

Abstract
We present a theory of the dependence on sequence of the three-dimensional size of large single-stranded (ss) RNA molecules. The work is motivated by the fact that the genomes of many viruses are large ssRNA molecules—often several thousand nucleotides long—and that these RNAs are spontaneously packaged into small rigid protein shells. We argue that there has been evolutionary pressure for the genome to have overall spatial properties—including an appropriate radius of gyration, Rg—that facilitate this assembly process. For an arbitrary RNA sequence, we introduce the (thermal) average maximum ladder distance (〈MLD〉) and use it as a measure of the “extendedness” of the RNA secondary structure. The 〈MLD〉 values of viral ssRNAs that package into capsids of fixed size are shown to be consistently smaller than those for randomly permuted sequences of the same length and base composition, and also smaller than those of natural ssRNAs that are not under evolutionary pressure to have a compact native form. By mapping these secondary structures onto a linear polymer model and by using 〈MLD〉 as a measure of effective contour length, we predict the Rg values of viral ssRNAs are smaller than those of nonviral sequences. More generally, we predict the average 〈MLD〉 values of large nonviral ssRNAs scale as N0.67±0.01, where N is the number of nucleotides, and that their Rg values vary as 〈MLD〉0.5 in an ideal solvent, and hence as N0.34. An alternative analysis, which explicitly includes all branches, is introduced and shown to yield consistent results.
Keywords: branched polymer, ladder distance, radius of gyration, secondary structure, viral RNA
Very little is known about the native size and conformation of large (103–104 nt) single-stranded (ss) RNA molecules, a category that includes the genomes of ssRNA viruses. This represents a challenging physical problem, because complementary base pairing gives rise to branched secondary structures whose complexity increases with length. Almost all theoretical and experimental studies of the structures of ssRNA sequences have been devoted to exploring the secondary and tertiary structures of smaller (101–102 nt) ssRNAs, such as tRNAs (1, 2) and ribozymes (3, 4), or of large ssRNAs that are complexed with proteins in ribosomal subunits (5, 6).
Yet the native structures of large ssRNAs are also of biological importance; the most prevalent form of viral genome is ssRNA, and these molecules are necessarily thousands of bases long to code for several proteins. There has been extensive work to determine the secondary and tertiary structures of small (102 nt) subsequences of ssRNA viral genomes, because of their importance in, for instance, genome replication or packaging (7, 8). Some studies have explored specific long-range tertiary interactions in these ssRNAs (9). By contrast, investigations of the overall native 3D sizes of viral-length ssRNAs have been very limited (10), and no theoretical models that predict the sizes of long ssRNAs from their primary sequences have yet appeared in the literature.
Spontaneous in vitro self-assembly has been demonstrated for several ssRNA viruses (11, 12). In each case, the infectious virions can form in a buffer solution containing only the capsid protein and the viral genome, indicating that there is no thermodynamic barrier to assembly. We therefore expect there cannot be a large disparity between the native size of a viral ssRNA genome and that of its capsid—and that, by optimizing genome size, there will be an enhancement in the efficiency of virion assembly, and thus of viral reproduction and infectivity. Accordingly, we argue that there has been selective pressure on the ssRNA genome to have a size appropriate to its protective shell.
The size of an ssRNA is determined by its tertiary structure, which is determined by its secondary structure, which is determined by its primary sequence. Consequently, it is natural that there are two levels of coding in the primary sequence of a viral ssRNA molecule. Not only do its individual genes need to code “in the usual way” for their protein products, but the overall (many-gene) sequence must give rise to a secondary/tertiary structure consistent with a size that enables the genome to be packaged within the capsid. Related arguments have been made in refs. 13–15. Because of these unique selective pressures, the size of ssRNAs of self-assembling viruses should be different from the average size of random (or other nonviral) ssRNAs having the same length and base composition.
Owing to their sequence-dependent branched structure, the sizes of ssRNAs cannot be understood by using the simple models available for linear homopolymers, such as dsDNA (see, however, ref. 5, in which RNA size and shape are described by the configurational statistics associated with an “equivalent” semiflexible polymer). The simplest model for a linear homopolymer is the freely jointed chain, in which the molecule is represented as a series of equal-length rigid links connected by flexible joints. In this model, the two intrinsic properties that determine the size of the molecule are the length of the links, or Kuhn length (b), and the contour length (L), which is b times the number of links. Treating the L ≫ b polymer as a statistical object yields a well known scaling relationship for the root-mean-square radius of gyration, Rg (16):
with ν ranging between one-third for poor solvents, where polymer–solvent interactions are unfavorable (leading to polymer collapse), to approximately three-fifths for good solvents, where polymer excluded volume effects dominate. In “ideal” solvents, the attractive and repulsive interactions between distant polymer segments cancel, and ν = 1/2.
For ssRNAs, L, of course, still plays a fundamental role; but because of the dependence of secondary structure on primary sequence, it is necessary to identify alternative intrinsic properties of this branched heteropolymer that determine its overall size. To address this problem, we propose a mapping between certain coarse-grained secondary structure features of large ssRNA molecules and those of linear homopolymers, thereby enabling a predictive correlation between primary sequence and 3D size. In particular, we associate with an arbitrary sequence an ensemble-average maximum ladder distance (〈MLD〉) and argue that the corresponding ssRNA molecule behaves like a linear polymer of contour length 〈MLD〉, and hence whose radius of gyration scales as
The angled brackets indicate a thermal (i.e., Boltzmann-weighted) average taken over the entire ensemble of possible structures. The MLD, which will be defined more precisely in Results, is a measure of the length of the longest direct path across an RNA secondary structure. We find that b is only weakly dependent on sequence, whereas the 〈MLD〉 values are significantly smaller for viral ssRNA genomes than for nonviral sequences, both random and evolved, of the same length and composition.
Methods
Because secondary structures of large ssRNAs are difficult to determine experimentally, and because we wish to calculate average properties of the thermodynamic ensemble of secondary structures associated with each of a large number of widely varying sequences, we use predictions of the secondary structure made by RNAsubopt, a program in the Vienna RNA Package, Version 1.7 (17). To evaluate robustness, we compare the results from RNAsubopt with those from three other RNA folding programs: RNAfold (also from the Vienna RNA Package); mfold, Version 3.1 (18); and a program we developed that employs a deliberately simplified energy model.
RNAsubopt, RNAfold, and mfold incorporate detailed empirically derived estimates of the free energy changes associated with loop closure and base stacking to estimate the free energies of nonpseudoknotted secondary structures formed from GC, AU, and GU base pairs; the restriction against pseudoknots means that, for any secondary structure in which base i is paired to base j, no base between i and j can pair with one outside that segment. Each base pair thus creates a domain that effectively isolates all bases between them from those lying outside. This “domain separation” is necessary for all programs that fold large RNA sequences, because it reduces an intractable problem to one whose computation time scales as N3 (19), where N is the number of bases. Base stacking energies are estimated from melting experiments on short oligoribonucleotide duplexes [double-stranded (ds) segments] and are incorporated into a nearest-neighbor model that takes into account the identity and orientation of adjoining base pairs. The free energies of ss loops are determined by the type of loop (hairpin, bubble, bulge, or multibranch); the base pair(s) closing the loop; the number of unpaired bases in the loop; and, often, the identity and sequence of those unpaired bases. Entropy is accounted for both explicitly, in the entropy penalties for loop closures, and implicitly, in the use of free energies rather than internal energies for base stacking. The simple folding program we developed incorporates only six stacking energies (GC:GC, AU:AU, GU:GU, GC:AU, GC:GU, and AU:GU; no distinctions are made for orientation or order), contains no pairing energies, and ignores loop entropy penalties and all other details.
RNAfold and mfold determine the best possible set of paired bases, i.e., the combination yielding the minimum free energy (MFE); reversing this process (“backtracking”) provides the structure. Even with the exclusion of pseudoknots, the number of possible secondary structures of a long RNA sequence is enormous (∼1.86N) (20), yielding an extremely high density of states. This, together with the close energy spacing of structures near the MFE, necessitates that RNA, at thermodynamic equilibrium, be viewed not as a single MFE secondary structure but instead as an ensemble of many secondary structures. By using RNAfold, we find that the frequency of appearance of the MFE structure within the ensemble is extraordinarily small, ∼10−0.01N for randomly permuted sequences [see supporting information (SI) Fig. S1].
McCaskill developed an algorithm that determines the equilibrium partition function for an ensemble of RNA secondary structures (21), exploiting the domain separation described above. This procedure, which has been incorporated into RNAfold, gives the pairing probability for every base pair that can be formed by a sequence. From this, one can obtain ensemble averages for any property that can be calculated directly from the pairing probabilities.
Some of the quantities we wish to determine, such as MLD, cannot be calculated from the pairing probabilities because they can only be measured from the individual secondary structures. Obtaining an exact value for the 〈MLD〉, therefore, would require measuring the MLD of every secondary structure in the ensemble and then thermally averaging. Because the number of secondary structures involved is so large, it is impossible to do this. However, an algorithm developed by Ding and Lawrence, first featured in their Sfold program (22) and incorporated subsequently into RNAsubopt (23), allows one to randomly generate secondary structures with probabilities in proportion to their Boltzmann weight. If a sufficient number of structures (we use 1,000) are created, one can accurately estimate the true ensemble average of any property by calculating the average value of this property within the generated subset. Thus, for any property X, its ensemble-average value, 〈X〉, is calculated as 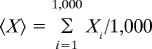 . To verify that these subsets are representative of the ensemble as a whole, properties were identified whose ensemble averages could be exactly determined by using RNAfold, e.g., the percentage of bases in pairs (PBP), the maximum average ladder distance (MALD) (a measure similar to 〈MLD〉), and the average ladder distance (ALD) (an alternate measure of size that explicitly includes branches). The exact thermally averaged values generated by RNAfold were, for each sequence, compared with the estimated thermal averages calculated from the representative subset generated by RNAsubopt. The differences were insignificant: For both the random ssRNAs of lengths 2,500–7,000 and the viral ssRNAs, the discrepancies in 〈PBP〉, MALD, and 〈ALD〉 averaged 0.03%, 0.3%, and 0.2%, respectively.
. To verify that these subsets are representative of the ensemble as a whole, properties were identified whose ensemble averages could be exactly determined by using RNAfold, e.g., the percentage of bases in pairs (PBP), the maximum average ladder distance (MALD) (a measure similar to 〈MLD〉), and the average ladder distance (ALD) (an alternate measure of size that explicitly includes branches). The exact thermally averaged values generated by RNAfold were, for each sequence, compared with the estimated thermal averages calculated from the representative subset generated by RNAsubopt. The differences were insignificant: For both the random ssRNAs of lengths 2,500–7,000 and the viral ssRNAs, the discrepancies in 〈PBP〉, MALD, and 〈ALD〉 averaged 0.03%, 0.3%, and 0.2%, respectively.
This thermal averaging is not available within mfold. Instead, after forming the MFE structure, mfold generates a list of all possible base pairs that can be formed by the sequence, excluding those present within the MFE structure. Then, for each of these base pairs, the lowest energy structure containing that base pair is determined. This results in a list of fewer than N2 structures, all higher in energy than the MFE structure. Mfold can be configured to output the 999 lowest energy structures from this set, and the MFE structure. We then calculate a Boltzmann-weighted average of any value X (AX) as:
 |
with ΔGi the free energy of the ith structure relative to that of the MFE one. Again, this average MLD (AMLD) does not represent a true ensemble average; rather, it is a thermal average over an arbitrary subset of the ensemble.
For all sequences, we generated both true ensemble-average pairing probabilities with RNAfold, and representative subsets of the thermal ensemble with RNAsubopt. To check for robustness, ensemble-average pairing probabilities were generated with our simplified energy model, and arbitrary subsets of the ensemble were generated with mfold. Viral ssRNA sequences were obtained from the National Center for Biotechnology Information Genome Database (www.ncbi.nlm.nih.gov). Randomly permuted ssRNA sequences were generated by using a Fisher–Yates shuffle driven by a Mersenne Twister random number generator (24) implemented in C++ (25). Yeast (Saccharomyces cerevisiae) genomic sequences were obtained from the Saccharomyces Genome Database (www.yeastgenome.org).
Results
The current RNA folding programs are known to have limited accuracy for long sequences (26). For our purposes, however, it is not necessary that all, or even most, of the individual pairings be correctly predicted. Rather, the predicted structures need only be sufficiently accurate to capture the coarse-grained features that determine 3D size. Our question therefore becomes the following: Can the relative sizes of large ssRNAs be predicted from computational estimates of appropriate properties of their secondary structures?
To make such estimates, we must identify a coarse-grained characteristic of the secondary structure that dictates 3D size. The single characteristic of a secondary structure that most obviously, and directly, meets this criterion is its “extendedness.” Fig. 1 A and B show, respectively, “typical-looking” viral and random ssRNAs of about the same length. It can be seen that the random ssRNA is strikingly more extended. The ssRNA in Fig. 1A is from a virus in the Leviviridae family. Additional representative structures, from the Bromovirus, Tymovirus and Tobamovirus genera, are shown in Figs. S2 and S3.
Fig. 1.

Predicted secondary structures of ssRNAs. (A) Enterobacteria phage Qß (in the Leviviridae family) ssRNA. (B) Randomly permuted ssRNA. Each is ≈4,000 nt in length and shown to the same scale. The MLDs of these structures are 221 and 368, respectively. (These are representative of their respective ensemble averages: The 〈MLD〉 of the phage Qß ssRNA is 240, and the 〈MLD〉 of 4,000-base random ssRNAs is 361.) The yellow overlays illustrate the paths associated with the MLDs (see text and the 50-nt example depicted in C). 〈MLD〉 values were calculated with RNAsubopt; figures were drawn with mfold.
This difference in the extendedness of secondary structures translates into a difference in 3D size. To evaluate extendedness as a candidate characteristic, a quantitative measure of this property is required. Bundschuh and Hwa introduced ladder distance as a measure of the distance between arbitrary bases in ssRNA secondary structures (27). The ladder distance, LDij, is the number of base pairs (“rungs” on a “ladder”) that are crossed along the most direct path in the secondary structure that connects bases i and j. Because ds sections are essentially stiff rods, whereas ss sections are floppy, only ds sections are counted in this measure of distance. To characterize the overall size of RNA secondary structures using a single quantity, we introduce maximum ladder distance (MLD), which is the largest value of LDij for all combinations of i and j. In other words, it is the ladder distance associated with the longest direct path across the secondary structure. This is illustrated in Fig. 1C, with an MFE secondary structure of an arbitrary 50-nt-long sequence, whose MLD happens to be 11. The MLD paths of this secondary structure and of those in Fig. 1 A and B are illustrated with yellow overlays.
To evaluate its usefulness as a predictive measure of size, we determined ensemble-average MLD (〈MLD〉) values in six viral taxa (listed in Table 1), all of whose virions consist simply of an ssRNA genome encased within a protein shell. The viruses of five of the taxa each have a fixed-radius spherical (T = 3 icosahedral) shell made up of 180 copies of a single gene product, the capsid protein. Their ssRNAs range in size from 3,000 to 7,000 nt, but the outer diameters of their capsids are all 26–28 nm (28, 29). By contrast, the viruses of the remaining taxon, the Tobamoviruses, assemble into cylindrical shells of fixed radius (18 nm) but variable length (averaging ≈300 nm). Thus, unlike the genomes of the icosahedral viruses, those of the Tobamoviruses are not required to fit into a shell of fixed size; longer ssRNA lengths simply lead to longer (fixed-diameter) cylinders (30). From our starting conjecture, one would predict that the Tobamoviruses are not under selective pressure to have RNAs that are particularly compact. In addition, because all five taxa of icosahedral viruses have capsids of approximately the same size, one would expect the divergence between the size of the viral and random ssRNAs to increase with sequence length.
Table 1.
Differences in 〈MLD〉s and 〈ALD〉s between viral and random sequences
| Viral taxon | No. seq.* | Mean N, nt | Mean Z score, 〈MLD〉† | Mean Z score, 〈ALD〉† |
|---|---|---|---|---|
| Bromoviridae RNA2 | 8 | 2,891 | −2.3 | −2.5 |
| Bromoviridae RNA1 | 8 | 3,265 | −1.4 | −1.9 |
| Leviviridae | 9 | 3,780 | −3.0 | −3.5 |
| Sobemovirus | 9 | 4,199 | −1.4 | −1.9 |
| Luteoviridae | 17 | 5,725 | −2.8 | −3.1 |
| Tymovirus | 9 | 6,300 | −2.7 | −3.5 |
| Tobamovirus | 22 | 6,425 | +0.6 | +0.1 |
*Number of sequences analyzed.
†The number of standard deviations separating the 〈MLD〉 or 〈ALD〉 of each viral ssRNA from the 〈MLD〉 or 〈ALD〉 predicted for random sequences of the same length, averaged for each taxon (RNAs 1 and 2 of the Bromoviridae are analyzed separately).
The average composition of the individual viral ssRNAs analyzed here (not including the Tymoviruses, whose compositions are atypical for the viruses examined in this study) is 24.0% G, 22.1% C, 26.9% A, and 27.0% U. However, we must account not only for the average composition, but also the average discrepancy in composition between bases potentially able to pair, i.e., G and C, A and U, and G and U. This composition discrepancy (again, not including the Tymoviruses) is 2.9 percentage points for %G − %C, 2.9 for %A − %U, and 4.0 for %G − %U (e.g., whether an individual viral ssRNA contained 22% G and 26% C, or 26% G and 22% C, its %G − %C difference would be 4 percentage points). To allow for a balance between these two averages—nucleotide percentages and their differences for pairing bases—we chose the “virus-like” composition 24% G, 22% C, 26% A, and 28% U for the randomly permuted sequences. With this composition, we generated and analyzed 500 random sequences of length 2,500 nt, 500 of length 3,000 nt, and 300 in each of the lengths 4,000, 5,000, 6,000, and 7,000 nt. The 〈MLD〉 of each viral and random sequence was determined with RNAsubopt.
The 〈MLD〉 values of the icosahedral viral RNAs are systematically smaller than those of the random RNAs, as can be seen in the log–log plot of 〈MLD〉 vs. sequence length displayed in Fig. 2. Each individual viral ssRNA is designated with a symbol indicating its taxon. The genomes of the Bromoviruses and Cucomoviruses are multipartite; they are divided among four different ssRNAs. Results are shown for the longest and second-longest of these, identified by convention as RNAs 1 and 2, which package into separate (but apparently identical) capsids. Also plotted are the average 〈MLD〉 (〈MLD〉) values of the various lengths of random sequences, and their standard deviations; the result is approximately linear (R2 = 0.993), with a slope indicating 〈MLD〉 ∼ N0.67±0.01 over this range.
Fig. 2.

Log–log plot of 〈MLD〉 vs. sequence length for viral and randomly permuted ssRNAs. The viral ssRNAs are identified by the symbols listed in the key (Inset). The Bromoviridae analyzed here are from the Bromovirus and Cucomovirus genera. The straight line is a least-squares fit to the 〈MLD〉 values computed for random sequences of lengths 2,500, 3,000, 4,000, 5,000, 6,000, and 7,000 nt; the vertical lines show the standard deviations. 〈MLD〉 values were calculated with RNAsubopt.
These scaling relationships for random ssRNAs are close to the N0.69 variation obtained numerically by Bundschuh and Hwa for a similar measure of distance, by using an energy model in which only Watson–Crick pairings are allowed, the interaction energy is the same for all pairs, and entropy is ignored (27). Their measure of distance is the ladder distance between the first and (N/2 + 1)th base, averaged over all structures in the ensemble for a random sequence of uniform composition and then over many sequences.
For each viral ssRNA, we calculated the Z score of the 〈MLD〉, i.e., the number of standard deviations separating its 〈MLD〉 from the predicted 〈MLD〉 values of random sequences of identical length. The latter is determined from the regression equation plotted in Fig. 2 (see SI Text). The mean Z score of each taxon is listed in Table 1. Those of the icosahedral viruses range from −1.4 to −3.0, indicating their RNAs have 〈MLD〉 values that are different from and smaller than the 〈MLD〉 values predicted for equal-length random RNAs. Further, a linear regression analysis of Z score vs. sequence length for the icosahedral viral RNAs shows a significant negative slope with a confidence interval >95%, implying that the relative compactness of these RNAs, all of which are required to fit into capsids of approximately the same size, increases with sequence length.
The average Z score of the 〈MLD〉 values of the Tobamovirus ssRNAs is +0.6. It is striking that these ssRNAs, which package into cylindrical capsids of variable length, have more extended secondary structures and larger 〈MLD〉 values than those of the icosahedral viruses. For both the icosahedral viruses and the Tobamoviruses, there appears to be a correspondence between the predicted secondary structures of their genomes (see Fig. S3) and the size and shape of the capsids into which the genomes must fit. We hypothesize that, to facilitate viral assembly, ssRNA sequences of self-assembling icosahedral viruses have evolved to have relatively small 〈MLD〉 values and that these smaller 〈MLD〉 values give rise to smaller Rg values.
These results suggest that the differences found between the viral and random RNAs do not occur simply because the viral RNAs are of biological origin (each is a positive-sense, directly translated messenger RNA); otherwise, one would not see a difference between the results for the icosahedral and cylindrical viruses. To examine this further, we analyzed 500 ssRNAs that are the transcripts of consecutive 3,000-base sections on yeast (S. cerevisiae) chromosomes XI and XII. These yeast-derived sequences were included to represent biological RNAs that, although evolved, have not been subjected to selective pressures to have a particular overall size and shape. Our findings, compiled in Table 2, show that the 〈MLD〉 values of the yeast-derived RNAs are approximately the same as those of the random RNAs, indicating that the differences between the random and viral ssRNAs do not result merely from the biological origin of the latter.
Table 2.
Composition-dependence of 〈MLD〉
| Type of ssRNA | No. seq. | N, nt | Composition,* % |
〈MLD〉 | |||
|---|---|---|---|---|---|---|---|
| G | C | A | U | ||||
| Random, viral-like composition | 500 | 3,000 | 24 | 22 | 26 | 28 | 299 ± 38 |
| Random, uniform composition | 500 | 3,000 | 25 | 25 | 25 | 25 | 296 ± 36 |
| Yeast-derived† | 500 | 3,000 | 19 | 19 | 31 | 31 | 300 ± 46 |
*The randomly permuted ssRNAs of each type are of identical composition; for the yeast ssRNAs, the mean composition is listed.
†These are ssRNA transcripts of successive 3,000-base sections of yeast (S. cerevisiae) chromosomes XI and XII.
As mentioned earlier, the composition of the random RNAs was chosen to match, on average, that of the viral RNAs as closely as possible. However, many individual viral RNAs differ significantly in composition from the random RNAs, raising the question of whether the same differences in 〈MLD〉 would be seen if the viral RNAs were each compared with random RNAs of identical composition. To test the sensitivity to composition of the 〈MLD〉 values of the random RNAs, we analyzed 3,000-base randomly permuted RNAs of uniform (25% G, 25% C, 25% A, 25% U) composition. The results, listed in Table 2, show that the 〈MLD〉 is insensitive to small composition changes. Further, the average composition of the yeast RNAs differs significantly from that of both sets of random RNAs, yet their 〈MLD〉 values are approximately the same.
How likely is it that the predicted differences in 〈MLD〉 between viral and nonviral RNAs are present in actual RNAs? RNAsubopt and all similar programs that predict RNA structure have the capability, in principle, to find all possible non-pseudoknotted structures. Thus, the accuracy of RNAsubopt (its ability to properly sample from the ensemble) depends not on what structures it is able to predict (it can predict all of them, barring those with pseudoknots), but rather on the energies it assigns to them, which are determined by its energy model. As mentioned earlier, we only require that RNAsubopt be sufficiently accurate to predict general coarse-grained features of the RNA secondary structure, such as 〈MLD〉. To evaluate whether our findings are specific to RNAsubopt (and therefore possibly an artifact of the particular energy model on which RNAsubopt is based), we compared viral and random ssRNAs by using mfold, which is similar to RNAsubopt but differs somewhat in both its energy model and the structures it samples from the ensemble. Whereas the 〈MLD〉 values generated by RNAsubopt are different from the AMLD values generated by mfold, both showed the same systematic difference in MLD between viral and random ssRNAs, and approximately the same scaling relationships for random sequences (AMLD ∼ N0.74±0.01 for mfold, see Fig. S4).
To further test the robustness of these predictions, we compared random and viral ssRNAs using our simplified RNA folding program. This program does not determine individual secondary structures, and consequently does not permit calculation of 〈MLD〉. However, it does determine pairing probabilities, which allows calculation of the maximum average ladder distance (MALD) of the entire ensemble of structures, which is the maximum value of the ensemble averages of the N2 ladder distances associated with each N-base sequence. We find that this program—like those discussed above, which are based on more realistic energy assignments—also predicts systematic differences between random and viral RNAs, giving smaller MALD values for viral sequences than for nonviral ones (see Fig. S5). Thus, even a highly simplified energy model that merely takes into account nearest-neighbor interactions is sufficient to reveal a fundamental difference between the secondary structures of viral and randomly permuted ssRNA sequences. With this simplified model, for random sequences of lengths 2,000–4,000, MALD ∼ N0.66±0.02.
The folding programs we employ cannot produce structures that contain pseudoknots. Although pseudoknots are known to occur in viral RNAs, such as those that form 3′-terminal tRNA-like structures (8), they are typically local (involving bases separated by <102 nt along the sequence); accordingly, ignoring them should not significantly affect our prediction of overall size. Evidence has been found for longer-range pseudoknots, such as kissing hairpins connecting bases separated by as many as 400 nt (31), but even these are close relative to the overall length of viral genomes. In any event, our aim is to develop a zeroth-order theoretical model that captures the determinants of overall size, with pseudoknots, kissing hairpins, and other details included later as necessary.
To translate 〈MLD〉 into Rg, it is useful to map the RNA secondary structures onto polymer models whose configurational statistics are well understood, such as ideal linear and “star” polymers. By using the simplest idealization, as in the freely jointed chain model discussed above, we can replace structures like the two shown in Fig. 1 A and B by linear chains whose effective contour lengths (Leff) are given by their 〈MLD〉 values. To complete this mapping, we model the duplex sections as the rigid links of the chain, and the ss bulges, bubbles, and multibranch loops as the flexible joints that connect them. The effective Kuhn length (beff) is thus the average duplex length in the ssRNA secondary structure, a property that is approximately the same (5 bp) for all sequences examined. This corresponds to an average RNA duplex length of 1–2 nm. Because the persistence length (a measure of the length scale at which bending is observed) of dsRNA is ≈60 nm (32), modeling the duplex sections as rigid bodies is an excellent approximation. The ss loops, on average, contain approximately six ss bases, and thus we estimate that a typical bubble has approximately three ss bases on each side; the persistence length of ssRNA is likely similar to that of ssDNA, approximately two bases (33).
From this mapping between secondary structures and effective linear polymers, it follows that the Rg of an ssRNA molecule with an arbitrary sequence should be determined by
Combining the last equation with our earlier result, 〈MLD〉 ∼ N0.67, yields
For a non-self-avoiding linear chain, ν = 0.5, in which case, Rg ∼ N0.34; for a self-avoiding linear chain, ν ≈ 0.6, giving Rg ∼ N0.40.
This approach can be broadened by mapping the ssRNA secondary structures onto an alternate polymer model system that accounts for all possible paths across the structure, and thus includes all branches. For any ideal polymer, linear or branched,
 |
where Lij is the distance along the backbone between monomers i and j (34). Proceeding as above, we obtain
 |
where Lij,eff has been replaced by LDij in the second step. The ALD is the average ladder distance, i.e., the average of the N2 pairwise ladder distances in an RNA secondary structure, and 〈ALD〉 is its ensemble average. By using values for 〈ALD〉 calculated exactly from the pairing probabilities generated by RNAfold, we have repeated the analysis shown in Fig. 2. The results are equivalent, with 〈ALD〉 ∼ N0.68±0.01 and Rg ∼ N0.34, and demonstrate that the differences between random and viral ssRNAs are preserved when branches are explicitly included (see Fig. 3 and the Z scores of the 〈ALD〉 values in the last column of Table 1). As with MLD, ALD is robust with respect to the energy model. Results obtained with the simplified folding program (〈ALD〉 ∼ N0.68±0.01) are shown in Fig. S6.
Fig. 3.

Same as Fig. 2, but with 〈ALD〉, calculated with RNAfold, replacing 〈MLD〉. 〈ALD〉 is a measure of size that explicitly includes all branches.
Discussion
Our goal has been to develop a generic, qualitative picture of how the 3D sizes of large ssRNAs depend on their sequences. Accordingly, we have identified coarse-grained features of RNA secondary structures (〈MLD〉 and 〈ALD〉) that can be used to predict variations in Rg that can be systematically compared with experimental measurements.
Although we have focused on the role of genome size on assembly, other properties, such as total charge (35), also play a role. It is clear, however, that the intrinsic size of the RNA in solution must be an important factor in determining the free energy of encapsidation and hence in controlling the degree of spontaneity of the process.
The smaller 〈MLD〉 values and 〈ALD〉 values of viral ssRNAs (relative to those of random sequences) cannot be explained by smaller values of 〈PBP〉. With the exception of the Tymoviruses, the 〈PBP〉 values of the individual viral ssRNAs are all close to (within one percentage point) or larger than the 〈PBP〉 values of random sequences. For random ssRNAs (of lengths 2,500–7,000), the overall average value of 〈PBP〉 is 62.0; for the viral ssRNAs the values of 〈PBP〉 are 63.3 (Bromovirus/Cucomovirus RNA2), 64.2 (Bromovirus/Cucomovirus RNA1), 68.4 (Leviviridae), 65.9 (Sobemovirus), 61.8 (Luteoviridae), 45.0 (Tymovirus), and 64.3 (Tobamovirus). Note also that the Tymovirus ssRNAs, despite their relatively low 〈PBP〉 values, exhibit approximately the same range of 〈MLD〉 and 〈ALD〉 values as those of the comparable-length Luteoviridae ssRNAs.
The 〈MLD〉 and 〈ALD〉 of a secondary structure result from its connectivity, which is in turn determined by its branching properties. The viral ssRNAs form more compact secondary structures than random ssRNAs in part because the former have significantly more (relative to sequence length) higher-order branches (those that are junctions for four or more duplexes). Among the viral ssRNAs, as the number of higher-order branches per unit sequence length increases, the Z scores of their 〈MLD〉 and 〈ALD〉 values become more negative. We are currently examining viral sequences to determine whether they share common patterns that give rise to the formation of these higher-order branches.
In predicting the native sizes of ssRNAs, we have assumed that their secondary structures are in thermodynamic equilibrium. Extensive in vitro studies indicate that, as ssRNAs are transcribed, they typically misfold into kinetically trapped states (36). However, more recent work, on the transcription of hairpin ribozyme sequences in yeast, has shown that not-yet-elucidated cofactors present in the nucleus strongly inhibit kinetic trapping in vivo, thereby increasing the importance of thermodynamic stability in determining the folded state of ssRNA (37). Similar factors may be operative in the cytoplasm of host cells infected by messenger–sense ssRNA genomes, from which viral ssRNA transcripts are synthesized by RNA-dependent viral replicases (as opposed to the usual DNA-dependent RNA polymerases). These considerations suggest that the thermodynamic ensembles we have used to estimate viral genome sizes are indeed relevant to overall size and hence to capsid packaging efficiencies.
Supplementary Material
Acknowledgments.
We thank Professors Jon Widom, Andrea Liu, and Paul van der Schoot for many helpful discussions throughout the course of this work and Dr. Nicholas Markham and Professors David Mathews and Ivo Hofacker for valuable assistance in understanding the various RNA folding programs we used. This research was supported by U.S. National Science Foundation Grants CHE04-00363 and CHE07-14411 (to W.M.G. and C.M.K.); U.S.–Israel Binational Science Foundation Grants 2002-75 and 2006-401 (to A.B.-S. and W.M.G.); Israel Science Foundation Grant 659/06 (to A.B.-S.); a University of California, Los Angeles Dissertation Year Fellowship (to A.M.Y.); and a Netherlands Organisation for Scientific Research Rubicon grant (to P.P.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0808089105/DCSupplemental.
References
- 1.Capriotti E, Marti-Renom MA. Computational RNA structure prediction. Curr Bioinf. 2008;3:32–45. [Google Scholar]
- 2.Lipfert J, Chu VB, Bai Y, Herschlag D, Doniach S. Low-resolution models for nucleic acids from small-angle X-ray scattering with applications to electrostatic modeling. J Appl Crystallogr. 2007;40:S229–S234. [Google Scholar]
- 3.Schultes EA, Spasic A, Mohanty U, Bartel DP. Compact and ordered collapse of randomly generated RNA sequences. Nat Struct Mol Biol. 2005;12:1130–1136. doi: 10.1038/nsmb1014. [DOI] [PubMed] [Google Scholar]
- 4.Chauhan S, et al. RNA tertiary interactions mediate native collapse of a bacterial group I ribozyme. J Mol Biol. 2005;353:1199–1209. doi: 10.1016/j.jmb.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Hyeon C, Dima RI, Thirumalai D. Size, shape, and flexibility of RNA structures. J Chem Phys. 2006;125:194905. doi: 10.1063/1.2364190. [DOI] [PubMed] [Google Scholar]
- 6.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 7.Lanchy JM, Lodmell JS. An extended stem-loop 1 is necessary for human immunodeficiency virus type 2 replication and affects genomic RNA encapsidation. J Virol. 2007;81:3285–3292. doi: 10.1128/JVI.02025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi YG, Dreher TW, Rao ALN. tRNA elements mediate the assembly of an icosahedral RNA virus. Proc Natl Acad Sci USA. 2002;99:655–660. doi: 10.1073/pnas.022618199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV. Long-range RNA–RNA interactions circularize the dengue virus genome. J Virol. 2005;79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zipper P, Durchschlag H. Modelling of bacteriophage capsids and free nucleic acids. J Appl Crystallogr. 2007;40:S153–S158. [Google Scholar]
- 11.Fraenkel-Conrat H, Williams RC. Reconstitution of active tobacco mosaic virus from its inactive protein and nucleic acid components. Proc Natl Acad Sci USA. 1955;41:690–698. doi: 10.1073/pnas.41.10.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bancroft JB. The self-assembly of spherical plant viruses. Adv Virus Res. 1970;16:99–134. doi: 10.1016/s0065-3527(08)60022-6. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto K, Yoshikura H. Relation between genomic and capsid structures in RNA viruses. Nucleic Acids Res. 1986;14:389–396. doi: 10.1093/nar/14.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beekwilder MJ, Nieuwenhuizen R, Vanduin J. Secondary structure model for the last two domains of single-stranded RNA phage Qβ. J Mol Biol. 1995;247:903–917. doi: 10.1006/jmbi.1995.0189. [DOI] [PubMed] [Google Scholar]
- 15.Kuznetsov YG, Daijogo S, Zhou J, Semler BL, McPherson A. Atomic force microscopy analysis of icosahedral virus RNA. J Mol Biol. 2005;347:41–52. doi: 10.1016/j.jmb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Flory PJ. Statistical Mechanics of Chain Molecules. New York: Hanser Publications; 1989. p. 11. [Google Scholar]
- 17.Hofacker IL, et al. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- 18.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nussinov R, Jacobson AB. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proc Natl Acad Sci USA. 1980;77:6309–6313. doi: 10.1073/pnas.77.11.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofacker IL, Schuster P, Stadler PF. Combinatorics of RNA secondary structures. Discrete Appl Math. 1998;88:207–237. [Google Scholar]
- 21.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofacker IL, Wuchty S, Fontana W. RNAsubopt - calculate suboptimal secondary structures of RNAs (Univ of Vienna) 2006 Available at www.tbi.univie.ac.at/∼ivo/RNA/RNAsubopt.html.
- 24.Matsumoto M, Nishimura T. Mersenne twister: A 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans Model Comput Simulat. 1998;8:3–30. [Google Scholar]
- 25.Wagner R. Mersenne twister random number generator (Univ of Michigan) 2003 Available at www-personal.umich.edu/∼wagnerr/MersenneTwister.html.
- 26.Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bundschuh R, Hwa T. Statistical mechanics of secondary structures formed by random RNA sequences. Phys Rev E. 2002;65 doi: 10.1103/PhysRevE.65.031903. 031903. [DOI] [PubMed] [Google Scholar]
- 28.Shepherd CM, et al. VIPERdb: A relational database for structural virology. Nucleic Acids Res. 2006;34:D386–D389. doi: 10.1093/nar/gkj032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. Virus Taxonomy: 8th Report of the International Committee on Taxonomy of Viruses; San Diego: Academic; 2005. [Google Scholar]
- 30.Whitfeld PR, Higgins TJV. Occurrence of short particles in beans infected with the cowpea strain of TMV. Virology. 1976;71:471–485. doi: 10.1016/0042-6822(76)90375-5. [DOI] [PubMed] [Google Scholar]
- 31.Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R. In vitro evidence for a long range pseudoknot in the 5′-untranslated and matrix coding regions of HIV-1 genomic RNA. J Biol Chem. 2002;277:5995–6004. doi: 10.1074/jbc.M108972200. [DOI] [PubMed] [Google Scholar]
- 32.Abels JA, Moreno-Herrero F, van der Heijden T, Dekker C, Dekker NH. Single-molecule measurements of the persistence length of double-stranded RNA. Biophys J. 2005;88:2737–2744. doi: 10.1529/biophysj.104.052811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha T. Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimm BH, Stockmayer WH. The dimensions of chain molecules containing branches and rings. J Chem Phys. 1949;17:1301–1314. [Google Scholar]
- 35.Belyi VA, Muthukumar M. Electrostatic origin of the genome packing in viruses. Proc Natl Acad Sci USA. 2006;103:17174–17178. doi: 10.1073/pnas.0608311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlenbeck OC. Keeping RNA Happy. RNA. 1995;1:4–6. [PMC free article] [PubMed] [Google Scholar]
- 37.Mahen EM, Harger JW, Calderon EM, Fedor MJ. Kinetics and thermodynamics make different contributions to RNA folding in vitro and in yeast. Mol Cell. 2005;19:27–37. doi: 10.1016/j.molcel.2005.05.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.