

Abstract
Shear stress increases NO production by endothelial cells, inner medullary collecting duct cells, and thick ascending limb. We postulated that the osmotic diuresis accompanying type 1 diabetes is associated with increased NOS activity and/or expression in the renal medulla. Diabetes was induced by injection of streptozotocin, with insulin provided to maintain moderate hyperglycemia (HYP) or euglycemia (EUG) for 3 weeks. SHAM rats received vehicle treatments. A separate group of rats (PHZ) received phlorizin to produce a glucose-dependent osmotic diuresis. Renal medullary NOS1 and NOS2 activities did not differ between groups, whereas NOS3 activity was significantly increased in HYP. Neither NOS1 nor NOS3 protein levels differed significantly between groups. Reduced phosphorylation of NOS3 at Thr495 and Ser633 was evident in medullary homogenates from HYP rats, with no difference apparent at Ser1177. Immunohistochemical analysis indicated prominent expression of pThr495NOS3 in the thick ascending limb and collecting duct of SHAM and PHZ rats. HYP rats displayed staining in the collecting duct but minimal thick ascending limb staining. Immunostaining with anti-pSer1177NOS3 was evident only in the thick ascending limb, with no apparent differences between groups. In summary, glucose-dependent osmotic diuresis alone did not alter NOS activity or expression in the renal medulla. Diabetic hyperglycemia increased medullary NOS3 activity without a concomitant increase in NOS3 protein levels; however, NOS3 phosphorylation was reduced at Thr495 and Ser633. Thus, changes in the phosphorylation of NOS at known regulatory sites might represent the primary mechanism underlying increased renal medullary NOS activity in diabetic hyperglycemia.
INTRODUCTION
An important physiological regulator of renal function, nitric oxide (NO) is synthesized as a byproduct of the conversion of l-arginine to l-citrulline. The greatest capacity for renal NO synthesis resides in the medulla (23;34), which expresses all three isoforms of NO synthase (NOS) (18). A paracrine NO system has been postulated to reside within the renal medulla, relying on cross-talk between epithelial and microvascular cells to match vasa recta blood flow to tubular metabolic transport activity (6). Accordingly, a decrease in NO production achieved by medullary interstitial infusion of NOS inhibitors leads to a reduction in medullary blood flow and salt retention, while infusion of NOS substrate increases medullary blood flow (15;28). A previous report from our laboratory revealed that NO production is modulated by shear stress in an inner medullary collecting duct cell line (IMCD-3), suggesting that tubular fluid flow may play a role in regulating sodium and water excretion by virtue of shear stress-mediated control of NO production (3). Moreover, Ortiz et al. (27) recently reported that flow stimulates translocation and phosphorylation of NOS3 and an associated increase in NO production by isolated thick ascending limb segments. However, it is unclear whether chronic increases in urine flow per se provoke an increase in NO synthesis and/or NOS expression in the renal medulla. One approach to examining this postulate is to determine if pathophysiological states associated with increased urine flow are associated with increased NOS activity in the renal medulla. For example, urine flow is significantly increased in type 1 diabetes mellitus (T1D) because of a glucose-dependent osmotic diuresis. Thus, if shear stress represents an important determinant of NO production in the thick ascending limb and/or collecting duct, one would predict that renal medullary NOS activity would be increased in T1D.
Most of the literature concerning the renal complications of T1D focuses on alterations in glomerular structure/function or impaired regulation of arteriolar tone/reactivity. However, recent studies have revealed that the early stage of T1D is accompanied by altered expression of multiple renal medullary transport proteins — e.g. the bumetanide-sensitive Na+-K+-2Cl− cotransporter; epithelial Na+ channel subunits α, β & γ; aquaporins 2 & 3; urea transporter UT-A1 (16;30). These changes should significantly influence salt and water balance, which also relies on appropriate NO regulation of medullary blood flow and transport in the distal nephron; however, little is known about the status of the medullary NO system in T1D. Shin et al. (29) detected increased NOS1 and NOS3 mRNA in the outer medulla of diabetic rats with no change evident in the inner medulla when compared with normal rats. These investigators also described a significant increase in NOS1 and NOS3 immunostaining in the proximal straight tubule and the medullary thick ascending limb of diabetic rats, with only a mild increase in NOS1 immunostaining observed in the outer medullary collecting duct and inner medullary collecting duct (29). Conversely, Choi et al. (4;5) found no change in the protein level of any NOS isoform in the renal medulla of rats studied 28 d after streptozotocin injection, although expression of all three isoforms was elevated in the cortex. To date, no studies have quantified NOS activity in the renal medulla of diabetic animals. The need to quantify NOS activity per se in this setting is underscored not only by the dearth of information available concerning renal medullary NOS protein levels in T1D but also by the fact that the activity of each NOS isoform is regulated by a complex array of post-transcriptional and post-translational events, any or all of which might be deranged in pathophysiological states. For example, exposure of endothelial cells to high glucose conditions results in O-glycosylation of NOS3 (7), which has the potential to impede serine/threonine phosphorylation of the enzyme at any of the numerous sites known to be important in regulating enzyme activity. Reduced NOS3 phosphorylation at Ser1177 has been documented in aortic endothelium from rats studied 16 weeks after onset of T1D (22); however, no information is available concerning the possibility that T1D alters NOS phosphorylation in the renal medulla.
The goal of the present study was to evaluate the hypothesis that increased flow through the distal nephron (as would occur in the polyuric states such as T1D) provokes an increase in renal medullary NOS activity. The strategy was to quantify NOS activity in rats with either a) streptozotocin-induced T1D, or b) polyuria resulting from phlorizin treatment of normal rats. Although increased medullary NOS3 activity was demonstrated in rats with T1D, phlorizin-induced osmotic diuresis yielding comparable urine flow was insufficient to provoke an increase in NOS activity. Accordingly, further experiments evaluated the contribution of increased NOS protein levels and/or post-translational events (phosphorylation at known regulatory sites) as mechanisms that might underlie increased renal medullary NOS activity in T1D.
METHODS
Animals
All animal procedures were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee and conducted according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Experiments were performed using male Sprague-Dawley rats (SAS:VAF strain) purchased from Charles River Laboratories (Wilmington, MA). At the beginning of the study (Day 0), blood glucose levels were measured (Accu-Check III model 766; Boehringer Mannheim, Indianapolis, IN) and each rat was weighed and assigned randomly to one of four groups: Normal rats receiving vehicle treatments (SHAM; n=17), diabetic rats with partial insulin replacement sufficient to maintain modest hyperglycemia (HYP; n=16), diabetic rats with insulin replacement to achieve euglycemia (EUG; n=7), and normal rats receiving phlorizin treatment to induce a glucose-dependent osmotic diuresis (PHZ; n=10).
Beginning on Day 0, rats in the PHZ group received 0.4 g/kg/day phlorizin delivered in twice daily subcutaneous injections of 40% phlorizin in propylene glycol. These animals were housed in pairs until the end of the study with free access to food and water containing 5% sucrose (to prevent weight loss due to chronic glucosuria).
Rats in the SHAM, HYP and EUG groups were anesthetized on Day 0 with methohexital sodium (50 mg/kg, ip) to facilitate intravenous injection of either 65 mg/kg streptozotocin (Sigma Chemical, St. Louis, MO; HYP and EUG rats) or vehicle (SHAM rats). These rats were allowed to recover from anesthesia and housed in pairs overnight with free access to food and water. The next day (Day 1), blood glucose levels were measured and the rats were anesthetized with methohexital sodium (50 mg/kg, ip) to facilitate subcutaneous insertion via a 16-G needle of either a 2.3×2.0 mm sustained-release insulin implant (HYP rats), a 5.25×2.0 mm sustained-release insulin implant (EUG rats) or a 2.3×2.0 mm micro-recrystallized palmitic acid implant (vehicle; SHAM rats). The insulin and vehicle implants were purchased from Linshin Canada (Scarborough, Ontario, Canada). After recovery from anesthesia, the animals were housed in pairs with free access to food and water until the end of the study.
Blood glucose measurements and body weight were measured in all rats at 3- to 4-d intervals for 19±1 days, and most animals were housed individually in metabolic cages (Nalge Nunc International, Rochester, NY) for 2 d just prior to sacrifice. The volume of urine collected (under oil) during the final 24-h period was determined gravimetrically and urine samples were centrifuged, filtered and stored at − 80°C until analysis. At the end of the study, rats were anesthetized with pentobarbital sodium (50 mg/kg, ip) and tracheotomized to facilitate spontaneous respiration. A blood sample was obtained from the abdominal aorta, centrifuged and the plasma stored at −80°C until analysis. Both kidneys were flushed with cold isotonic saline via the aorta, harvested, weighed, and the medulla was snap frozen in liquid N2 and stored at −80°C until analysis.
Analyses
NOS Activity
Total NOS activity in the renal medulla was determined on the basis of the rate of l-[3H]citrulline formation from l-[3H]arginine, as described previously (13). Briefly, the medulla of the left kidney was weighed, minced and homogenized in ice-cold buffer containing 50 mmol/L Tris, 0.1 mmol/L EGTA, 0.1 mmol/L EDTA, 10% glycerol, 0.1% β-mercaptoethanol, 1 μmol/L aprotinin, 1 μmol/L pepstatin A, 2 μmol/L leupeptin, and 1 mmol/L PMSF (pH 7.4). The homogenate was centrifuged at 100,000×g and separated into cytosolic and particulate fractions. The particulate fraction was resuspended in half of the original homogenization buffer along with the original concentration of inhibitors and homogenized. Aliquots of cytosolic and particulate fractions were incubated with [3H]arginine (71 Ci/mmol, 10 μmol/L final arginine concentration; Perkin-Elmer Life and Analytical Sciences, Shelton, CT) in the presence of 1 mmol/L NADPH, 30 nmol/L calmodulin, 3 μmol/L tetrahydrobiopterin, 2 mmol/L CaCl2, 3 mmol/L valine, 1 μmol/L FAD and 1 μmol/L flavin mononucleotide in a final volume of 50 μL for 30 min at room temperature. NOS activity was normalized to mg of protein (pmol/30 min/total protein). Total NOS activity was defined as [3H]arginine-to-[3H]citrulline conversion that was inhibited by the nonselective NOS inhibitor Nω-nitro-l-arginine (LNNA, 1 mmol/L). The activity of each NOS isoform was determined based on the actions of isoform-specific inhibitors: N5-(1-imino-3-butenyl)-l-ornithine (VNIO, 1 μmol/L) was employed as a specific NOS1 inhibitor, and 1400W dihydrochloride (1400W, 100 nmol/L) was used as a NOS2-specific inhibitor. NOS1 and NOS2 activities were defined as the VNIO-sensitive and 1400W-sensitive components of total NOS activity, respectively. NOS3 activity was estimated as Total NOS activity – (NOS1 specific activity + NOS2 specific activity).
Western Blotting
The Western blot protocol was performed as described previously (13;31). Briefly, proteins (300 μg) were separated on 7.5% SDS-PAGE and transferred to nitrocellulose by wet blotting (Bio-Rad Laboratories, Hercules, CA) for 55 min. The blots were allowed to air-dry for 20 min and blocked with 5% nonfat dry milk diluted in Tris buffered saline (blocking buffer) for 1 hr at room temperature. The blots were incubated overnight with primary antibody against NOS isoforms (NOS3, 1:1000; NOS1, 1:2500; Transduction Laboratories, Lexington, KY) or phosphorylation site-specific primary antibody (pSer1177NOS3, 1:500, Cell Signaling Technologies, Beverly, MA; pSer633NOS3, 1;500, Upstate Biotechnology, Lake Placid, NY; pThr495NOS3, 1:500; Upstate). The blots were incubated with the secondary antibody (horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit; 1:2000; Amersham Biosciences, Piscataway, NJ) for 1 hr at room temperature, followed by 5 washes with Tris-buffered saline. The specific bands were detected by use of the enhanced chemiluminescence (Supersignal Chemiluminescent Substrate; Pierce Biotechnology, Rockford, IL). Densitometry was used to quantify the bands with an Alpha Innotech Imaging System (San Leandro, CA). The Bradford assay was used to assess protein concentrations. Equal protein loading was verified by stripping with ReBlot Plus Stripping Solution (Chemicon International, Temecula, CA) and reprobing the immunoblots for β-actin (1:5000; Sigma-Aldrich Immunochemicals, St. Louis, MO).
Immunohistochemical Analysis
In some rats, kidneys were cleared of blood under pentobarbital anesthesia with heparinized saline via an aortic cannula and then perfused with 4% paraformaldehyde in 100 mmol/L dibasic sodium phosphate buffer. Kidneys were removed and immersed in 4% paraformaldehyde solution overnight at room temperature, transferred to 20% sucrose for 24 hours at 4°C and paraffin-embedded. The kidneys were sectioned at a thickness of 4 μm onto Superfrost plus slides. Endogenous peroxidase was blocked by exposure to 3% H2O2 for 15 min followed by washing successively in deionized, distilled H2O and phosphate-buffered saline for 5 min. Tissues were processed for specific staining by first utilizing Target Retrieval Solution (DakoCytomation, Carpinteria, CA), rinsing in phosphate-buffered saline, and incubating in the absence or presence of primary antibody (pThr495NOS3, 1:100; or pSer1177NOS3, 1:50) in humidity chambers overnight at 4°C. Slides were rinsed with phosphate-buffered saline followed by incubation with peroxidase-conjugated donkey anti-rabbit IgG (1:300, Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 min at room temperature. Specific staining was detected with diaminobenzamidine (DAKO liquid DAB substrate chromogen system, DakoCytomation, Carpinteria, CA), counterstained with Mayers hematoxylin and coverslipped with Cytoseal 60 mounting medium (Richard-Allan Scientific, Kalamazoo, MI). The stained sections were viewed with an Olympus BX40 microscope (Olympus America, Melville, NY) on brightfield setting fitted with a digital camera (Olympus DP12, Olympus America, Melville, NY).
Plasma and Urine Analyses
Electrolyte concentrations were determined by ion-selective electrodes (Synchron EL-ISE; Beckman Instruments, Brea, CA). Urinary cGMP concentration was measured by a scintillation proximity method, as described previously (14). ELISA kits were utilized to quantify urinary microalbumin concentration (Nephrat, Exocell, Inc., Philadelphia, PA) and plasma insulin concentration (ALPCO Diagnostics, Windham, NH). Urinary glucose concentration was determined by the hexokinase method using the Liquid Glucose Reagent Set (Pointe Scientific, Lincoln Park, MI).
Statistics
All values are expressed as means±SE (n=number of animals). Statistical comparisons were performed using one-way ANOVA followed by the Holm-Sidak test for multiple comparisons. If the data failed a normality test, the Kruskal-Wallis one-way ANOVA on ranks was applied in concert with Dunn's method for multiple comparisons. P-values <0.05 were considered significant.
RESULTS
Animal Characteristics
On Day 0 (prior to any experimental manipulation), the rats utilized in this study weighed 317±4 g and had blood glucose levels averaging 84±2 mg/dl (n=50). Twenty-four hours after streptozotocin injection, blood glucose levels were significantly elevated in HYP and EUG rats (332±14 mg/dl; n=23), confirming the onset of T1D prior to implantation of sustained release insulin pellets. The efficacy of the two insulin replacement regimens is indicated by significantly reduced plasma insulin levels and increased blood glucose concentration averaging approximately 350 mg/dl in HYP rats (Table 1), while EUG rats exhibited only a minimal increase in blood glucose concentration with plasma insulin levels that did not differ significantly from those observed in SHAM rats. Table 1 also summarizes other characteristics of the animals as evident approximately 3 wk after induction of T1D or initiation of phlorizin treatment. Relative to SHAM rats, animals in the HYP group displayed renal hypertrophy, polyuria, natriuresis, kaliuresis, and increased urinary excretion of cGMP. None of these parameters differed significantly between SHAM and EUG rats. The ability of the PHZ rat model to mimic the diuresis of T1D is evidenced by the fact that urine flow did not differ significantly in PHZ vs HYP rats, averaging 4−5 times that observed in SHAM rats. In addition, urinary glucose excretion was markedly increased in the PHZ group, although not to the extent observed in HYP rats (P=0.025). However, PHZ rats did not differ from SHAM rats with regard to any other of these basic characteristics.
Table 1.
Characteristics of SHAM, Euglycemic, Hyperglycemic and Phlorizin-treated rats
|
STZ-treated rats |
||||
|---|---|---|---|---|
| SHAM | EUG | HYP | PHZ | |
| BW, g | 385±13 (n=17) |
397±5 (n=7) |
346±9* (n=16) |
385±6 (n=10) |
| Left KW, mg | 1381±62 (n=12) |
1342±63 (n=7) |
1689±66* (n=12) |
1428±44 (n=10) |
| Left KW/BW, mg/g | 3.38±0.09 (n=12) |
3.38±0.15 (n=7) |
4.66±0.16* (n=12) |
3.71±0.09 (n=10) |
| Plasma [Insulin], μg/L | 2.75±0.33 (n=9) |
2.96±0.64 (n=7) |
1.09±0.12* (n=8) |
1.88±0.30 (n=10) |
| Blood [Glucose], mg/dl | 86±2 (n=17) |
100±11 (n=7) |
355±12* (n=16) |
78±1 (n=10) |
| Urine Flow, ml/h | 0.8±0.1 (n=9) |
1.1±0.1 (n=7) |
3.8±0.6* (n=8) |
3.2±0.3* (n=10) |
| Glucose Excretion, mg/h | 0.11±0.04 (n=9) |
20±9 (n=7) |
331±67* (n=8) |
188±18* (n=10) |
| Na+ Excretion, μmol/h | 66±4 (n=9) |
78±4 (n=7) |
93±10* (n=8) |
54±5 (n=6) |
| K+ Excretion, μmol/h | 217±8 (n=9) |
227±8 (n=7) |
323±28* (n=8) |
223±11 (n=10) |
| Microalbumin Excretion, μg/h | 55±15 (n=9) |
70±18 (n=7) |
113±53 (n=5) |
97±21 (n=7) |
| cGMP Excretion, pmol/h | 2720±470 (n=9) |
2786±484 (n=7) |
5369±425* (n=8) |
3028±647 (n=10) |
SHAM, rats receiving vehicle treatments; HYP, STZ-treated rats receiving partial insulin replacement; EUG, STZ-treated rats receiving insulin replacement sufficient to restore euglycemia; PHZ, rats receiving chronic phlorizin treatment; BW, body weight on date of sacrifice; KW, kidney weight. Blood [Glucose] values represent averages obtained from measurements made every 3−4 days from Day 1 until sacrifice. Excretory data were derived from 24-h urine collections in metabolic cages within 48 h of sacrifice. Plasma samples were obtained under anesthesia immediately prior to sacrifice. Values are means±SE (n=number of rats)
P<0.05 vs SHAM.
NOS Activity
Total NOS activity in the renal medulla was measured as l-NNA-sensitive l-[3H]citrulline formation from l-[3H]arginine in the presence of NOS cofactors. In tissue from SHAM rats (n=9), total NOS activity in the cytosolic fraction averaged 17.9±2.8 pmol/30min/mg protein, while activity in the particulate fraction averaged 16.1±2.6 pmol/30min/mg protein (Figure 1A & 1B). Based on the effects of isoform-specific inhibitors, ∼75% of total NOS activity in both the cytosolic and particulate fractions could be attributed to NOS3, with ∼20% of total activity reflecting NOS1 and ∼5% attributed to NOS2. This general trend was apparent in each of the animal groups.
Figure 1.

Renal medullary NOS activity in kidneys from rats receiving vehicle treatments (SHAM), streptozotocin-treated rats receiving partial insulin replacement (HYP), streptozotocin-treated rats receiving insulin replacement to restore euglycemia (EUG), and normal rats receiving chronic phlorizin treatment (PHZ). A: Total NOS, NOS1, NOS2 and NOS3 activities in the cytosolic fraction of renal medulla. B: Total NOS, NOS1, NOS2 and NOS3 activities in the particulate fraction of renal medulla. Activities of each isoform were determined based on effects of isoform-specific inhibitors (see text for details). Values are means±SE; n=7−10. *P<0.05 vs SHAM.
Total NOS activity in the cytosolic fraction of renal medulla from HYP rats was twice that observed in samples from SHAM rats (P<0.05), while values observed in tissue from EUG and PHZ rats did not differ significantly from SHAM (Figure 1A). This phenomenon was mirrored by an increase in NOS3 activity in the cytosolic fraction prepared from HYP rats (approximately double that observed in SHAM rats; P<0.05), while PHZ and EUG rats displayed NOS3 activities similar to SHAM. NOS1 activity in the cytosolic fraction tended to be greater in HYP than in SHAM; however, its value was numerically similar to EUG, and ANOVA failed to verify a significant difference in this parameter between the 4 treatment groups (P=0.22). Moreover, NOS2 activity in the cytosolic fraction did not differ between the treatment groups (P=0.59). In the particulate fraction, none of the treatment groups differed with regard to either total NOS activity or the activity of any single NOS isoform (Figure 1B; all P values >0.22). Thus, HYP (but not PHZ) rats displayed increased total NOS activity in the cytosolic fraction of renal medulla and this effect could be attributed primarily to an increase in NOS3 activity.
NOS Protein Levels
Renal medullary NOS1 and NOS3 protein levels were assessed by Western blot in the four groups of rats. Two immunoreactive bands for NOS1 were detected in the immunoblots of renal medullary tissue. Additional immunoblots were performed with two other NOS1 specific primary antibodies with the same results (data not shown). In all four groups, NOS1 protein levels were markedly higher in the cytosolic fraction than in the particulate fraction (Figure 2). There was a tendency toward increased NOS1 levels in medullary cytosolic fractions from HYP and PHZ rats; however, the densitometric data were quite variable. Accordingly, ANOVA failed to verify a significant difference between the 4 treatment groups with regard to NOS1 levels in either the cytosolic fraction (P=0.09) or the particulate fraction (P=0.19).
Figure 2.

NOS1 protein levels in cytosolic (C) and particulate (P) fractions of renal medulla from SHAM, EUG, HYP and PHZ rats. A: Representative Western blots. B: Summary of densitometric data, normalized to values observed in cytosolic fraction from SHAM rats. Values are means±SE. No significant between-group difference in NOS1 protein was apparent in either the cytosolic or the particulate fraction.
In contrast with NOS1 distribution, NOS3 levels tended to be greater in the particulate fraction than in the cytosolic fraction (Figure 3). SHAM, HYP, EUG and PHZ rats did not differ significantly with regard to NOS3 protein levels in either fraction (cytosolic P=0.07; particulate P=0.50). Thus, the immunoblotting data failed to provide convincing evidence that an increase in NOS3 protein levels underlies the elevated NOS3 activity apparent in the renal medulla of HYP rats.
Figure 3.

NOS3 protein levels in cytosolic (C) and particulate (P) fractions of renal medulla from SHAM, EUG, HYP and PHZ rats. A: Representative Western blots. B: Summary of densitometric data, normalized to values observed in cytosolic fraction from SHAM rats. Values are means±SE. No significant between-group difference in NOS3 protein was apparent in either the cytosolic or the particulate fraction.
NOS Phosphorylation
Phosphorylation of NOS3 at known regulatory sites was evaluated in medullary homogenates (without separation into cytosolic and particulate fractions) from SHAM and HYP rats by Western blot. As shown in Figure 4, pThr495NOS3/total-NOS3 in medullary tissue from HYP rats was only 30% of that detected in SHAM rats (P=0.002, n=8). pSer633NOS3/total-NOS3 in HYP tissue also averaged 30% of values evident in medullary homogenates from SHAM rats (P=0.02, n=8). However, this analysis failed to reveal a significant difference between SHAM and HYP rats with regard to pSer1177NOS3/total-NOS3 (P=0.16, n=8). Thus, decreased NOS3 phosphorylation at Thr495 and Ser633 is apparent in the renal medulla of HYP rats.
Figure 4.

Effect of T1D on NOS3 phosphorylation in renal medulla. A: Representative Western blots of medullary homogenates (without separation into cytosolic and membrane fractions). Phosphorylated and total NOS3 in each homogenate were probed on separate gels. As an equal loading control, each NOS3 and phospho-NOS3 blot was stripped and reprobed for β-actin (not shown). B: Densitometric values for phosphorylation site-specific NOS3 and NOS3 (each normalized to β-actin) were utilized to determine pNOS3/NOS3 for each phosphorylation site. Values are means±SE. n=8 rats in each group. *P<0.05 vs SHAM.
Immunohistochemical analysis
Immunohistochemical analysis was performed to localize pThr495NOS3 and pSer1177NOS3 in the renal medulla of SHAM, HYP and PHZ rats. As illustrated in Figure 5, pThr495NOS3 immunostaining is evident in the thick ascending limb and in both the outer and inner medullary segments of the collecting duct in kidneys from SHAM rats. Anti-pThr495NOS3 immunostaining of the collecting duct cells appears cytosolic and diffuse, in striking contrast with the predominant apical localization of staining in the thick ascending limb. Immunostaining for pThr495NOS3 is also apparent in the inner medullary vasa recta, as well as the endothelium of intrarenal arteries. In kidneys from HYP rats, pThr495NOS3 immunostaining is obvious in the outer and inner medullary segments of the collecting duct; however, minimal staining is evident in the thick ascending limb. PHZ rats demonstrated renal medullary pThr495NOS3 immunostaining patterns similar to that observed in SHAM rats (data not shown). Sections processed in the absence of primary antibody showed no immunostaining.
Figure 5.
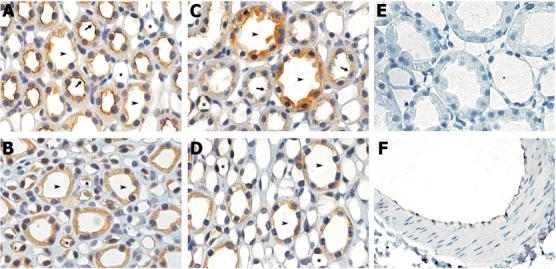
Representative immunohistochemical localization of pThr495NOS3 in the renal medulla from SHAM rats (A, B) and HYP rats (C - F). A, C & E: Outer medulla. B & D: Inner medulla. F: Intrarenal artery. Arrowheads, collecting duct; Arrows, thick ascending limb; Asterisks, vasa recta. Original magnification = 100×.
Immunostaining of pSer1177NOS3 in the renal medulla of SHAM and HYP rats (Figure 6) showed no demonstrable differences between groups. Immunostaining was present in the apical membrane of the thick ascending limb, but little or no staining was evident in the collecting duct. PHZ rats displayed similar patterns of medullary immunostaining for pSer1177NOS3 (data not shown).
Figure 6.
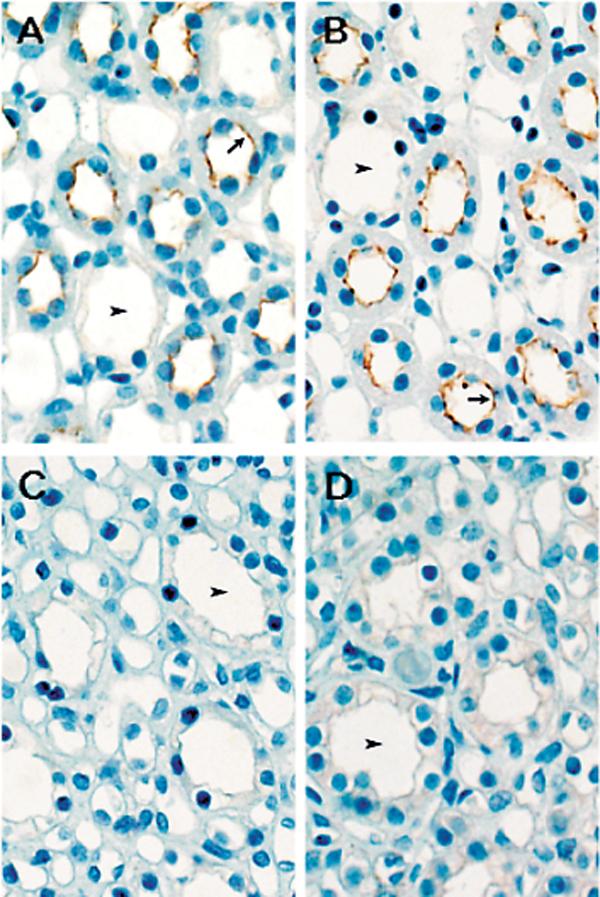
Immunohistochemical localization of pSer1177NOS3 in the renal medulla from SHAM rats (A & C) and HYP rats (B & D). A & B: Outer medulla. C & D: Inner Medulla. Arrowheads, collecting duct; Arrows, thick ascending limb. Original magnification = 100×.
DISCUSSION
The roles of NO and NOS in the diabetic kidney are controversial. A number of studies have produced contradictory findings, reviewed recently by Komers & Anderson (17), most likely reflecting differing levels of glycemic control and the phase of nephropathy. This report focuses on the early, hyperfiltration phase of T1D in a rat model with moderate hyperglycemia maintained by exogenous insulin therapy, thus resembling the clinically relevant state that ultimately engenders diabetic nephropathy. The major findings from the present study are: 1) NOS activity is increased in the cytosolic fraction of the renal medulla of diabetic rats; 2) this phenomenon can be attributed primarily to an increase in NOS3 activity; 3) the increased NOS activity accompanying T1D is prevented by insulin therapy sufficient to maintain euglycemia, but is not mimicked by a phlorizin-induced osmotic diuresis; 4) medullary NOS1 and NOS3 protein levels are not significantly altered in T1D and phlorizin-induced osmotic diuresis; 5) kidneys of diabetic rats exhibit decreased phosphorylation of renal medullary NOS3 at Thr495 and Ser633, but not at Ser1177; and 6) the reduced phosphorylation of NOS3 at Thr495 appears most evident in the thick ascending limb.
Shear stress or flow can increase NO production by cultured inner medullary collecting duct cells (IMCD-3) and isolated thick ascending limbs, suggesting that tubular fluid flow might influence sodium and water excretion via alterations in NO-dependent events in these nephron segments (3;27). However, no previous studies have explored the possibility that NOS activity in these (or other) medullary structures might be increased in response to a chronic in vivo change from conditions of normal tubular fluid flow to conditions of high flow. Accordingly, the present study addressed the hypothesis that the glucose-dependent osmotic diuresis associated with T1D, with the attendant chronic increase in tubular fluid flow through the thick ascending limb and collecting duct, would evoke changes in renal medullary NOS activity. Changes in medullary NOS activity in T1D might result not only from polyuria per se, but also as a direct effect of hyperglycemia and/or hypoinsulinemia on medullary structures. Hence, our approach was to determine if a chronic glucose-dependent osmotic diuresis, induced in otherwise normal rats, might mimic the effects of T1D on renal medullary NOS activity. Phlorizin, a competitive inhibitor of Na+-glucose co-transport (SGLT), was administered to normal rats to simulate the osmotic diuresis and high tubular fluid flow condition seen during the early stage of T1D. Indeed, while blood glucose and plasma insulin levels were similar in SHAM and PHZ rats, glucose excretion was markedly elevated in both HYP and PHZ rats, relative to SHAM and EUG rats. However, glucose excretion was significantly higher in HYP than in PHZ rats. As the HYP and PHZ rats had similar urine flows in this setting, the full diuretic effect of phlorizin might not be solely due to the glucose-dependent osmotic diuresis, but may also be promoted by effects of phlorizin on other renal transporters such as the Cl−/HCO −3 exchanger (21) or the Na+-myo-inositol transporter (8). Despite uncertainty regarding the full mechanism underlying the phlorizin-induced increase in urine flow, the primary effect is widely considered to represent a glucose-dependent osmotic diuresis. In the present study, with similar urine flows evident in HYP and PHZ rats, renal medullary NOS activity was increased in the HYP rats but not in the PHZ rats. Hsieh et al. (12) recently reported increased NOS1 mRNA in the cortex, outer medulla, and inner medulla of T1D rats and utilized phlorizin-treated normal and diabetic rats to provide evidence that renal NOS1 mRNA levels are correlated to high blood glucose, rather than high urinary glucose excretion. However, these authors did not determine whether a similar correlation was evident with regard to NOS1 enzymatic activity or protein expression, nor did they examine NOS2 and NOS3. Our data extend these observations by indicating that chronic glucose-dependent osmotic diuresis, with the attendant increase in tubular fluid flow through the loop of Henle and collecting duct, is insufficient to account for the increase in renal medullary NOS (mainly NOS3) activity accompanying T1D.
The present study demonstrates that the NOS3 activity and expression in the renal medulla are cytosolic as well as associated with membrane structures. In contrast, NOS3 is localized to plasma membrane caveolar domains and Golgi membranes in endothelial cells due to post-translational fatty acylation, specifically myristoylation and palmitoylation, at the NH2 terminus of the protein (32). Future experiments are needed to clarify whether or not NOS3 in the kidney is regulated by fatty acylation similar to aortic endothelial cells. This process might have functional implications with respect to the pool of enzyme available for stimulation or the bioavailability of NO produced by NOS3 associated with cytosolic or membrane structures in the renal medulla.
This study also demonstrates that the hyperglycemic condition increases cytosolic NOS3 activity without a concomitant increase in protein expression. Insulin treatment that lowers blood glucose levels normalized the increase in NOS activity. We previously reported an increase in total NOS activity in renal cortical homogenates without a corresponding upregulation in any of the three NOS isoforms (13); however, that study did not evaluate medullary NOS or the subcellular localization of NOS isoforms in T1D. Omer et al. (24) reported an increase in Ca2+-dependent NOS activity measured in whole kidney homogenates of female rats studied 2 wk after streptozotocin-induced T1D. Choi et al. (4;5) reported unaltered expression of all three NOS isoforms in the renal medulla after 7 and 28 days of streptozotocin-induced T1D, a result similar to the present study. On the other hand, Shin et al. (29) demonstrated increased NOS1 and NOS3 mRNA in the outer medulla of rats with streptozotocin-induced T1D. Furthermore, these investigators reported that NOS1 and NOS3 immunoreactive staining was substantially increased in the proximal straight tubule and medullary thick ascending limb of diabetic rats, with slight increases evident in the collecting duct (29). The work by Shin et al. (29) was conducted 6 wks post-streptozotocin, which may account for the difference with our study performed after 3 wks of diabetes.
Post-translational phosphorylation/dephosphorylation of NOS3 plays an active role in the stimulation of NO production in aortic endothelial cells by shear stress, insulin, and various other agonists (2;9). The five sites on NOS3 known to be regulated by serine/threonine phosphorylation in aortic endothelial cells are Ser116, Ser617, Ser633, Ser1177 and Thr495 (human amino acid sequence numbering). Relatively little is known about the roles of Ser116 and Ser617 in regulating NOS3 activity; however, studies of aortic endothelial cells suggest that NOS3 is activated by phosphorylation of Ser633 and Ser1177 and/or dephosphorylation of Thr495 (1). Ortiz and coworkers (27) recently provided the first evidence that NOS3 phosphorylation at Ser1177 is associated with increased NO production in the kidney, specifically in the thick ascending limb. The results of the present study provide further evidence that NOS3 phosphorylation at known regulatory sites is altered in the renal medulla under conditions associated with changes in NOS3 activity.
The immunoblotting analysis revealed decreased NOS3 phosphorylation at Ser633 and Thr495 in the renal medulla of HYP rats, with no significant change apparent at Ser1177. Dephosphorylation at Thr495 would favor NOS3 activation, while dephosphorylation at Ser633 would favor a decline in NOS3 activity (1). Given that NOS3 activity was increased in HYP rats (Figure 1), one might speculate that the phosphorylation state of Thr495 might be more predominant than that of Ser633 as a determinant of renal medullary NOS3 activity. Moreover, different pools of NOS3 (e.g. localized in specific medullary structures) could exist, with each potentially subject to distinct phosphoregulatory mechanisms. In an initial attempt to explore this postulate, immunostaining was employed to localize the renal medullary structures that display NOS3 phosphorylation at specific regulatory sites. As immunostaining with anti-pSer633NOS3 was unsuccessful using paraffin-embedded kidneys in this study, further research is necessary to localize renal medullary structures exhibiting NOS3 phosphorylation at Ser633, particularly those subject to dephosphorylation in diabetic rats. However, immunohistochemical analysis of pThr495NOS3 and pSer1177NOS3 proved to be informative.
Strong pThr495NOS3 immunostaining was apparent at the apical membrane of the thick ascending limb in kidneys from SHAM rats, with diffuse staining evident in both outer and inner medullary segments of the collecting duct. In the HYP group, pThr495NOS3 immunostaining was reduced only in the thick ascending limb, with staining maintained in the collecting duct. Thus, the reduced pThr495NOS3 levels evident in the immunoblotting data are likely a reflection of events occurring in the thick ascending limb. Localization of pThr495NOS3 in the apical membrane of the thick ascending limb in SHAM rats and the corresponding loss of phosphorylation at this site in the HYP rat kidneys should translate to an increase in NOS3 activity (Figure 1) without a concomitant increase in expression (Figure 3). Recently, Lin et al. (20) utilized mutant NOS3 transfectants to demonstrate that the dephosphorylation of Thr495 may be a “switch” that determines whether NOS3 produces NO or superoxide. At the present time, it is unknown whether dephosphorylation at Thr495 causes increased NO or superoxide production by NOS3 in the thick ascending limb.
Ortiz et al (27) described a doubling of Ser1177 phosphorylation at the apical membrane when isolated thick ascending limbs were introduced to flow conditions (27). The immunohistochemical results of the present study confirm the prominent localization of pSer1177NOS3 at the apical membrane of the thick ascending limb under conditions of normal flow (SHAM), as well as during chronic elevations in tubular fluid flow (HYP), with minimal pSer1177NOS3 apparent in the collecting duct. However, the immunoblotting analyses of the present study failed to reveal a difference between SHAM and HYP with regard to phosphorylation at Ser1177. Thus, NOS3 phosphorylation at Ser1177 in the thick ascending limb may be differently regulated within varied flow ranges (no flow-to-normal flow vs. normal flow-to-high flow) and/or in response to acute vs. chronic flow alterations.
Dephosphorylation of Thr495 and Ser633 may occur directly through activation of phosphatases and/or inactivation of kinase pathways under hyperglycemic conditions. Protein phosphatases 1 and 2A have been shown to be active in the thick ascending limb, where they may play a role in regulating Na+-K+-ATPase activity (19). Although protein phosphatase 2A has been implicated in the control of NOS3 dephosphorylation in endothelial cells (2;9), no data are available concerning whether or not this process contributes to regulation of NOS3 activity in the thick ascending limb. Inhibition of protein phosphatase 2B (calcineurin) blocks bradykinin-mediated dephosphorylation of Thr495NOS3 and NO production in cultured aortic endothelial cells (11). Moreover, protein phosphatase 2B activation is evident in the renal cortex during the early phase of T1D and is implicated in the renal and glomerular hypertrophy in diabetic rats (10). An inhibitory effect of hyperglycemia on activation of Akt kinase and subsequent NOS3 phosphorylation at Ser1177 has been reported to occur in endothelial cells (22); however, no previous reports have determined the impact of hyperglycemia on the phosphorylation status of Ser1177 in the kidney or the phosphorylation status of Thr495 or Ser633 in any cell type during hyperglycemia. Our observations implicate NOS3 dephosphorylation at Ser633 and Thr495 as a potentially important mechanism contributing to the renal complications of T1D; however, further studies are warranted to clarify how hyperglycemia alters the post-translational modifications of NOS3 in the renal medulla.
The physiological relevance of renal medullary NOS3 activation could have implications in the regulation of sodium and water transport and medullary blood flow. Renal medullary NOS3 activation in T1D likely involves, at least in part, dephosphorylation of Thr495NOS3 residing in the apical membrane of the thick ascending limb. Increased NO production by the thick ascending limb reduces Na+ reabsorption by that nephron segment (25), which would increase Na+ delivery to more distal segments of the nephron, thereby promoting natriuresis. However, the increased solute delivery to the macula densa would tend to suppress GFR via the tubuloglomerular feedback mechanism, a phenomenon that might temper the magnitude of diabetic hyperfiltration. Indeed, alterations in distal tubular fluid/solute delivery have been proposed to contribute to the hyperfiltration state through a tubuloglomerular feedback-dependent mechanism (33). On the other hand, if dephosphorylation at Thr495 acts as a molecular switch (20), it could increase superoxide production. An increase in superoxide production by the thick ascending limb would reduce Na+ delivery to the macula densa and other distal nephron segments (i.e. changes that are the opposite of those evoked by NO). These changes might arise as a consequence of decreased NO bioavailability (due to formation of peroxynitrite) and/or the direct effect of superoxide to increase Na+ reabsorption by the thick ascending limb (26). Moreover, it is also possible that increased NO production by the thick ascending limb would exert a paracrine influence on neighboring cells, altering transport function in the collecting duct and/or increasing blood flow through outer medullary vasa recta (6;15;28). As an additional caveat, the lack of information concerning the medullary structure(s) exhibiting dephosphorylation of NOS3 at Ser633 during T1D makes it impossible to speculate on the functional consequences of this phenomenon. Given the complexity and interactive nature of NO-dependent parameters in the renal medulla, having both epithelial and vascular effectors, it is evident that a thorough investigation of the biochemical and functional consequences of changes in NOS3 phosphorylation at specific regulatory sites will be required in order to unveil the impact of these processes on solute and volume homeostasis during T1D.
ACKNOWLEDGEMENTS
Rachel W. Fallet and William J. Langer provided valuable technical assistance. This work was supported by the NIH (DK63416 to PKC & JSP; HL60653-S1 Postdoctoral Supplement to JSP for support of DLL) and the American Heart Association (Predoctoral fellowship to JMS; Established Investigator Awards to DMP and JSP).
REFERENCES
- 1.Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, Sessa WC. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278:14841–14849. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- 2.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 3.Cai Z, Xin J, Pollock DM, Pollock JS. Shear stress-mediated NO production in inner medullary collecting duct cells. Am J Physiol Renal Physiol. 2000;279:F270–F274. doi: 10.1152/ajprenal.2000.279.2.F270. [DOI] [PubMed] [Google Scholar]
- 4.Choi KC, Kim NH, An MR, Kang DG, Kim SW, Lee JU. Alterations of intrarenal renin-angiotensin and nitric oxide systems in streptozotocin-induced diabetic rats. Kidney Int. 1997;51:S23–S27. [PubMed] [Google Scholar]
- 5.Choi KC, Lee SC, Kim SW, Kim NH, Lee JU, Kang YJ. Role of nitric oxide in the pathogenesis of diabetic nephropathy in streptozotocin-induced diabetic rats. Korean J Intern Med. 1999;14:32–41. doi: 10.3904/kjim.1999.14.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowley AW, Jr., Mori T, Mattson D, Zou A-P. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regulatory Integrative Comp Physiol. 2003;284:R1355–R1369. doi: 10.1152/ajpregu.00701.2002. [DOI] [PubMed] [Google Scholar]
- 7.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eladari D, Chambrey R, Pezy F, Podevin RA, Paillard M, Leviel F. pH dependence of Na+/myo-inositol cotransporters in rat thick limb cells. Kidney Int. 2002;62:2144–2151. doi: 10.1046/j.1523-1755.2002.00690.x. [DOI] [PubMed] [Google Scholar]
- 9.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regulatory Integrative Comp Physiol. 2003;284:R1–R12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 10.Gooch JL, Barnes JL, Garcia S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol. 2003;284:F144–F154. doi: 10.1152/ajprenal.00158.2002. [DOI] [PubMed] [Google Scholar]
- 11.Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–16591. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh MC, Wu CH, Chen CL, Chen HC, Chang CC, Shin SJ. High blood glucose and osmolality, but not high urinary glucose and osmolality, affect neuronal nitric oxide synthase expression in diabetic rat kidney. J Lab Clin Med. 2003;141:200–209. doi: 10.1067/mlc.2003.21. [DOI] [PubMed] [Google Scholar]
- 13.Ishii N, Patel KP, Lane PH, Taylor T, Bian K, Murad F, Pollock JS, Carmines PK. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J Am Soc Nephrol. 2001;12:1630–1639. doi: 10.1681/ASN.V1281630. [DOI] [PubMed] [Google Scholar]
- 14.Jackson RW, Treiber FA, Harshfield GA, Waller JL, Pollock JS, Pollock DM. Urinary excretion of vasoactive factors are correlated to sodium excretion. Am J Hypertens. 2001;14:1003–1006. doi: 10.1016/s0895-7061(01)02169-0. [DOI] [PubMed] [Google Scholar]
- 15.Kakoki M, Zou A-P, Mattson DL. The influence of nitric oxide synthase 1 on blood flow and interstitial nitric oxide in the kidney. Am J Physiol Regulatory Integrative Comp Physiol. 2001;281:R91–R97. doi: 10.1152/ajpregu.2001.281.1.R91. [DOI] [PubMed] [Google Scholar]
- 16.Kim D, Sands JM, Klein JD. Changes in renal medullary transport proteins during uncontrolled diabetes mellitus in rats. Am J Physiol Renal Physiol. 2003;285:F303–F309. doi: 10.1152/ajprenal.00438.2002. [DOI] [PubMed] [Google Scholar]
- 17.Komers R, Anderson S. Paradoxes of nitric oxide in the diabetic kidney. Am J Physiol Renal Physiol. 2003;284:F1121–F1137. doi: 10.1152/ajprenal.00265.2002. [DOI] [PubMed] [Google Scholar]
- 18.Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am J Physiol Renal Physiol. 1997;272:F561–F578. doi: 10.1152/ajprenal.1997.272.5.F561. [DOI] [PubMed] [Google Scholar]
- 19.Li D, Aperia A, Celsi G, da Cruz e Silva EF, Greengard P, Meister B. Protein phosphatase-1 in the kidney: evidence for a role in the regulation of medullary Na+-K+-ATPase. Am J Physiol. 1995;269:F673–F680. doi: 10.1152/ajprenal.1995.269.5.F673. [DOI] [PubMed] [Google Scholar]
- 20.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, Jr., Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- 21.Meyer G, Porta C, Garavaglia M, Cremaschi D. Inhibitors of the Cl−/HCO3− exchanger activate an anion channel with similar features in the epithelial cells of rabbit gallbladder: patch-clamp analysis. Pflugers Arch. 2001;441:467–473. doi: 10.1007/s004240000419. [DOI] [PubMed] [Google Scholar]
- 22.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser1179. J Biol Chem. 2001;276:30392–30398. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 23.Nava E, Llinás MT, González JD, Salazar FJ. Nitric oxide synthase activity in renal cortex and medulla of normotensive and spontaneously hypertensive rats. Am J Hyperten. 1999;9:1236–1239. doi: 10.1016/s0895-7061(96)00325-1. [DOI] [PubMed] [Google Scholar]
- 24.Omer S, Shan J, Varma DR, Mulay S. Augmentation of diabetes-associated renal hyperfiltration and nitric oxide production by pregnancy in rats. J Endocr. 1999;161:15–23. doi: 10.1677/joe.0.1610015. [DOI] [PubMed] [Google Scholar]
- 25.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol. 2002;282:F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz PA, Garvin JL. Superoxide stimulates NaCl absorption by the thick ascending limb. Am J Physiol Renal Physiol. 2002;283:F957–F962. doi: 10.1152/ajprenal.00102.2002. [DOI] [PubMed] [Google Scholar]
- 27.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. Am J Physiol Renal Physiol. 2004;287:F274–F280. doi: 10.1152/ajprenal.00382.2003. [DOI] [PubMed] [Google Scholar]
- 28.Pallone TL, Mattson DL. Role of nitric oxide in regulation of the renal medulla in normal and hypertensive kidneys. Curr Opin Nephrol Hypertens. 2002;11:93–98. doi: 10.1097/00041552-200201000-00014. [DOI] [PubMed] [Google Scholar]
- 29.Shin SJ, Lai FJ, Wen JD, Hsiao PJ, Hsieh MC, Tzeng TF, Chen HC, Guh JY, Tsai JH. Neuronal and endothelial nitric oxide synthase expression in outer medulla of streptozotocin-induced diabetic rat kidney. Diabetologia. 2000;43:649–659. doi: 10.1007/s001250051354. [DOI] [PubMed] [Google Scholar]
- 30.Song J, Knepper MA, Verbalis JG, Ecelbarger CA. Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am J Physiol Renal Physiol. 2003;285:F1125–F1137. doi: 10.1152/ajprenal.00143.2003. [DOI] [PubMed] [Google Scholar]
- 31.Sullivan JC, Pollock DM, Pollock JS. Altered nitric oxide synthase 3 distribution in mesenteric arteries of hypertensive rats. Hypertension. 2002;39:597–602. doi: 10.1161/hy0202.103286. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan JC, Pollock JS. NOS 3 subcellular localization in the regulation of nitric oxide production. Acta Physiol Scand. 2003;179:115–122. doi: 10.1046/j.1365-201X.2003.01181.x. [DOI] [PubMed] [Google Scholar]
- 33.Thomson SC, Vallon V, Blantz RC. Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol. 2004;286:F8–F15. doi: 10.1152/ajprenal.00208.2003. [DOI] [PubMed] [Google Scholar]
- 34.Wu F, Park F, Cowley AW, Jr., Mattson DL. Quantification of nitric oxide synthase activity in microdissected segments of the rat kidney. Am J Physiol Renal Physiol. 1999;276:F874–F881. doi: 10.1152/ajprenal.1999.276.6.F874. [DOI] [PubMed] [Google Scholar]