However, it is critical to note that the dilator response to 30 μM NS1619 was prevented by 1 mM TEA and completely reversed by 50 μM CbTX. These observations provide compelling support for the contention that the afferent vasodilator response to 30 μM NS1619 arises predominantly via opening of BKCa channels in our experimental setting. Accordingly, this concentration of NS1619 was employed in all further experiments involving pharmacological BKCa channel activation.
Imig et al (21) were the first to report that 1 mM TEA produces afferent arteriolar constriction (∼10% decline in diameter) in the juxtamedullary microvasculature studied during perfusion with physiological salt solution. Quantitatively similar responses (−15 ± 3% decrease in afferent diameter; n = 12 arterioles pooled from the ANG II and AVP responsiveness experiments) were evident in the present study using the blood-perfused juxtamedullary nephron technique. These observations confirm a contribution of BKCa channels to basal tone of afferent arterioles in this experimental setting that preserves myogenic, tubuloglomerular feedback, and paracrine influences on the microvasculature. The stimulus underlying significant tonic BKCa channel activity under these conditions remains uncertain. In the isolated perfused hydronephrotic rat kidney, afferent arteriolar smooth muscle membrane potential is −40 mV (28). Previous reports indicate that BKCa channel blockers have no effect on membrane potential in the absence of induced hyperpolarization (4, 17), and that a background (cGMP-dependent) dilator influence is necessary to allow an effect of BKCa channel inhibition on mesangial cell function (40). Our experimental setting exhibits a significant NO-dependent tonic dilator influence on both afferent and efferent arterioles, as evidenced by constrictor responses to NO synthase inhibition (20, 35). Assuming that the juxtamedullary arteriolar dilator impact of NO arises, at least in part, via a cGMP-dependent mechanism (1), activation of BKCa channels by cGMP-dependent protein kinase (38) may contribute to the tonic impact of BKCa channels on afferent arteriolar resistance. BKCa channel activation counteracting myogenic tone may also occur under our experimental conditions, in accord with the ability of 1 mM TEA to potentiate the afferent arteriolar myogenic response in the isolated perfused hydronephrotic kidney (30). Regardless of the mechanism underlying tonic BKCa channel activation in the afferent arteriole, the capacity for further augmentation of the BKCa channel influence on afferent arteriolar tone is revealed by the CbTX- and TEA-sensitive dilator response to 30 μM NS1619. Thus, the results of the present study support the contention that BKCa channels exert a tonic dilator influence on the juxtamedullary afferent arteriole and that pharmacological opening of BKCa channels can further dilate this vascular segment. Efferent arteriolar diameter was unaffected by either TEA or NS1619, in accord with the relative insensitivity of this vascular segment to changes in membrane potential (7, 29).
Further evidence of that BKCa channels can influence afferent arteriolar function is provided by our observations concerning the effect of NS1619 on ANG II-induced vasoconstrictor responsiveness. The afferent arteriolar smooth muscle response to ANG II involves membrane depolarization (28) and Ca2+ influx through voltage-gated channels (8); hence, pharmacological opening of BKCa channels to promote membrane hyperpolarization can be expected to temper the constrictor response to ANGII. Indeed, to the extent that the effects of 30 μM NS1619 can be attributed to opening of BKCa channels, the ability of this agent to significantly attenuate afferent arteriolar ANG II responsiveness suggests that BKCa channels are capable of modulating agonist-induced vasoconstriction of this vascular segment. Efferent arteriolar responses to ANG II were unaffected by NS1619, in accord with the absence of ANG II-induced membrane depolarization (28) and minimal involvement of voltage-gated Ca2+ influx in the response of this vascular segment to ANG II (8).
While the effect of NS1619 on afferent arteriolar ANG II responsiveness suggests that resident BKCa channels are capable of influencing vasoconstrictor responsiveness, it does not indicate the extent to which this phenomenon occurs physiologically. Indeed, ANG II and other peptide agonists that provoke membrane depolarization and increases in [Ca2+]i may either inhibit BKCa channel activity to promote constriction or activate BKCa channels to temper constriction. We reasoned that pharmacological BKCa channel blockade should inhibit ANG II-induced constriction if BKCa channel closure normally contributes to ANGII-induced membrane depolarization. Conversely, if BKCa channels activate in a negative-feedback fashion to temper vasoconstriction, pharmacological BKCa channel blockade should allow an exaggerated vasoconstrictor response to ANG II. Because pharmacological BKCa channel blockade did not curtail AngII-induced vasoconstriction, inhibition of BKCa channels does not appear to be a prominent mechanism for eliciting ANG II-induced depolarization of afferent arteriolar smooth muscle. This conclusion is in accord with evidence that ANG II-induced depolarization of preglomerular microvascular smooth muscle arises primarily via opening of Ca2+-activated Cl− channels (6, 23, 24). To our surprise, pharmacological blockade of BKCa channels (TEA, IbTX or CbTX) did not allow exaggerated afferent arteriolar constrictor responses to either ANG II or AVP. These observations argue against a significant negative-feedback role of BKCa channels in tempering afferent arteriolar constrictor responses to depolarizing and Ca2+ mobilizing peptide agonists in our experimental setting.
Given the known effects of membrane potential and [Ca2+]i on the BKCa channel, and the effects of ANG II and AVP to provoke depolarization and increase [Ca2+]i in preglomerular microvascular smooth muscle, our observations might be explained if these peptides provoke additional signaling events that act to prevent or counteract negative feedback activation of the BKCa channel. 20-HETE is released from isolated perfused kidney exposed to ANG II (9) and is known to inhibit BKCa channels in renal microvascular smooth muscle (44); hence, this P-450 arachidonic acid metabolite may act to prevent negative feedback BKCa channel activation in response to ANG II. However, this mechanism is not likely involved in the response to AVP, which does not stimulate renal release of 20-HETE (9). Recently, protein kinase C (PKC) has been demonstrated to inhibit BKCa channels (2). Both ANG II and AVP stimulate PKC activity in rat vascular smooth muscle (11), and PKC blockade attenuates afferent arteriolar contractile and [Ca2+]i responses to ANG II (31, 39). The ability of ANG II and AVP to provoke afferent arteriolar vasoconstriction may critically rely on the receptor-dependent PKC activation and subsequent phosphorylation events that prevent negative feedback activation of BKCa channels. Nelson et al (33) have suggested that agonist-induced sparks of Ca2+ release from intracellular stores cause spatially-localized transient increases [Ca2+]i that, while not influencing the contractile apparatus or global [Ca2+]i, provoke opening of nearby BKCa channels to temper membrane depolarization. If this represents the primary mechanism responsible for negative feedback activation of BKCa channels, the strong dependence of afferent arteriolar ANG II and AVP responsiveness on membrane depolarization and Ca2+ influx, with lesser involvement of Ca2+ mobilization from intracellular stores, may generate insufficient Ca2+ spark activity to activate negative-feedback BKCa channel activation. These postulates require further investigation at the single cell level.
Because endothelial cells possess BKCa channels, the possibility exists that TEA acts on the endothelium in a manner that masks the impact of BKCa channel blockade in afferent arteriolar smooth muscle. Blockade of endothelial BKCa channels should depolarize the membrane, decrease Ca2+ influx through effects on the electrochemical driving force, decrease NO release and provoke vasoconstriction (13). If TEA influences the juxtamedullary microvasculature by decreasing NO release, it should mimic the effect of NO synthase inhibition. In our experimental setting, NO synthase inhibition provokes afferent and efferent arteriolar constriction and allows exaggerated vasoconstrictor responses to ANG II and AVP in both vascular segments (20). In contrast, TEA constricts only afferent arterioles and does not modify vasoconstrictor responses to ANG II or AVP in either vascular segment. Thus, it is unlikely that the effects of TEA on arteriolar diameter in our experimental setting result from a diminution of NO release, although we cannot rule out the possibility that endothelial BKCa blockade has other consequences that might mask direct effects of TEA on arteriolar smooth muscle function.
In summary, the in vitro blood-perfused juxtamedullary nephron technique was employed to explore the impact of BKCa channels on renal arteriolar function. Pharmacological opening of BKCa channels provoked afferent arteriolar dilation and reduced constrictor responsiveness to ANG II. Blockade of BKCa channels caused afferent arteriolar constriction but did not alter vasoconstrictor responsiveness to either ANG II or AVP. Efferent arteriolar diameter and vasoconstrictor responsiveness was unaltered by pharmacological manipulation of BKCa channel activity. These observations indicate that the BKCa channels influence afferent arteriolar tone, presumably by virtue of their effect on smooth muscle membrane potential. However, changes in BKCa channel activity do not appear to be prominently involved in engendering or modulating afferent arteriolar constrictor responses to ANG II and AVP under our experimental conditions.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant DK39202. The Nebraska Affiliate of the American Heart Association provided fellowship support for H. Ikenaga (postdoctoral) and J. P. Bast (predoctoral).
REFERENCES
- 1.Archer SL, Huang JMC, Hampl V, Nelson DP, Shultz PJ, Weir KE. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci USA. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonev AD, Jaggar JH, Rubart M, Nelson MT. Activators of protein kinase C decrease Ca2+ spark frequency in smooth muscle cells from cerebral arteries. Am J Physiol Cell Physiol. 1997;273:C2090–C2095. doi: 10.1152/ajpcell.1997.273.6.C2090. [DOI] [PubMed] [Google Scholar]
- 3.Brayden JE. Potassium channels in vascular smooth muscle. Clin Exp Pharmacol Physiol. 1996;23:1069–1076. doi: 10.1111/j.1440-1681.1996.tb01172.x. [DOI] [PubMed] [Google Scholar]
- 4.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 5.Carl A, Lee HK, Sanders KM. Regulation of ion channels in smooth muscles by calcium. Am J Physiol Cell Physiol. 1996;271:C9–C34. doi: 10.1152/ajpcell.1996.271.1.C9. [DOI] [PubMed] [Google Scholar]
- 6.Carmines PK. Segment-specific effect of chloride channel blockade on rat renal arteriolar contractile responses to angiotensin II. Am J Hypertens. 1995;8:90–94. doi: 10.1016/0895-7061(94)00170-G. [DOI] [PubMed] [Google Scholar]
- 7.Carmines PK, Fowler BC, Bell PD. Segmentally distinct effects of depolarization on intracellular [Ca2+] in renal arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1993;265:F677–F685. doi: 10.1152/ajprenal.1993.265.5.F677. [DOI] [PubMed] [Google Scholar]
- 8.Carmines PK, Navar LG. Disparate effects of Ca channel blockade on afferent and efferent arteriolar responses to ANG II. Am J Physiol Renal Fluid Electrolyte Physiol. 1989;256:F1015–F1020. doi: 10.1152/ajprenal.1989.256.6.F1015. [DOI] [PubMed] [Google Scholar]
- 9.Carroll MA, Balazy M, Margiotta P, Huang DD, Falck JR, McGiff JC. Cytochrome P-450-dependent HETEs: Profile of biological activity and stimulation by vasoactive peptides. Am J Physiol Regulatory Integrative Comp Physiol. 1996;271:R863–R869. doi: 10.1152/ajpregu.1996.271.4.R863. [DOI] [PubMed] [Google Scholar]
- 10.Casellas D, Navar LG. In vitro perfusion of juxtamedullary nephrons in rats. Am J Physiol Renal Fluid Electrolyte Physiol. 1984;246:F349–F358. doi: 10.1152/ajprenal.1984.246.3.F349. [DOI] [PubMed] [Google Scholar]
- 11.Chardonnens D, Lang U, Capponi AM, Vallotton MB. Comparison of the effects of angiotensin II and vasopressin on cytosolic free calcium concentration, protein kinase C activity, and prostacyclin production in cultured rat aortic and mesenteric smooth muscle cells. J Cardiovasc Pharmacol. 1989;14(Suppl. 6):S39–S44. [PubMed] [Google Scholar]
- 12.Clark SG, Fuchs LC. Role of nitric oxide and Ca++-dependent K+ channels in mediating heterogeneous microvascular responses to acetylcholine in different vascular beds. J Pharmacol Exp Ther. 1997;282:1473–1479. [PubMed] [Google Scholar]
- 13.Demirel E, Rusko J, Laskey RE, Adams DJ, van Breemen C. TEA inhibits ACh-induced EDRF release: Endothelial Ca2+- dependent K+ channels contribute to vascular tone. Am J Physiol Heart Circ Physiol. 1994;267:H1135–H1141. doi: 10.1152/ajpheart.1994.267.3.H1135. [DOI] [PubMed] [Google Scholar]
- 14.Edwards G, Niederste-Hollenberg A, Schneider J, Noack T, Weston AH. Ion channel modulation by NS 1619, the putative BKCa channel opener, in vascular smooth muscle. Br J Pharmacol. 1994;113:1538–1547. doi: 10.1111/j.1476-5381.1994.tb17171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gebremedhin D, Kaldunski M, Jacobs ER, Harder DR, Roman RJ. Coexistence of two types of Ca2+-activated K+ channels in rat renal arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F69–F81. doi: 10.1152/ajprenal.1996.270.1.F69. [DOI] [PubMed] [Google Scholar]
- 16.Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine renal artery: novel mechanism for agonist-induced membrane depolarization. Circ Res. 1995;77:121–130. doi: 10.1161/01.res.77.1.121. [DOI] [PubMed] [Google Scholar]
- 17.Gokina NI, Wellman TD, Bevan RD, Walters CL, Penar PL, Bevan JA. Role of Ca2+-activated K+ channels in the regulation of membrane potential and tone of smooth muscle in human pial arteries. Circ Res. 1996;79:881–886. doi: 10.1161/01.res.79.4.881. [DOI] [PubMed] [Google Scholar]
- 18.Gordienko DV, Clausen C, Goligorsky MS. Ionic currents and endothelin signaling in smooth muscle cells from rat renal resistance arteries. Am J Physiol Renal Fluid Electrolyte Physiol. 1994;266:F325–F341. doi: 10.1152/ajprenal.1994.266.2.F325. [DOI] [PubMed] [Google Scholar]
- 19.Holland M, Langton PD, Standen NB, Boyle JP. Effects of the BKCa channel activator, NS1619, on rat cerebral artery smooth muscle. Br J Pharmacol. 1996;117:119–129. doi: 10.1111/j.1476-5381.1996.tb15163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikenaga H, Fallet RW, Carmines PK. Basal nitric oxide production curtails arteriolar vasoconstrictor responses to ANG II in rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;271:F365–F373. doi: 10.1152/ajprenal.1996.271.2.F365. [DOI] [PubMed] [Google Scholar]
- 21.Imig JD, Zou A-P, Stec DE, Harder DR, Falck JR, Roman RJ. Formation and actions of 20-hydroxyeicosatetraenoic acid in rat renal arterioles. Am J Physiol Regulatory Integrative Comp Physiol. 1996;270:R217–R227. doi: 10.1152/ajpregu.1996.270.1.R217. [DOI] [PubMed] [Google Scholar]
- 22.Jackson WF, Blair KL. Characterization and function of Ca2+-activated K+ channels in arteriolar muscle cells. Am J Physiol Heart Circ Physiol. 1998;274:H27–H34. doi: 10.1152/ajpheart.1998.274.1.H27. [DOI] [PubMed] [Google Scholar]
- 23.Jensen BL, Ellekvist P, Skott O. Chloride is essential for contraction of afferent arterioles after agonists and potassium. Am J Physiol Renal Physiol. 1997;272:F389–F396. doi: 10.1152/ajprenal.1997.272.3.F389. [DOI] [PubMed] [Google Scholar]
- 24.Jensen BL, Skott O, Temdrup C. Blockade of chloride channels by DIDS stimulates renin release and inhibits contraction of afferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F718–F727. doi: 10.1152/ajprenal.1996.270.5.F718. [DOI] [PubMed] [Google Scholar]
- 25.Kaczorowski GJ, Garcia ML. Pharmacology of voltage-gated and calcium-activated potassium channels. Curr Opin Chem Biol. 1999;3:448–458. doi: 10.1016/S1367-5931(99)80066-0. [DOI] [PubMed] [Google Scholar]
- 26.Kim CJ, Kim KW, Park JW, Lee JC, Zhang JH. Role of Ca2+-dependent K+ channels in erythrocyte lysate-induced contraction of rabbit cerebral artery. Neurol Res. 1999;21:705–711. doi: 10.1080/01616412.1999.11741002. [DOI] [PubMed] [Google Scholar]
- 27.Langton PD, Nelson MT, Huang Y, Standen NB. Block of calcium-activated potassium channels in mammalian arteriolar myocytes by tetraethylammonium ions. Am J Physiol Heart Circ Physiol. 1991;260:H927–H934. doi: 10.1152/ajpheart.1991.260.3.H927. [DOI] [PubMed] [Google Scholar]
- 28.Loutzenhiser R, Chilton L, Trottier G. Membrane potential measurements in renal afferent and efferent arterioles: actions of angiotensin II. Am J Physiol Renal Physiol. 1997;273:F307–F314. doi: 10.1152/ajprenal.1997.273.2.F307. [DOI] [PubMed] [Google Scholar]
- 29.Loutzenhiser R, Hayashi K, Epstein M. Divergent effects of KCl-induced depolarization on afferent and efferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1989;257:F561–F564. doi: 10.1152/ajprenal.1989.257.4.F561. [DOI] [PubMed] [Google Scholar]
- 30.Loutzenhiser RD, Parker MJ. Hypoxia inhibits myogenic reactivity of renal afferent arterioles by activating ATP-sensitive K+ channels. Circ Res. 1994;74:861–869. doi: 10.1161/01.res.74.5.861. [DOI] [PubMed] [Google Scholar]
- 31.Nagahama T, Hayashi K, Ozawa Y, Takenaka T, Saruta T. Role of protein kinase C in angiotensin II-induced constriction of renal microvessels. Kidney Int. 2000;57:215–223. doi: 10.1046/j.1523-1755.2000.00822.x. [DOI] [PubMed] [Google Scholar]
- 32.Navar LG, Inscho EW, Majid DSA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- 33.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 34.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 35.Ohishi K, Carmines PK, Inscho EW, Navar LG. EDRF-angiotensin II interactions in rat juxtamedullary afferent and efferent arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1992;263:F900–F906. doi: 10.1152/ajprenal.1992.263.5.F900. [DOI] [PubMed] [Google Scholar]
- 36.Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca2+-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol. 1994;251:53–59. doi: 10.1016/0014-2999(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 37.Prior HM, Yates MS, Beech DJ. Functions of large conductance Ca2+-activated (BKCa), delayed rectified (Kv) and background K+ channels in the control of membrane potential in rabbit renal arcuate artery. J Physiol (Lond) 1998;511:159–169. doi: 10.1111/j.1469-7793.1998.159bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 1993;265:C299–C303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- 39.Salomonsson M, Kornfeld M, Gutierrez AM, Magnusson M, Persson AE. Effects of stimulation and inhibition of protein kinase C on the cytosolic calcium concentration in rabbit afferent arterioles. Acta Physiol Scand. 1997;161:271–279. doi: 10.1046/j.1365-201X.1997.d01-1962.x. [DOI] [PubMed] [Google Scholar]
- 40.Sansom SC, Stockand JD. Physiological role of large, Ca2+-activated K+ channels in human glomerular mesangial cells. Clin Exp Pharmacol Physiol. 1996;23:76–82. doi: 10.1111/j.1440-1681.1996.tb03066.x. [DOI] [PubMed] [Google Scholar]
- 41.Stockand JD, Sansom SC. Large Ca2+-activated K+ channels responsive to angiotensin II in cultured human mesangial cells. Am J Physiol Cell Physiol. 1994;267:C1080–C1086. doi: 10.1152/ajpcell.1994.267.4.C1080. [DOI] [PubMed] [Google Scholar]
- 42.Takenaka T, Harrison-Bernard LM, Inscho EW, Carmines PK, Navar LG. Autoregulation of afferent arteriolar blood flow in juxtamedullary nephrons. Am J Physiol Renal Fluid Electrolyte Physiol. 1994;267:F879–F887. doi: 10.1152/ajprenal.1994.267.5.F879. [DOI] [PubMed] [Google Scholar]
- 43.Toro L, Amador M, Stefani E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. Am J Physiol Heart Circ Physiol. 1990;258:H912–H915. doi: 10.1152/ajpheart.1990.258.3.H912. [DOI] [PubMed] [Google Scholar]
- 44.Zou A-P, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. 20-HETE is an endogenous inhibitor of the large-conductance Ca2+-activated K+ channel in renal arterioles. Am J Physiol Regulatory Integrative Comp Physiol. 1996;270:R228–R237. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 45.Zou A-P, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K+-channel activity. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F822–F832. doi: 10.1152/ajprenal.1996.270.5.F822. [DOI] [PubMed] [Google Scholar]