

Abstract
Cytochrome P450 (CYP) 3A4 contributes to the metabolism of approximately 50% of commercial drugs by oxidizing a large number of structurally diverse substrates. Like other endoplasmic reticulum-localized P450s, CYP3A4 contains a membrane-anchoring N-terminal helix and a significant number of hydrophobic domains, important for the interaction between CYP3A4 and the membrane. Although the membrane affects specificity of CYP3A4 ligand binding, the structural details of the interaction have not been revealed so far because x-ray crystallography studies are available only for the soluble domain of CYP3A4. Here we report sample preparation and initial magic-angle spinning (MAS) solid-state NMR (SSNMR) of CYP3A4 (Δ3−12) embedded in a nanoscale membrane bilayer, or Nanodisc. The growth protocol yields ∼2.5 mg of the enzymatically active, uniformly 13C, 15N-enriched CYP3A4 from a liter of growth medium. Polyethylene glycol 3350-precipitated CYP3A4 in Nanodiscs yields spectra of high resolution and sensitivity, consistent with a folded, homogeneous protein. CYP3A4 in Nanodiscs remains enzymatically active throughout the precipitation protocol as monitored by bromocriptine binding. The 13C line widths measured from 13C-13C 2D chemical shift correlation spectra are ∼0.5 ppm. The secondary structure distribution within several amino acid types determined from 13C chemical shifts is consistent with the ligand-free x-ray structures. These results demonstrate that MAS SSNMR can be performed on Nanodisc-embedded membrane proteins in a folded, active state. The combination of SSNMR and Nanodisc methodologies opens up new possibilities for obtaining structural information on CYP3A4 and other integral membrane proteins with full retention of functionality.
Membrane proteins participate in numerous important biological processes and are targets of most modern pharmaceuticals (1, 2). Significant progress has been made in protein structure determination by both x-ray crystallography and solution NMR in recent years. However, membrane proteins remain the least structurally characterized of all proteins. Solid-state NMR (SSNMR) is a powerful technique for obtaining site-specific structural information without requiring proteins to be soluble at high concentrations for solution NMR or to form single crystals for diffraction studies. Recent advances in magic-angle spinning (MAS) NMR techniques have enabled high-resolution structures of several small globular proteins to be determined in the microcrystalline state (3-6). These studies benefited from improved instrumentation and pulse sequence techniques, as well as sophisticated isotopic labeling schemes. MAS NMR studies on membrane proteins are especially interesting since a variety of membrane mimetics are compatible with SSNMR techniques, allowing investigations into the structure-function relationships of both lipid and protein (7). Historically, many studies of membrane proteins have been performed with site-specific labeling, and such techniques continue to be valuable for providing insights, for example, into ligand-protein interactions (8). More recently, efforts have increasingly focused on samples isotopically labeled to a high percentage, enabling sequential assignments to be performed. To date, several membrane proteins have been addressed in this manner, including the light-harvesting complex 2 (9, 10), sensory rhodopsin (11) and DsbB (12), expanding the possibilities for solving full structures of these proteins.
An important challenge for functional and structural studies of membrane proteins is developing suitable membrane mimetics that are not only compatible with the intended studies, but also capable of retaining native protein structure and function. For most solution NMR studies of membrane proteins performed so far, extensive screening of detergents and phospholipids has been required to identify micelle or bicelle conditions approximating the native state (13, 14). Likewise, the preparation of lipid bilayer samples for oriented sample solid-state NMR works best for proteins that are mostly embedded in the membrane. These approaches have been successful in a number of cases, but may not be extendable to all integral membrane proteins. An elegant and promising alternative membrane mimetic that has proved valuable for studies of variety of membrane proteins is the Nanodisc system in which discoidal phospholipid bilayers are encircled by two molecules of amphipathic helical membrane scaffold protein (MSP). Nanodiscs have been well-characterized (15, 16) and extensively used to stabilize and study membrane proteins in fully functional states, since self-assembly of target membrane proteins into the Nanodisc provides the ability to control the lipid constitution and protein oligomerization state (17-19). Nanodiscs are monodisperse (16), stable over a variety of conditions including a large range of temperatures (20, 21), and amenable to incorporation of a variety of lipids. Besides Nanodiscs consisting of a single type of lipid such as POPC (16, 18), DMPC and DPPC (21), Nanodiscs with POPC and varying concentrations of POPS (22), POPC/POPG mixtures (23), and total E. coli lipids (23) have been prepared and utilized for membrane protein studies. These properties of Nanodiscs have made them quite useful in extending functional studies of a number of membrane proteins, as recently reviewed (24).
Nanodiscs have been demonstrated to be especially valuable for the study of cytochrome P450 monooxygenases (25, 26), including CYP3A4 (18), the most abundant P450 in the human liver, which metabolizes approximately 50% of xenobiotics encountered by man (27). CYP3A4 is a promiscuous enzyme with a large, adaptive active site, capable of accommodating two to three smaller substrates (28, 29), and often exhibiting allosteric kinetic behavior (18, 19, 30, 31). Some studies suggest that the presence of external factors such as phospholipids may stabilize some of the CYP3A4 kinetically distinguishable conformations and affect substrate affinity (32, 33). Further characterization of CYP3A4 interactions with its ligands is driven by the fact that development of effective drug therapies is often hampered by unwanted drug-drug interactions between substrates and inhibitors (34, 35). The crystal structure of substrate-free CYP3A4 has been solved to 2.05 Å resolution (36), along with the several substrate and inhibitor-bound structures of CYP3A4 at lower resolution (37, 38). The available structures obtained on CYP3A4 with the N-terminal lipid anchor domain absent (Δ3−23) are extremely valuable, but because they were obtained in the absence of lipids they do not provide insights into the effects of membrane interactions on the CYP3A4 structure and its context-specific affinity for various ligands. Incorporation of CYP3A4 into Nanodiscs yields homogeneous samples of monomeric CYP3A4 in a ∼10 nm physiologically relevant phospholipid bilayer. This construct allows structural and functional characterization of CYP3A4 void of complications induced by conformational heterogeneity caused by different oligomeric states (39), or by interactions between P450 and detergents used for solubilization (40). This system has also been characterized structurally by small angle x-ray scattering (16, 18), and functionally by a number of biophysical techniques (18, 19, 41).
In this work, we demonstrate the first SSNMR study of an integral membrane protein embedded in a Nanodisc. The employed sample preparation protocol retains the activity of CYP3A4. We further demonstrate that the microscopic structural homogeneity of CYP3A4 in Nanodiscs is well suited for high-resolution MAS NMR studies. Secondary structure analysis based on 13C chemical shift distribution within the same amino acid type validates the structural integrity of CYP3A4 in precipitated Nanodiscs. The experimental data agree well with the spectrum predicted from the highest resolution ligand-free CYP3A4 x-ray structure (36), indicating that the global fold of precipitated CYP3A4 in a Nanodisc remains similar to that of the truncated crystallized protein prepared in the absence of lipids. These results demonstrate that SSNMR in combination with the model membrane system, Nanodisc, is an excellent technique complementary to crystallography, for the purposes of examining ligand-specific binding events, allosteric regulation, and the impact of membrane interactions on CYP3A4 activity.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human CYP3A4 from the NF-14 construct in the pCWOri+ vector (Δ3−12), with a C-terminal pentahistidine tag (provided by Dr. Guengerich and co-workers, Vanderbilt University, Nashville, TN) (42) was co-expressed in the DH5α strain with groEL and groES chaperone plasmids (43) purchased from Fisher Scientific (Hampton, NH). Chaperone plasmid pG-KJE8 has groES and groEL under control of the Pzt-1 promoter inducible by tetracycline. High-yield expression of CYP3A4 was achieved with a modified version of the Marley et al. protocol (44); cells were grown in 1 L Terrific broth (TB) enhanced with BactoPeptone (2 g/L), supplemented with trace metals, 50 mg/L ampicillin and 20 mg/L chloramphenicol, at 240 rpm, in a New Brunswick Scientific Series 25 incubator shaker (Edison, NJ) at 37°C to OD600 ∼ 0.7−1. The cells were then centrifuged at ∼3500 × g and transferred to 250 ml of 13C, 15N-enriched M9 minimal media in a 2 L baffled flask for induction. Each liter of 13C, 15N-enriched M9 minimal media contained 100 ml of 10× M9 salts (67.8 g Na2HPO4, 30 g KH2PO4, and 5 g NaCl in 1 L of autoclaved H2O, pH 7.4 adjusted using 5 M KOH), and 2 g 15NH4Cl, 4 g 13C glucose, 0.011 g of CaCl2•2H2O, 0.246 g MgSO4, 0.02 g thiamine, 50 mg/L ampicillin, and 20 mg/L chloramphenicol, and was sterile filtered before use. Isotopically labeled materials were obtained from Cambridge Isotope Laboratories (Andover, MA). After a one-hour adaptation period, expression was induced at a new shaking rate of 190 rpm, 28−30°C, by adding 1 mM IPTG and 1 mM δ-aminolevulinic acid (Frontier Scientific, Logan, UT) to help promote heme synthesis and holoenzyme formation. Cells were collected 48 h after induction and CYP3A4 was purified as described by Denisov et al. (41). Purified CYP3A4 was quantified using carbon monoxy (CO)-bound ferrous difference spectra with an extinction coefficient of 91 mM−1 cm−1 (45). The use of the Nanodisc system for solubilization of CYP3A4 and incorporation into nanoscale bilayers has been described in detail in several previous publications (18, 19, 24, 41).
Precipitation of CYP3A4 Nanodiscs
CYP3A4 Nanodiscs were concentrated to 60−80 μM and batch-precipitated with a volume equivalent of 40% PEG 3350. The mixture was incubated overnight at 4°C, and centrifuged at 8000 rpm in an Eppendorf 5415 centrifuge for 2 h. The supernatant was removed, and the pellet was transferred into a 4.0 mm limited speed rotor (Varian, NMR, Palo Alto, CA) with working volumes of ∼80 μl and confined to active sample region (central 60 μl) of the rotor by Kel-F and rubber spacers as described previously (46). The total mass of the material packed into the rotor was ∼60 mg. The sample contained ∼3 mg of CYP3A4 in Nanodiscs as determined by difference of protein quantities in the solution before precipitation and the supernatant after precipitation. The amount of protein was also confirmed by comparison in intensity of one-dimensional (1D) 13C spectra to standard proteins of known quantity.
To test the integrity of CYP3A4 in precipitated Nanodiscs, a sample were prepared with natural abundance CYP3A4 and subjected to the same precipitation treatment as the isotopically labeled CYP3A4 in Nanodiscs for SSNMR. Bromocriptine was added prior to precipitation and a spectrum of the precipitated pellet was acquired, which indicated that the protein remains active in the precipitated state (see Supporting Information). This sample could not be fully resuspended, preventing accurate analysis of the resuspended pellet by UV-Vis spectra. We prepared another sample with addition of 20% glycerol prior to precipitation, which promoted full resuspension of the pellet. The supernatant was removed, and the precipitated pellet was resuspended in 130 μl of 0.1 M potassium phosphate buffer (pH 7.5). Sample integrity in both cases was confirmed by bromocriptine-binding induced high-spin shift. Furthermore, SSNMR data were acquired on the samples containing (data not shown) and lacking glycerol, and they are identical within the experimental errors, indicating that glycerol does not affect CYP3A4 conformation. All experiments in this work were performed on a CYP3A4 Nanodisc sample not containing glycerol, since this sample has a higher packing density.
SSNMR Spectroscopy
SSNMR experiments were performed on a 600 MHz InfinityPlus four-channel Varian NMR spectrometer. The spectrometer was equipped with a 4 mm 1H-13C-15N Balun probe. Spectra were acquired at a MAS rate of 10 kHz, and the temperature of the sample cooling gas was maintained at −25 ± 3°C unless otherwise noted. Pulse sequences were implemented with tangent ramped cross-polarization (CP) (47) with ∼ 60 kHz on 1H and ∼ 83 kHz on 13C and two pulse phase modulation (TPPM) 1H decoupling (48) at ∼65−75 kHz. The typical π/2 pulse widths were 2.3 μs on 1H and 3.2 μs on 13C. Data were processed with NMRPipe (49) with back linear prediction and polynomial baseline (frequency domain) correction applied to the direct dimension. Zero filling and Lorentzian-to-Gaussian apodization were employed for each dimension before Fourier transformation. Additional acquisition and processing parameters for each spectrum are included in the figure captions. Chemical shifts were referenced externally with adamantane (50).
RESULTS AND DISCUSSION
Protein Expression and Precipitation of Nanodisc-Embedded CYP3A4
The expression of functional CYP3A4 with yields sufficient for SSNMR structural studies required development of a high-yield expression protocol for this membrane protein. Growths performed on minimal media, 1% Bioexpress 1000-supplemented minimal media, or 100% Bioexpress 1000 (Cambridge Isotopes, MA), failed to yield significant quantities of natively folded protein. Rather, the use of the TB starter culture according to the Marley method (44), in combination with the chaperone groEL and groES co-expression system was instrumental in obtaining sufficient quantities of the isotopically labeled CYP3A4, as shown for other P450s (51). Cell growth was initiated in the modified TB media to an OD600 of 0.8−1, and then the cells were transferred to a quarter-volume of minimal media in order to produce milligram/liter quantities of natively folded CYP3A4 (as defined by absorbance at 450 nm using CO-difference spectroscopy). Under these conditions, supplementation of 10 ml of Bioexpress 1000 to 1 L of minimal media did not significantly increase CYP3A4 expression. Also, alternate host strains such as E. coli C41 and C43 (Avidis, France) (52) did not improve the protein yield. For the highest level of protein expression, 2 L baffled flasks were utilized during the growth period in minimal media for optimal aeration; volumes smaller than 250 ml in such flasks resulted in decreased protein yield. Purification of the labeled CYP3A4 proceeded as in previous studies (41).
MAS NMR experiments require samples to be microscopically ordered and macroscopically immobilized on the NMR timescale. Based on our previous study of Nanodiscs by SSNMR (53), and use of PEG as the precipitant in the reported CYP3A4 crystallography studies (36, 38), we screened a small range of PEG concentrations to identify a robust condition for the precipitation of the CYP3A4-Nanodisc complex. The best conditions were highly similar to those optimal for the precipitation of Nanodiscs without an embedded membrane protein.
We tested the sample integrity before and after CYP3A4 Nanodisc precipitation by bromocriptine binding, an assay that reports not only the integrity of CYP3A4 itself, but that of the CYP3A4-Nanodisc complex. As previously reported (41, 54), functional CYP3A4 in solution exhibits only a partial low spin to high spin shift upon binding of bromocriptine, while CYP3A4 in Nanodiscs displays a full spin shift. The CO difference spectrum of purified CYP3A4 in buffer containing Emulgen 913 reveals functional CYP3A4 with all of the protein absorbing at 450 nm, and none at 420 nm (Supporting Information). Bromocriptine binding of CYP3A4 in Emulgen 913 results in a partial spin shift, shifting the absorbance from ∼420 nm to ∼390 nm (Figure 1A). In contrast, the complete low spin to high spin shift is obtained upon bromocriptine binding to CYP3A4-Nanodiscs (Figure 1B). The spectral features observed prior to precipitation (Figure 1B) are replicated after resolubilization in phosphate buffer (Figure 1C). The bromocriptine-binding spectra before and after precipitation confirm that the precipitated CYP3A4-Nanodiscs sample contains the active protein, since a refolding event restoring activity upon dilution into phosphate buffer is not feasible.
Figure 1.

Bromocriptine binding to CYP3A4. A) Purified CYP3A4 in Emulgen 913 containing buffer in the presence of equal molar amount of bromocriptine. B) CYP3A4 Nanodiscs in the presence of 23 μM bromocriptine prior to PEG 3350 precipitation. C) CYP3A4 Nanodiscs in the presence of 27 μM bromocriptine after precipitation with PEG 3350. Precipitation pellet was resuspended in 130 μl of 0.1 M potassium phosphate buffer pH 7.5. The concentration of CYP3A4 Nanodisc sample is ∼5 μM for B) and ∼ 6 μM for C) as calculated from the absorbance at 417 nm, and CYP3A4 extinction coefficient taken as 115 cm−1M−1.
2D 13C-13C Correlation Spectra and Spin System Identification
To determine the optimal temperature for multi-dimensional experiments of CYP3A4 Nanodiscs, 1D CP-MAS 13C spectra were acquired in the temperature range of −10°C to −40°C (Figure 2). Resolution was optimal at the high end of this temperature range, but for purposes of sample longevity, 2D correlation spectra were acquired at −25°C, where the resolution as judged by the 1D spectra is only compromised slightly.
Figure 2.
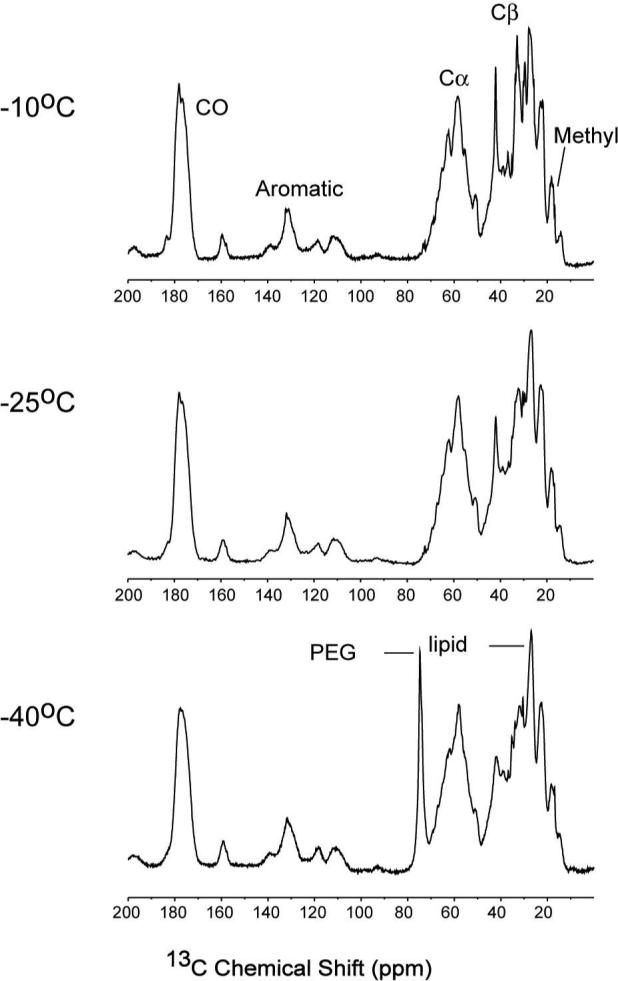
CP-MAS 1D 13C spectra of [U-13C, 15N] CYP3A4 in Nanodiscs at −10°C, −25°C and −40°C. CP and decoupling conditions were optimized at each temperature; other acquisitions parameters were the same. Spectra were acquired on ∼50 nmol (∼3 mg) CYP3A4, with 10.24 ms acquisition time (1024 × 10 μs), ∼75 kHz 1H TPPM decoupling (6.7 μs, 16.5°), 1024 scans and 3 s pulse delay. The CP contact time was 0.7 ms for all of the temperatures (optimized for total signal intensity). The data were processed without apodization and zero filled to 16384 complex points prior to Fourier transform.
2D 13C-13C homonuclear correlation spectra of uniformly 13C,15N-labeled CYP3A4 embedded in Nanodiscs were acquired at 600 MHz 1H frequency. The spectrum with 25 ms dipolar-assisted rotational resonance (DARR) mixing time (55) (Figure 3A) shows 13C line widths of ∼0.5 ppm from individually resolved cross peaks, demonstrating that high-resolution SSNMR spectra can be obtained with the Nanodisc sample preparation protocol. The observed narrow line widths (see Supporting Information) are comparable to those observed in SSNMR studies of 2D microcrystals (56) and membrane proteins in a liposome mimetic (11), and indicate that the protein is microscopically well-ordered. Although the very large number of amino acid spin systems (498) in this ∼57 kDa protein cannot yet be assigned, the 2D 13C-13C homonuclear correlation spectra allow for identification of signals corresponding uniquely to individual residues in the protein. Analysis of chemical shifts yields information about the secondary structure due to the strong dependence of Cα and Cβ chemical shifts on not only amino acid type but also backbone conformation (57, 58).
Figure 3.
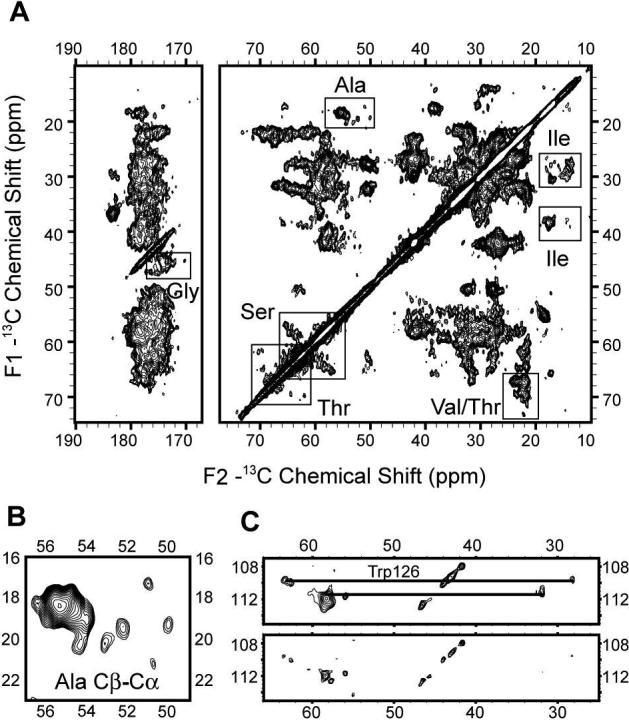
A) 2D 13C-13C chemical shift correlation spectrum of [U-13C, 15N] CYP3A4 in Nanodiscs at 600 MHz 1H frequency with 25 ms DARR mixing time. B) Enlarged Ala Cα-Cβ crosspeak region from the same spectrum demonstrating the secondary structure distribution. C) Tentatively assigned Trp 126 spin system enlarged from the same data set, processed with 0.66 ppm (100 Hz) (top panel), and with 0.33 ppm (60 Hz) (bottom panel) net line broadening (Lorentzian-to-Gaussian apodization) in both dimensions. Data were acquired for 48 h. Other acquisition parameters are 0.75 ms 1H-13C CP contact time, ∼72 kHz 1H TPPM decoupling (6.9 μs, 16°), 6.4 ms t1 acquisition time (640 × 10 μs) with TPPI detection, 15.36 ms t2 acquisition time (1536 × 10 μs), 3 s pulse delay. The data were processed with 0.33 ppm (50 Hz) net line broadening (Lorentzian-to-Gaussian apodization) in the F1, and 0.26 ppm (40 Hz) net line broadening (Lorentzian-to-Gaussian apodization) in the F2 dimension, ∼ 81° phase shifted sine bell functions in each dimension, and zero filled to 8192 (F2) × 4096 (F1) complex points before Fourier transformation.
Ser, Thr, Gly, Ala, and Ile are among the most easily identifiable spin systems. As shown in Figure 3B, six isolated Ala Cα-Cβ cross peaks can be observed (∼32%), with the chemical shifts corresponding to either β-strand or random coil, assigned according to secondary chemical shifts determined from the statistics of re-referenced protein chemical shift database (RefDB) (59). Integration of the remaining Ala Cα-Cβ intensity using Sparky (Goddard and Kneller, Sparky 3.112, University of California, San Francisco), in the chemical shift range characteristic for Ala in a helical secondary structure (53.5−56.8 ppm for Cα, and 17.2−19.6 ppm for Cβ) yielded a peak count of 13 (∼68%), which is consistent with the number of alanines in the Δ3−12 CYP3A4. It should be noted that due to the statistical nature of the database, this approach could only be used to give an estimate of the secondary structure distribution within a spectrum, and correct secondary structure determination requires assignments of a stretch of amino acids. We anticipate that with improvements in sensitivity, assignments of this type will be possible in future studies. To compare our experimental data with x-ray structures of CYP3A4, we chose the ligand-free CYP3A4 structure with the highest resolution (PDB ID: 1TQN) and assigned the secondary structure using the program DSSPcont (60). The distribution of Ala secondary structure (∼38% β-strand and coil, and ∼62% helix) is in good agreement with the experimental data. Spectra were further analyzed for other amino acids with easily identifiable chemical shifts. Similar integration of Gly Cα-C’ cross-peak region and Thr Cβ-Cγ cross-peak region (analyzed from a 2D 13C-13C correlation spectrum with polarization transferred by SPC-5 recoupling scheme (61), which transfers significant polarization only between directly bonded atoms at the mixing time used) yielded estimates of 22 out of 26 peaks for Gly, and 24 out of 26 for Thr. Overall, the agreement of secondary structure between the experimental SSNMR data and the ligand-free crystal structure further proves that CYP3A4 is in its folded state within the precipitated Nanodisc complex.
As another example of individually resolved spin systems, three Trp have been identified in the same 2D 13C-13C spectrum (Figure 3C), consistent with the number of Trp in CYP3A4 sequence. One Trp (ζ2 at 109.8 ppm, Cα at 63.5 ppm and Cβ at 28.1 ppm) can be tentatively assigned to Trp 126 in the middle of helix C, based upon chemical shift patterns consistent with a helical conformation, which is observed for only one tryptophan (Trp 126) in the crystal structure. The interactions of heme propionates with the side chain of this Trp (they are within 5 Å of each other) (37) could cause a further downfield shift in the Cα of this Trp. The other two Trp residues have 13C chemical shifts consistent with β-strand or random coil conformations and can be assigned to the remaining two Trp residues (Trp 72 and Trp 408). They are individually resolved in the spectrum processed for improved resolution (only 0.33 ppm, or 60 Hz, line broadening applied in each dimension).
Comparison with Crystal Structures Based on Predicted Chemical Shifts
To assess at low resolution whether the SSNMR experimental data is consistent with the available crystal structures, we used ShiftX (62) to calculate the Cα and Cβ chemical shifts semi-empirically, using both the highest resolution ligand-free CYP3A4 x-ray structure (36), as well as the ketaconazole-bound CYP3A4 structure (38). We restricted our analysis to the Cα and Cβ chemical shifts, for which ShiftX gives the highest correlation factors between predicted chemical shifts and experimental values (0.98 for Cα and 0.996 for Cβ ), with reported RMS errors of 0.98 ppm for Cα, and 1.1 ppm for Cβ chemical shifts. The predicted chemical shifts of the two structures were then compared with the 2D 13C-13C correlation spectrum incorporating SPC-5 polarization transfer. The overall agreement for most spectral regions is quite good between either of the structures and the experimental spectrum (Figure 4). However, some differences are clear between predicted chemical shifts for both CYP3A4 structures and the experimental data, as well as between the chemical shifts of the two different CYP3A4 structures. These differences are most readily observed among Leu, Asp, Ser, and Phe Cα-Cβ chemical shifts, and they indicate the sensitivity of chemical shifts to small structural differences. For example, in the ketoconazole-bound CYP3A4 structure, residues 210−213 (two Leu, Arg, and Phe) adopt a helix-like shape from random coil in the ligand-free structure, resulting in predicted chemical shift changes of more than 3 ppm for some (Cβ of Leu210, for example). In similar manner, it is expected that future studies of CYP3A4 in Nanodisc will reveal details about changes that take place upon the interaction of CYP3A4 with the lipid membrane.
Figure 4.
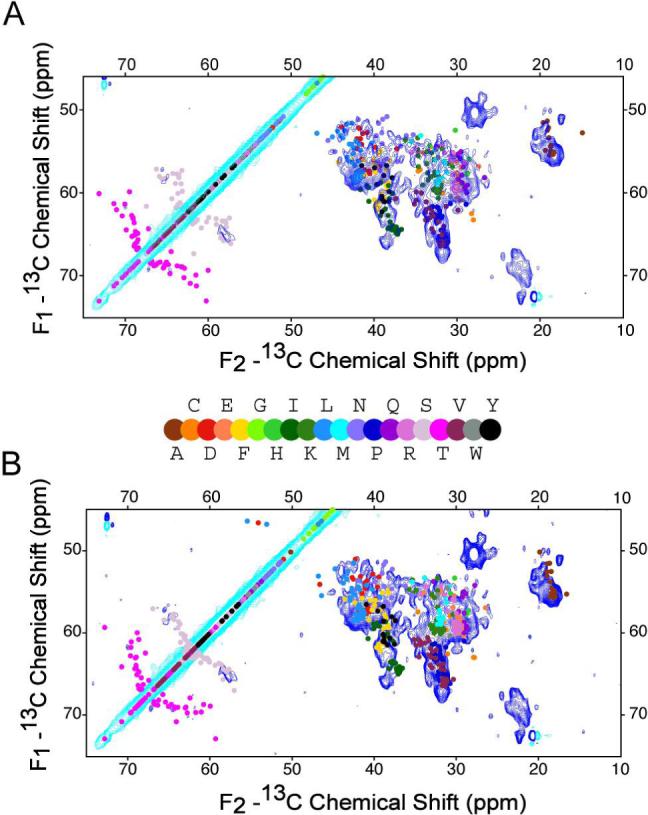
A) 2D 13C-13C chemical shift correlation spectrum of [U-13C, 15N] CYP3A4 (∼50 nmol) in a Nanodisc overlaid with the 2D 13C-13C spectrum predicted from a CYP3A4 crystal structure (PDB ID 1TQN). The contours were cut at 5 σ noise level with positive contours in turquoise, and negative contours in blue. The data were acquired with SPC-5 recoupling scheme for ∼36 hr. The other acquisition parameters are 0.5 ms 1H-13C CP contact time, ∼72 kHz 1H TPPM decoupling (6.9 μs, 16.6°), 6.4 ms t1 acquisition time (640 × 10 μs) with TPPI detection, 15.36 ms t2 acquisition time (1536 × 10 μs) and 3 s pulse delay. The data were processed with 0.26 ppm (40 Hz) net line broadening (Lorentzian-to-Gaussian apodization in both dimensions), 67.5° phase shifted sine bell functions, and zero filled to 8192 (F2) × 2048 (F1) complex points prior to Fourier transform. B) Overlay of the simulated 2D 13C-13C spectrum predicted from ketoconazole bound CYP3A4 structure (PDB ID 2J0CB) and the same experimental spectrum. The chemical shifts used to generate the simulated spectra are predicted by program ShiftX (62).
The construct used in these structural studies has ten amino acids from the N-terminal helix of the full length CYP3A4, deleted, but it retains the membrane-anchoring domain. From previous studies it is known that this construct is fully active (18, 19, 42, 63). Studies conducted on other P450s with the same global fold (64, 65) show that human P450s contain other hydrophobic patches that represent secondary membrane binding domains (66). These membrane interacting domains include the F/G and the B/C loops (64, 65, 67), which possibly orient a substrate-access channel towards the membrane. They are also consistent with the available crystallographic data, but the effect of the membrane bilayer and its properties on protein conformation leading to proper orientation of the substrate channel remains to be determined (37).
CONCLUSIONS
We have demonstrated methods for preparation of active CYP3A4 for SSNMR studies in a nanoscale membrane bilayer. The optimized growth protocol for uniform 13C,15N-labeling includes use of pG-KJE8 plasmid for co-expression with groEL and groES chaperones. Incorporation of CYP3A4 into Nanodiscs yields milligrams of enzymatically active CYP3A4 in a membrane environment, which remains intact and active when precipitated with PEG. The complex prepared in this manner yields 2D MAS SSNMR spectra with narrow line widths and high sensitivity. Chemical shift analysis demonstrates that the secondary structure of CYP3A4 in Nanodiscs is consistent with that reported by crystallography. Although the 2D spectra alone are not yet sufficient to determine site-specific assignments, they indicate that this new sample preparation method is viable for full assignments and structure determination.
As demonstrated in many previous studies, Nanodiscs provide a number of advantages for biophysical studies of membrane proteins, including functional relevance, free choice of lipid constitution, robustness, homogeneity, stability, and ease of assembly and membrane protein incorporation. They have proven as an ideal membrane system for structural studies of CYP3A4, a protein that makes conformationally and functionally inequivalent oligomers in other membrane mimetics (39). Approaches presented here are very likely to be applicable to numerous other membrane proteins, particularly those that are strongly affected by lipid-protein interactions, and suffering from similar obstacles for such studies as CYP3A4. Combining the unique features of the Nanodisc with MAS SSNMR opens a number of possibilities for conducting functional and structural studies of membrane proteins in common, near-native bilayer conditions, long an outstanding problem in membrane protein biochemistry.
Supplementary Material
Acknowledgment
The authors thank Heather Frericks, Dr. Deborah Berthold, and Yelena V. Grinkova for various aspects of sample preparation advice, Sanjeewa Rupasinghe and Dr. Mary Schuler for providing chaperone plasmids and discussions on CYP3A4 expression, Dr. Ilia Denisov for assistance in analyzing bromocriptine-bound pellet spectra, and Amy Shih for assistance with VMD graphics.
Abbreviations
- CP
cross-polarization
- CYP
cytochrome P450
- DARR
dipolar assisted rotational resonance
- MAS
magic-angle spinning
- PEG
polyethylene glycol
- SSNMR
solid-state nuclear magnetic resonance
- TPPM
two-pulse phase modulation
Footnotes
This work was supported by the University of Illinois (startup funds to C. M. Rienstra), and the National Institutes of Health grants GM33775 and GM31756 (to S.G. Sligar) and GM79530 (to C.M. Rienstra and S.G. Sligar).
Supporting Information Available. A figure showing CO difference spectrum of purified uniformly 13C, 15N labeled CYP3A4, spectrum of bromocriptine-bound precipitated CYP3A4 Nanodiscs, a figure for line width comparison of microcrystalline protein preparation and CYP3A4 Nanodisc complex, and a table of line widths from the 13C-13C correlation spectrum. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 2.von Heijne G. The membrane protein universe: what's out there and why bother? J. Intern. Med. 2007;261:543–557. doi: 10.1111/j.1365-2796.2007.01792.x. [DOI] [PubMed] [Google Scholar]
- 3.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle- spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 4.Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: application to microcrystalline ubiquitin. J. Am. Chem. Soc. 2005;127:8618–8626. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- 5.Lange A, Becker S, Seidel K, Giller K, Pongs O, Baldus M. A concept for rapid protein-structure determination by solid-state NMR spectroscopy. Angew. Chem. Int. Edit. 2005;44:2089–2092. doi: 10.1002/anie.200462516. [DOI] [PubMed] [Google Scholar]
- 6.McDermott AE. Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr. Opin. Struct. Biol. 2004;14:554–561. doi: 10.1016/j.sbi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Durr UHN, Waskell L, Ramamoorthy A. The cytochromes P450 and b5 and their reductases --Promising targets for structural studies by advanced solid-state NMR spectroscopy. Biochim. Biophys. Acta. 2007 doi: 10.1016/j.bbamem.2007.08.007. in press. [DOI] [PubMed] [Google Scholar]
- 8.Jovanovic T, McDermott AE. Observation of ligand binding to cytochrome P450 BM-3 by means of solid-state NMR spectroscopy. J. Am. Chem. Soc. 2005;127:13816–13821. doi: 10.1021/ja0438314. [DOI] [PubMed] [Google Scholar]
- 9.Egorova-Zachernyuk TA, Hollander J, Fraser N, Gast P, Hoff AJ, Cogdell R, de Groot HJM, Baldus M. Heteronuclear 2D-correlations in a uniformly [C-13, N-15] labeled membrane-protein complex at ultra-high magnetic fields. J. Biomol. NMR. 2001;19:243–253. doi: 10.1023/a:1011235417465. [DOI] [PubMed] [Google Scholar]
- 10.van Gammeren AJ, Hulsbergen FB, Hollander JG, Groot HJ. Residual backbone and side-chain 13C and 15N resonance assignments of the intrinsic transmembrane light-harvesting 2 protein complex by solid-state Magic Angle Spinning NMR spectroscopy. J. Biomol. NMR. 2005;31:279–293. doi: 10.1007/s10858-005-1604-8. [DOI] [PubMed] [Google Scholar]
- 11.Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2006;46:459–462. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. Partial (13)C and (15)N chemical-shift assignments of the disulfide-bond-forming enzyme DsbB by 3D magic-angle spinning NMR spectroscopy. ChemBioChem. 2007;8:434–442. doi: 10.1002/cbic.200600484. [DOI] [PubMed] [Google Scholar]
- 13.Vinogradova O, Sonnichsen F, Sanders CR. On choosing a detergent for solution NMR studies of membrane proteins. J. Biomol. NMR. 1998;11:381–386. doi: 10.1023/a:1008289624496. [DOI] [PubMed] [Google Scholar]
- 14.Sorgen PL, Cahill SM, Krueger-Koplin RD, Krueger-Koplin ST, Schenck CC, Girvin ME. Structure of the Rhodobacter sphaeroides light-harvesting 1 beta subunit in detergent micelles. Biochemistry. 2002;41:31–41. doi: 10.1021/bi011576j. [DOI] [PubMed] [Google Scholar]
- 15.Bayburt TH, Grinkova YV, Sligar SG. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002;2:853–856. [Google Scholar]
- 16.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 17.Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 18.Baas BJ, Denisov IG, Sligar SG. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch. Biochem. Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J. Biol. Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 20.Shaw AW, McLean MA, Sligar SG. Phospholipid phase transitions in homogeneous nanometer scale bilayer discs. FEBS Lett. 2004;556:260–264. doi: 10.1016/s0014-5793(03)01400-5. [DOI] [PubMed] [Google Scholar]
- 21.Denisov IG, McLean MA, Shaw AW, Grinkova YV, Sligar SG. Thermotropic phase transition in soluble nanoscale lipid bilayers. J. Phys. Chem. B. 2005;109:15580–15588. doi: 10.1021/jp051385g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J. Biol. Chem. 2007;282:6556–6563. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 23.Alami M, Dalal K, Lelj-Garolla B, Sligar SG, Duong F. Nanodiscs unravel the interaction between the SecYEG channel and its cytosolic partner SecA. EMBO J. 2007;26:1995–2004. doi: 10.1038/sj.emboj.7601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- 25.Civjan NR, Bayburt TH, Schuler MA, Sligar SG. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. Biotechniques. 2003;35:556–560. 562–553. doi: 10.2144/03353rr02. [DOI] [PubMed] [Google Scholar]
- 26.Duan H, Civjan NR, Sligar SG, Schuler MA. Co-incorporation of heterologously expressed Arabidopsis cytochrome P450 and P450 reductase into soluble nanoscale lipid bilayers. Arch. Biochem. Biophys. 2004;424:141–153. doi: 10.1016/j.abb.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Guengerich FP. A malleable catalyst dominates the metabolism of drugs. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13565–13566. doi: 10.1073/pnas.0606333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domanski TL, He YA, Khan KK, Roussel F, Wang Q, Halpert JR. Phenylalanine and tryptophan scanning mutagenesis of CYP3A4 substrate recognition site residues and effect on substrate oxidation and cooperativity. Biochemistry. 2001;40:10150–10160. doi: 10.1021/bi010758a. [DOI] [PubMed] [Google Scholar]
- 29.Lu P, Lin Y, Rodrigues AD, Rushmore TH, Baillie TA, Shou M. Testosterone, 7-benzyloxyquinoline, and 7-benzyloxy-4-trifluoromethyl-coumarin bind to different domains within the active site of cytochrome P450 3A4. Drug Metab. Dispos. 2001;29:1473–1479. [PubMed] [Google Scholar]
- 30.Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 31.Hutzler JM, Tracy TS. Atypical kinetic profiles in drug metabolism reactions. Drug Metab. Dispos. 2002;30:355–362. doi: 10.1124/dmd.30.4.355. [DOI] [PubMed] [Google Scholar]
- 32.Nath A, Grinkova YV, Sligar SG, Atkins WM. Ligand binding to cytochrome P450 3A4 in phospholipid nanodiscs: The effect of model membranes. J. Biol. Chem. 2007 doi: 10.1074/jbc.M703568200. in press. [DOI] [PubMed] [Google Scholar]
- 33.Kim KH, Ahn T, Yun CH. Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4. Biochemistry. 2003;42:15377–15387. doi: 10.1021/bi035280k. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka E. Clinical importance of non-genetic and genetic cytochrome P450 function tests in liver disease. J. Clin. Pharm. Ter. 1998;23:161–170. doi: 10.1046/j.1365-2710.1998.00135.x. [DOI] [PubMed] [Google Scholar]
- 35.Wilkinson GR. Cytochrome P4503A (CYP3A) metabolism: prediction of in vivo activity in humans. J. Pharmacokinet. Biopharm. 1996;24:475–490. doi: 10.1007/BF02353475. [DOI] [PubMed] [Google Scholar]
- 36.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 37.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 38.Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davydov DR, Fernando H, Baas BJ, Sligar SG, Halpert JR. Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: heterogeneity of the enzyme caused by its oligomerization. Biochemistry. 2005;44:13902–13913. doi: 10.1021/bi0509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hosea NA, Guengerich FP. Oxidation of nonionic detergents by cytochrome P450 enzymes. Arch. Biochem. Biophys. 1998;353:365–373. doi: 10.1006/abbi.1998.0659. [DOI] [PubMed] [Google Scholar]
- 41.Denisov IG, Grinkova YV, Baas BJ, Sligar SG. The ferrousdioxygen intermediate in human cytochrome P450 3A4. Substrate dependence of formation and decay kinetics. J. Biol. Chem. 2006;281:23313–23318. doi: 10.1074/jbc.M605511200. [DOI] [PubMed] [Google Scholar]
- 42.Gillam EM, Baba T, Kim BR, Ohmori S, Guengerich FP. Expression of modified human cytochrome P450 3A4 in Escherichia coli and purification and reconstitution of the enzyme. Arch. Biochem. Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- 43.Kagawa N, Hori H, Waterman MR, Yoshioka S. Characterization of stable human aromatase expressed in E. coli. Steroids. 2004;69:235–243. doi: 10.1016/j.steroids.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 45.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. solubilization, purification, and properties. J. Biol. Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 46.Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM. Magic-angle spinning solid-state NMR spectroscopy of the beta1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J. Am. Chem. Soc. 2005;127:12291–12305. doi: 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- 47.Hediger S, Meier BH, Kurur ND, Bodenhausen G, Ernst RR. NMR cross-polarization by adiabatic passage through the Hartmann-Hahn condition (APHH) Chem. Phys. Lett. 1994;223:283–288. [Google Scholar]
- 48.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995;103:6951–6958. [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe - a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 51.Rupasinghe SG, Duan H, Frericks Schmidt HL, Berthold DA, Rienstra CM, Schuler MA. High-yield expression and purification of isotopically labeled cytochrome P450 monooxygenase for solid-state NMR spectroscopy. Biochim. Biophys. Acta. 2007 doi: 10.1016/j.bbamem.2007.09.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Kijac AZ, Sligar SG, Rienstra CM. Structural analysis of nanoscale self-assembled discoidal lipid bilayers by solid-state NMR spectroscopy. Biophys. J. 2006;91:3819–3828. doi: 10.1529/biophysj.106.087072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davydov DR, Halpert JR, Renaud JP, Hui Bon Hoa G. Conformational heterogeneity of cytochrome P450 3A4 revealed by high pressure spectroscopy. Biochem. Biophys. Res. Commun. 2003;312:121–130. doi: 10.1016/j.bbrc.2003.09.247. [DOI] [PubMed] [Google Scholar]
- 55.Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- 56.Hiller M, Krabben L, Vinothkumar KR, Castellani F, van Rossum BJ, Kuhlbrandt W, Oschkinat H. Solid-state magic-angle spinning NMR of outer-membrane protein G from Escherichia coli. ChemBioChem. 2005;6:1679–1684. doi: 10.1002/cbic.200500132. [DOI] [PubMed] [Google Scholar]
- 57.Iwadate M, Asakura T, Williamson MP. C alpha and C beta carbon-13 chemical shifts in proteins from an empirical database. J. Biomol. NMR. 1999;13:199–211. doi: 10.1023/a:1008376710086. [DOI] [PubMed] [Google Scholar]
- 58.Spera S, Bax A. Empirical correlation between protein backbone conformation and C-alpha and C-beta C-13 nuclear-magnetic-resonance chemical-shifts. J. Am. Chem. Soc. 1991;113:5490–5492. [Google Scholar]
- 59.Zhang H, Neal S, Wishart DS. RefDB: a database of uniformly referenced protein chemical shifts. J. Biomol. NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 60.Carter P, Andersen CA, Rost B. DSSPcont: Continuous secondary structure assignments for proteins. Nucleic Acids Res. 2003;31:3293–3295. doi: 10.1093/nar/gkg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hohwy M, Rienstra CM, Jaroniec CP, Griffin RG. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. J. Chem. Phys. 1999;110:7983–7992. [Google Scholar]
- 62.Neal S, Nip AM, Zhang H, Wishart DS. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 63.Gillam EM. Human cytochrome P450 enzymes expressed in bacteria: reagents to probe molecular interactions in toxicology. Clin. Exp. Pharmacol. Physiol. 1998;25:877–886. doi: 10.1111/j.1440-1681.1998.tb02338.x. [DOI] [PubMed] [Google Scholar]
- 64.Deng H, Wu J, So SP, Ruan KH. Identification of the residues in the helix F/G loop important to catalytic function of membrane-bound prostacyclin synthase. Biochemistry. 2003;42:5609–5617. doi: 10.1021/bi026749z. [DOI] [PubMed] [Google Scholar]
- 65.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol. Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 66.Johnson EF, Stout CD. Structural diversity of human xenobioticmetabolizing cytochrome P450 monooxygenases. Biochem. Biophys. Res. Commun. 2005;338:331–336. doi: 10.1016/j.bbrc.2005.08.190. [DOI] [PubMed] [Google Scholar]
- 67.Cosme J, Johnson EF. Analyzing binding of N-terminal truncated, microsomal cytochrome P450s to membranes. Methods Enzymol. 2002;357:116–120. doi: 10.1016/s0076-6879(02)57671-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.