

Abstract
Immunoglobulin (Ig) class switch recombination (CSR) deficiencies are rare primary immunodeficiencies characterized by the lack of switched isotype (IgG/IgA/IgE) production. In some cases, CSR deficiencies can be associated with abnormal somatic hypermutation. Analysis of CSR deficiencies has helped reveal the key functions of CSR-triggering molecules, i.e., CD40L, CD40, and effector molecules such as activation-induced cytidine deaminase and uracil N-glycosylase. We report a new form of B cell–intrinsic CSR deficiency found in three patients with deleterious, homozygous mutations in the gene encoding the PMS2 component of the mismatch repair machinery. CSR was found partially defective in vivo and markedly impaired in vitro. It is characterized by the defective occurrence of double-strand DNA breaks (DSBs) in switch regions and abnormal formation of switch junctions. This observation strongly suggests a role for PMS2 in CSR-induced DSB generation.
Ig class switch recombination (CSR) deficiencies are rare primary immunodeficiencies characterized by normal or increased serum IgM levels and a contrasting, marked decrease or absence of IgG, IgA, and IgE. As a result of this molecular defect, defective CSR may be associated with faulty generation of somatic hypermutations (SHMs) in the Ig variable (V) region. The molecular identification and analysis of several CSR deficiencies has made it possible to better describe the mechanisms underlying CSR and SHM, which are both key elements in the maturation of antibody responses.
CSR results in the production of antibodies of different isotypes (IgG, IgA, and IgE) with the same V(D)J specificity and, therefore, the same antigen affinity. SHM primarily introduces stochastic mutations into the V region of the Ig, a genetic modification followed by positive selection of B cells harboring a B cell receptor with high antigen affinity.
Mutations in the gene encoding CD40L (1) or CD40 (2) result in a CSR deficiency generally associated with reduced SHM generation, demonstrating the essential role of the B cell's CD40 signaling pathway for both CSR and SHM. Other CSR deficiencies are a consequence of an intrinsic defect in the CSR machinery. The activation-induced cytidine deaminase (AID) is characterized by impairment of both CSR and SHM (2). This finding, together with the description of AID−/− mice, has demonstrated the master role of AID in antibody maturation. In fact, AID selectively changes cytosines into uracils in the switch (S) and V regions. Uracil N-glycosylase (UNG; mutated in another CSR deficiency) (3) removes uracils introduced into DNA by AID (4) and, thus, produces an abasic site that is cleaved by a specific endonuclease. This event ultimately leads to scattered, single-strand DNA breaks (SSBs).
AID- and UNG-induced DNA lesions are differently repaired in S and V regions. In S regions, CSR-induced double-strand DNA break (DSB) repair requires phosphorylation of the H2AX histone (γH2AX), and the presence of the MRE11–RAD50–NBS1 complex, 53BP1, MDC1, and some of the molecules in the nonhomologous end-joining repair pathway. It is very probable that other DNA repair factors are required (5, 6). Mutations in the MRE11 (leading to ataxia-like disease), Nijmegen breakage syndrome (NBS1), and ataxia telangiectasia (ATM) genes lead to CSR deficiencies, revealing the role of these molecules in S region DNA repair in humans (7).
The mismatch repair system (MMR) is known to play a role in CSR in mice, as shown by abnormal switched isotype levels and switch junctions (8, 9). There are two main MMR components: the MutS homologue (MSH1–6) and the MutL homologue (PMS2/MLH1/PMS1). The MSH2–MSH6 complex appears to recognize AID-induced DNA mismatch in the absence of UNG, leading to backup CSR and SHM, as shown by the phenotype of a double UNG-MSH2 knockout mutant (10). Recently, it has also been reported that MSH5 variants in humans can be associated with common variable immunodeficiency (CVID) and IgA deficiency phenotypes, including abnormal switch junctions that are characteristic of DNA repair defects (11). The role of the PMS2–MLH1 complex is less clear. Recently, it has been proposed that the MMR system can convert DNA SSBs into DSBs (12). Interestingly, fewer blunt DSBs were observed in mouse PMS2- than in MSH2-deficient B cells.
The error-prone repair of SHM-induced DNA breaks requires the MMR and error-prone DNA polymerases. The MSH2–MSH6 complex is essential in SHM for recognizing the AID-induced U/G mismatch, and recruiting exonuclease (EXO1) and polymerase η. The role of the PMS2–MLH1 complex in SHM remains subject to debate (13, 14). In this paper, we report that human PMS2 deficiency is associated with a variable but significant CSR defect, as observed in three patients carrying mutations in the PMS2 gene.
RESULTS AND DISCUSSION
PMS2 gene mutations and protein expression
PMS2 gene mutations.
Three patients with Ig-CSR deficiency (see the following paragraph) were found to carry homozygous mutations in the PMS2 gene. Recurrent infections and café-au-lait skin spots observed in patient 1 (P1) were evocative of MMR defect. First-degree consanguinity enabled us to perform a genome scan using polymorphic markers flanking the MLH1, PMS1, PMS2, MSH2, and MSH6 genes. A large, homozygous, 107-bp region including the PMS2 gene was observed on chromosome 7p22. The PMS2 gene was thus sequenced on genomic DNA, and a homozygous deletion of exons 11–14 was observed (p.N412DfsX6). All other exons and their flanking regions had a normal sequence. Parents were found to be heterozygous for the deletion, whereas the healthy sister had a wild-type homozygous PMS2 sequence. The homozygous deletion of exons 11–14 was confirmed on the patient's cDNA, with an 18-nucleotide frameshift insertion and a stop codon at the very beginning of exon 15. This abnormality was found in cDNA from both fibroblasts and PBLs before and after CSR induction by sCD40L/IL-4 co-stimulation (Fig. 1).
Figure 1.

Induction of RNA transcripts in control, P1, and P2 lymphocytes undergoing in vitro CSR. RNA transcripts of the genes encoding CD19, AID, UNG2, PMS2, MLH1, PMS1, MSH2, MSH6, and IgE (germline and functional) were analyzed using RT-PCR in PBLs from controls (n = 5), and P1 and P2 (n = 1) both before and after a 5-d activation with sCD40L plus IL-4. As is the case for AID and UNG2, MMR transcripts were induced during CSR activation. Germline IgE transcripts were normally expressed after activation, in contrast to the lack of functional transcripts in P1 and low level in P2. (D0, day 0; D5, day 5).
The PMS2 deletion was predicted to lead to a truncated protein lacking the MLH1 dimerization domain, the endonuclease domain, the putative nuclear localization signal, and the nuclear export signal (Fig. 2 A) (15). As previously reported, a homozygous PMS2 nonsense mutation predicting a defect downstream from the endonuclease domain, at the very end of the domain of interaction with MLH1, was found in P2 (p.R802X) (16). P3 carried a homozygous frameshift mutation in PMS2, leading to a premature stop codon and the loss of exons 12–15 and most of exon 11 (p.N412DfsX6; Fig. 2 A) (17).
Figure 2.

PMS2 deficiency. (A) Mutations in the PMS2 gene. Mutations in PMS2 gene and predicted proteins. (B) PMS2 protein expression in patients' fibroblasts and EBV B cell lines by Western blot analysis. Antibody raised against the C terminus of PMS2 failed to detect PMS2 in P1 fibroblasts (n = 1), whereas an antibody raised against the N terminus revealed a truncated 46-kD protein (not found in control cells) in P1 fibroblasts (n = 3) and the EBV B cell line (n = 1). The antibody specific for the N terminus of PMS2 revealed no detectable protein in P2 EBV B cells (n = 3). MLH1 expression was found to be lower in P1 fibroblasts and in P1 and P2 EBV B cell lines (n = 3) compared with control cells. (C) Subcellular localization of PMS2. Primary fibroblasts from a control and P1 were labeled with anti–N-terminal PMS2 and anti-MLH1 antibodies, followed by secondary antibody (Alexa Fluor 488). PMS2 was predominantly observed in the cytoplasm in P1 fibroblasts, contrasting with a predominantly nuclear localization in control fibroblasts. No difference was observed in terms of the subcellular localization of MLH1, although MLH1 expression appeared to be slightly lower in P1 cells (n = 3). Bar, 20 μm.
PMS2 protein expression.
Western blot analysis performed on protein extracts from fibroblasts (P1) and EBV B cell lines (P1 and P2) using a monoclonal antibody against the protein N terminus revealed, respectively, a truncated PMS2 protein in P1 cells (46 kD, instead of the 110-kD protein for wild-type PMS2) and an absence of protein in P2. A monoclonal antibody against the C-terminal part of PMS2 did not reveal PMS2 in both patients' cell lines nor in P3 colic carcinoma cells (17). MLH1 was detected in patients' fibroblasts (P1) and EBV B cells (P1 and P2), but the intensity of the signal was clearly lower than that in control cells (Fig. 2 B).
Because the C-terminal dimerization domain in PMS2 and MLH1 has been shown to promote the nuclear import of their complex (MutLα) (18), we analyzed the subcellular localization of MLH1 and the truncated form of PMS2 in P1 fibroblasts by using an antibody against the N-terminal part of PMS2. As shown in Fig. 2 C, PMS2 was primarily detected in the cytoplasm of the patient's fibroblasts, contrasting with the nuclear localization observed in control fibroblasts. MLH1 was predominantly found in the nuclei of P1 fibroblasts, although its intensity appeared to be slightly lower than that in control nuclei. These results indicate that the truncated PMS2 product (lacking the MLH1 interaction domain and nuclear import signal) is indeed retained in the cytoplasm. In the absence of PMS2, MLH1 can nevertheless still localize to the nucleus, perhaps by heterodimerization with PMS1 or MLH3 (18). Reduced MLH1 expression may be a consequence of the protein's inability to interact with PMS2.
CSR deficiency
In P1, the serum Ig levels determined at 9 yr of age, before the occurrence of cancer and administration of chemotherapy, were typical of a CSR deficiency, with high serum IgM levels, profoundly low serum IgG levels, and an absence of IgA. This immunodeficiency subsequently required Ig substitution. Normal serum Ig levels were detected in both parents. In P2 and P3, low levels of IgG2 and a lack of IgG4 were recorded. In P3, at 9 yr of age, a decrease in IgA level was also observed. It was not found later on (17 yr; Table I). It is noteworthy that serum Ig determination was performed at some distance after chemotherapy (6 yr in P2 and 8 yr in P3). The variability of Ig levels could result from an in vivo accumulation of switched memory B cells with age (P1 is the youngest patient), as observed in mice deficient for UNG or for several factors in the MMR pathway (10, 19).
Table I.
Immunological data of patients
| Ig levels (g/liter)
|
||||||||
|---|---|---|---|---|---|---|---|---|
| IgM | IgG | IgG1 | IgG2 | IgG3 | IgG4 | IgA | CD27+/CD19+ (%)a | |
| P1b (9 yr) |
2.5b | 3.5b | ND | ND | ND | ND | <0.06b | 4 |
| P2 (22 yr) |
1 | 7.4 | 4.6 | 0.3 | 0.4 | 0.002 | 2.6 | 6 |
| P3 (9 yr) |
0.8 | 8.7 | ND | ND | ND | ND | 0.3 | ND |
| P3 (17 yr) |
2.5 | 8.9 | 6.2 | 0.1 | 0.3 | 0.001 | 0.8 | 23 |
| Controls (9 yr) |
0.5–1.5 | 5.8–11.5 | 0.5–1.6 | 13–58 | ||||
| Controls (17–22 yr) |
0.5–2.1 | 6.6–12.7 | >4 | >0.6 | >0.2 | >0.01 | 0.7–3.4 | 13–58 |
Bolded numbers indicate abnormal values. ND, not determined.
Gated on CD19+ B cells.
At age of immunodeficiency diagnosis before IgG substitution.
Blood CD19+ B lymphocyte numbers were normal in all three patients. CD27+ memory B cells were low in P1 and P2 and normal in P3 (Table I). In all three patients, virtually all CD19+CD27+ B cells were IgM+IgD+ (Fig. 3 A), and very few IgA+ (switched) B cells were detected (P3; 2 vs. 8% in an age-matched control; Fig. 3 A), pointing to a defective in vivo CSR toward IgA. In vitro activation of P1, P2, and P3 B cells with sCD40L plus IL-4 or sCD40L plus IL-10 led to a poorly efficient CSR toward IgE and IgA, respectively, although potent B cell proliferation could be detected when tested for (Fig. 3 B). These observations point to a CSR defect located downstream of the transcription step, because IgE germline transcripts were normally expressed in CSR-activated B cells from P1 and P2 (Fig. 1).
Figure 3.

In vivo and in vitro Ig-CSR deficiency in patients. (A) B cell phenotype. IgM and IgD expression was studied on CD19+CD27+ gated PBLs. Very few IgM+IgD+ cells were observed in P1, P2, and P3 (n = 1) compared with controls (n = 7). A reduction of IgA+ B cells on CD19+-gated PBLs was observed in P3 compared with an age-matched control (n = 2). (B) Defective IgE and IgA production by CSR activated patients' PBLs. PBLs were activated by sCD40L plus IL-4 for 12 d or sCD40L plus IL-10 for 10 d for CSR toward IgE (P1, n = 2; P2 and P3, n = 1; controls, n = 10) and IgA (P1 and P3, n = 1; controls, n = 5), respectively. Proliferation was assessed after a 5-d stimulation with sCD40L plus IL-4 by measuring [3H]thymidine incorporation. Error bars represent one SD (P1 and P3, n = 1; controls, n = 10).
We next sought to investigate and delineate the mechanism of this B cell–intrinsic CSR deficiency: CD40 expression, gene sequence, and function (proliferation) were all found to be normal in P1. Likewise, AID and UNG transcripts were normally expressed after in vitro CSR induction (Fig. 1), whereas no mutations were found in the encoding genes. The previously described (6) CSR deficiency associated with impaired DNA repair was also ruled out, because the sensitivity of P1 skin fibroblasts to ionizing radiation was within the normal range (not depicted).
Because uracil excision by UNG and abasic site–specific endonuclease activities appear to be essential for CSR (as shown by the UNG-deficient phenotype) (3), and even though the UNG1 and UNG2 sequences were found to be normal, we decided to assess uracil excision and cleavage in fibroblasts from P1 and from age-matched controls. Whole protein lysates from control and P1 fibroblasts were equally able to cleave a double-stranded DNA probe containing a U/G mismatch, thus ruling out abnormal activity of UNG and the APEX1 and APEX2 endonucleases, which were recently reported as being essential in CSR in mice (20). The assay's specificity was confirmed by using the UNG inhibitor Ugi or UNG-deficient cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20080789/DC1).
As an attempt to localize the precise step of the CSR defect, we checked for the occurrence of DSBs in Sμ regions of sCD40L plus IL-4–activated P1 B cells. Sμ DSBs could not be detected, even in the presence of T4 polymerase, suggesting a defect in the generation of both scattered and blunt DSBs (Fig. 4 A). One cannot formally exclude that DSBs were not detected because of the low number of available B cells; nevertheless, our data indicate that Sμ DSB generation was at least markedly decreased in PMS2-deficient B cells as compared with controls.
Figure 4.

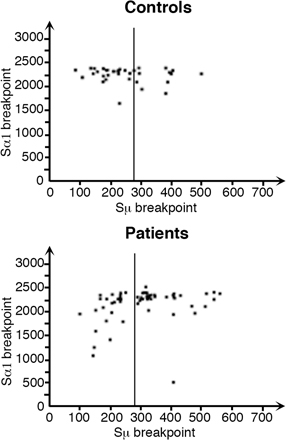
Defective DSB occurrence and junction repair. (A) DSB occurrence in the Sμ regions of DNA agarose plugs from 20,000 B cells (CD19+) or non–B cells (CD19−) were treated or not with T4 polymerase. After ligation with the linker, ligation-mediated PCR products were transferred and Southern blotting performed using a 32P-labeled Sμ probe. DSBs were detected in the Sμ regions of control (n = 4) but not of P1 (n = 1) B cells. Amplification of Pinx1 was used as a control of DNA integrity and input. (B) Abnormal pattern of Sμ–Sα1 junctions in patients. Representation of the length of the switch junctions is shown (controls, n = 38; P1, n = 29; P2, n = 16; and P3, n = 15). Statistically significant differences are indicated (*, P < 0.05; and ***, P < 0.001 using the χ2 test).
To assess the nature of the switch junctions, we sequenced the in vivo–generated Sμ–Sα switch junctions in patients. Most were found to have been repaired using microhomology, as shown in Fig. 4 B and Fig. S2 (available at http://www.jem.org/cgi/content/full/jem.20080789/DC1). When compared with controls, a significantly lower number of blunt junctions was observed in P1–3 (24, 31, and 27%, respectively, vs. 62% in controls; P < 0.05 using a χ2 test), whereas a significant increase of 7 bp or more in microhomology usage was found (52% in P1, 50% in P2, and 47% in P3 compared with 8% in controls; P < 0.001 using a χ2 test). Significantly more Sμ breakpoints in patients' B cells were located in the section of the Sμ region that shows the highest degree of homology with Sα1 (positions 275–760; 50% as compared with 19% in control B cells; P < 0.01; Fig. S3). Such an observation was previously reported in PMS2 knockout mice (8). Another feature was the reduction in the number of insertions observed at the Sμ–Sα junctions from patients (5%) as compared with controls (29%; P < 0.01).
Because it has been suggested by analysis of the DNA-dependent protein kinase catalytic subunit–deficient mice that DNA repair mechanisms might differ in the process of CSR toward IgA and IgG, Sμ–Sγ1 junctions were also analyzed. Interestingly, these junctions were identically repaired in patients' and controls' B cells, and no microhomology usage was observed (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20080789/DC1), a result reminiscent of that observed in ligase IV–deficient human B cells (21). This observation could be the consequence of a different mechanism involved in Sμ–Sα and Sμ–Sγ1 junction repair. Also, the fact that Sμ and Sα1 regions share more microhomology sequences as compared with Sμ and Sγ1 regions could account for it. No Sμ–Sγ2 junctions could be amplified in patients, in correlation with the strong decrease of IgG2. Collectively, these results indicate that the CSR deficiency, which varied in intensity from one patient to another, likely results from a defect in the CSR-induced generation of DSBs.
To delineate the PMS2 defect's role in the patients' CSR deficiency, we tested PMS2 induction during CSR activation in PBLs and purified B cells from controls. Indeed, PMS2, PMS1, and MLH1 transcripts (all weakly detectable in unactivated PBLs or B cells) were induced as strongly as AID and UNG2 transcripts after sCD40L/IL-4 activation of PBLs and B cells. Furthermore, MSH2/MSH6 transcripts (detectable in unstimulated conditions) were induced by CSR activation, as recently reported by others (12). A similar induction of MMR transcripts was observed in stimulated PBLs from patients P1 and P2 (Fig. 1).
SHMs in CD19+CD27+ B cells
The occurrence of SHM was studied in the VH3-23 of IgM on purified CD19+CD27+ B cells from P1 and P2; these cells accounted for 0.5 and 0.6% of the patients' total PBLs, respectively. Nine and eight mutated clones were amplified in P1 and P2, respectively, and seven of these were different and analyzed. Compared with age-matched controls, the mutation frequency was found to be slightly lower in P1 (1.9 vs. 2.3–6.5% in controls; P < 0.001) and normal in P2 (3.3%). A normal nucleotide substitution pattern was observed in both patients (Table S1, available at http://www.jem.org/cgi/content/full/jem.20080789/DC1). However, only few mutations could be analyzed because of the strong reduction of patients' CD19+CD27+ B cells. We cannot, therefore, exclude a mild defect in the pattern of SHM generation. The reduced number of memory B cells and decreased SHM frequency could be caused by a shorter lifespan of highly proliferating B cells in germinal centers (14).
Relationship between the patients' CSR and PMS2 deficiencies
Although not strictly demonstrated, the relationship between the CSR and PMS2 deficiencies appears very likely because no other known causes of CSR defects could be found, and deleterious consequences of PMS2 mutations were detected in all three cases. The similarity of the disease with the PMS2 deficiency in mice also suggests a relationship: PMS2-deficient mice have a partial in vivo and in vitro CSR defect, characterized by the usage of microhomology in switch junctions (8, 9), whereas SHM appears to be normal in terms of the nucleotide substitution pattern. A similar phenotype is reported in MLH1-deficient mice (14). Given that PMS2 deficiency also led to a reduction in MLH1 expression, one cannot strictly exclude the possibility that the partial quantitative MLH1 deficiency has an impact on the Ig-CSR deficiency observed in patients. However, some translocation of MLH1 to the nucleus could be detected in P1 cell line.
In mice, deficiencies in either MSH2/MSH6 differ from those in MLH1 or PMS2 (even though the former can also lead to a partial CSR defect) (22), because in the former, the DSB occurrence in S region is relatively preserved (12), the switch junctions are normally repaired (9), and the SHM presents with a skewed pattern to G/C mutations (23). The role of MSH2/MSH6 in CSR and SHM has been recently confirmed by analysis of the phenotype of UNG/MSH2 and UNG/MSH6 double knockout mice (10, 19). In these animals, the absence of SHM on A:T residues and of CSR strongly suggests that a dual pathway is involved in the repair of AID-induced U/G mismatches, i.e., either UNG and base excision repair or MSH2/MSH6 recognition and repair (10, 19).
Sparse data suggest that MMR exerts a role in antibody maturation in humans. IgA deficiency has indeed been reported in a few MSH2- or MSH6-deficient patients (24), but no data are available about IgG levels, in vitro CSR, or SHM generation. More recently, it has been reported that genetic variation in the MSH5 gene is associated with IgA deficiency and CVID (11). The MSH5 variants were shown to bind weakly to MSH4 and were associated with defective S region junctions in CVID.
Our data provide evidence that PMS2 plays an important role in CSR in humans. The fact that a very severe CSR defect occurs in UNG-deficient patients (3) indicates that the PMS2-initiated mismatch repair does not represent an alternative pathway for base excision repair in human CSR. PMS2's function is likely to be situated downstream of UNG in the same pathway. Stavnezer et al. have proposed that PMS2 is involved in converting the AID/UNG-induced SSBs into the DSBs required for CSR (7). Several observations support this hypothesis, because the cleavage of a DNA probe containing a U/G mismatch reveals that SSBs normally occur in patients in the absence of PMS2 (thus situating the defect downstream of UNG/APEX activities), mouse MMR–deficient B cells (and PMS2-deficient B cells in particular) (12) exhibit a low level of blunt CSR-induced DSBs in Sμ regions (as observed in the studied patients), and the increased usage of microhomology in switch junction repair observed in PMS2-deficient mice (8, 9) and our patients is compatible with defective generation of CSR-induced DSBs in S regions.
PMS2 has been described as having an endonuclease activity necessary for efficient MMR (25–27). It has been shown in in vitro experiments that PMS2 is recruited by the MSH2–MSH6 complex on a U/G mismatch occurring close to a DNA nick located at its 3′ side, and can induce the DNA cleavage on the 5′ site of the mismatch, thus allowing EXO1 to exert its 5′–3′ exonucleasic activity (25). A defect in this endonuclease activity of PMS2 can thus account for the decreased frequency of DSBs in Sμ region observed in patients. MLH3, which also possesses an endonuclease active site, could replace the defective PMS2 but likely not efficiently enough to correct the defect in DNA cleavage.
Alternatively, data from the literature indicate that the MutLα complex (PMS2–MLH1) inhibits MMR-induced EXO1 processivity and terminates EXO1-catalyzed excision (28). One can thus propose that in the absence of MLH1/PMS2, increased EXO1 activity induced by MHS2/MSH6 recruited close to a nick located 5′ from the mismatch leads to the generation of single-strand overhangs. The latter could be preferentially processed by microhomology whenever possible (Sμ–Sα junctions). In summary, we have described another cause of Ig-CSR deficiency in humans, highlighting a subtle role for PMS2 in Ig-CSR likely at the DSB generation step.
MATERIALS AND METHODS
Case reports.
P1, a 12-yr-old Turkish female, was born to a first-degree consanguineous family. She presented with multiple café-au-lait skin spots and suffered from recurrent and severe infections from the age of 1, leading to an immunodeficiency diagnosis at 9 yr of age. 1 yr later, she developed a colorectal adenocarcinoma. We selected P2 and P3 on the basis of their PMS2 deficiency, as previously reported (16, 17). Blood samples were obtained after provision of informed consent by P2 and by the parents of P1 and P3. This study was approved by the local institutional review board (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale n°05632 Paris Saint Antoine; approval dated 20 May 2005).
Sequence analysis of human PMS2.
Genomic DNA (PMS2 ENSG-0000122512) was amplified in a long-range PCR as described by Clendenning (29).
PMS2 and MLH1 protein expression.
Immunoblotting was performed using anti-MLH1 (Santa Cruz Biotechnology, Inc.), anti–Cter-PMS2 (Santa Cruz Biotechnology, Inc.), anti–Nter-PMS2 (Abcam), and anti-Ku70 (Neo Markers).
Immunofluorescence experiments.
The immunofluorescence staining procedure adapted from Wu et al. (15) was performed using anti-MLH1, anti–Nter-PMS2, and goat anti–mouse Alexa Fluor 488 (Invitrogen).
Expression of CD19, AID, UNG, IgE, and MMR transcripts.
CD19, AID, UNG2, Iε-Cε germline, and VH-Cε functional transcripts were assessed as previously described (2, 3). PMS2, MLH1, PMS1, MSH2, and MSH6 transcripts were amplified with specific primers.
Flow cytometry analysis of B cells.
PBLs were stained with allophycocyanin anti-CD19 (BD Biosciences), FITC anti-CD27 (BD Biosciences), biotin anti-IgM (Jackson ImmunoResearch Laboratories), streptavidin-PerCP antibody (BD Biosciences), and R-PE anti-IgD (Harlan Sera-Laboratory) or R-PE anti-IgA (SouthernBiotech).
In vitro B cell proliferation and CSR toward IgE and IgA production.
Proliferation and CSR toward IgE and IgA were assessed on PBLs after activation with 500 ng/ml sCD40L (Amgen), and 100 U/ml IL-4 (R&D Systems) or 2.9 × 103 U/ml IL-10 (R&D Systems), as previously described (3).
Uracil incision assay.
Uracil incision assay was performed as previously described (6).
Analysis of CSR-induced DSB occurrence in Sμ regions.
DNA isolation and detection of DNA DSBs by ligation-mediated PCR were performed as previously described (30), with the difference that half of each DNA agarose plug was treated with T4 DNA polymerase (Promega) before ligation. Amplification of the gene encoding Pinx1 was used as a control of DNA integrity and input.
Study of switch junctions.
Amplification of switch junctions was performed using a nested PCR on genomic DNA from patients' and controls' PBLs (available from GenBank/EMBL/DDBJ under accession nos. X54713 [Sμ], L19121 [Sα1], and U39737 [Sγ1]) (21).
Analysis of SHM in variable Ig gene.
SHM in the VH3-23 IgM gene in CD19+CD27+-sorted B cells was assessed as previously described (2).
Online supplemental material.
Fig. S1 shows the efficient uracil incision activity in fibroblasts of P1. Fig. S2 shows all analyzed sequences of Sμ–Sα1 junctions from controls and patients. Fig. S3 shows the breakpoints in Sμ–Sα1 junctions from controls and patients. Fig. S4 shows the pattern and all analyzed sequences of Sμ–Sγ1 junctions from controls and patients. Table S1 shows SHM in the V region of IgM in CD27+ B cells from P1 and P2. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080789/DC1.
Supplementary Material
Acknowledgments
We are indebted to M. Forveille and F. Selz for excellent technical assistance, and to P. Revy for helpful discussions. We are grateful to Amgen for providing sCD40L.
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale, the Association de la Recherche contre le Cancer, the Association Nationale pour la Recherche, and EURO-PADnet. S. Péron is supported by the Fondation pour la Recherche Médicale.
The authors have no conflicting financial interests.
References
- 1.Korthauer, U., D. Graf, H.W. Mages, F. Briere, M. Padayachee, S. Malcolm, A.G. Ugazio, L.D. Notarangelo, R.J. Levinsky, and R.A. Kroczek. 1993. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 361:539–541. [DOI] [PubMed] [Google Scholar]
- 2.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 3.Imai, K., G. Slupphaug, W.I. Lee, P. Revy, S. Nonoyama, N. Catalan, L. Yel, M. Forveille, B. Kavli, H.E. Krokan, et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4:1023–1028. [DOI] [PubMed] [Google Scholar]
- 4.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 5.Yan, C.T., C. Boboila, E.K. Souza, S. Franco, T.R. Hickernell, M. Murphy, S. Gumaste, M. Geyer, A.A. Zarrin, J.P. Manis, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 449:478–482. [DOI] [PubMed] [Google Scholar]
- 6.Peron, S., Q. Pan-Hammarstrom, K. Imai, L. Du, N. Taubenheim, O. Sanal, L. Marodi, A. Bergelin-Besancon, M. Benkerrou, J.P. de Villartay, et al. 2007. A primary immunodeficiency characterized by defective immunoglobulin class switch recombination and impaired DNA repair. J. Exp. Med. 204:1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stavnezer, J., J.E. Guikema, and C.E. Schrader. 2008. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 26:261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrenstein, M.R., C. Rada, A.M. Jones, C. Milstein, and M.S. Neuberger. 2001. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 98:14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrader, C.E., J. Vardo, and J. Stavnezer. 2002. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J. Exp. Med. 195:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 16:163–171. [DOI] [PubMed] [Google Scholar]
- 11.Sekine, H., R.C. Ferreira, Q. Pan-Hammarstrom, R.R. Graham, B. Ziemba, S.S. de Vries, J. Liu, K. Hippen, T. Koeuth, W. Ortmann, et al. 2007. Role for Msh5 in the regulation of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 104:7193–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrader, C.E., J.E. Guikema, E.K. Linehan, E. Selsing, and J. Stavnezer. 2007. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J. Immunol. 179:6064–6071. [DOI] [PubMed] [Google Scholar]
- 13.Phung, Q.H., D.B. Winter, R. Alrefai, and P.J. Gearhart. 1999. Hypermutation in Ig V genes from mice deficient in the MLH1 mismatch repair protein. J. Immunol. 162:3121–3124. [PubMed] [Google Scholar]
- 14.Kim, N., G. Bozek, J.C. Lo, and U. Storb. 1999. Different mismatch repair deficiencies all have the same effects on somatic hypermutation: intact primary mechanism accompanied by secondary modifications. J. Exp. Med. 190:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu, X., J.L. Platt, and M. Cascalho. 2003. Dimerization of MLH1 and PMS2 limits nuclear localization of MutLalpha. Mol. Cell. Biol. 23:3320–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Vos, M., B.E. Hayward, R. Charlton, G.R. Taylor, A.W. Glaser, S. Picton, T.R. Cole, E.R. Maher, C.M. McKeown, J.R. Mann, et al. 2006. PMS2 mutations in childhood cancer. J. Natl. Cancer Inst. 98:358–361. [DOI] [PubMed] [Google Scholar]
- 17.Kratz, C.P., C.M. Niemeyer, E. Juttner, M. Kartal, A. Weninger, A. Schmitt-Graeff, U. Kontny, M. Lauten, S. Utzolino, J. Radecke, et al. 2007. Childhood T-cell non-Hodgkin's lymphoma, colorectal carcinoma and brain tumor in association with cafe-au-lait spots caused by a novel homozygous PMS2 mutation. Leukemia. 22:1078–1080. [DOI] [PubMed] [Google Scholar]
- 18.Cannavo, E., B. Gerrits, G. Marra, R. Schlapbach, and J. Jiricny. 2007. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 282:2976–2986. [DOI] [PubMed] [Google Scholar]
- 19.Shen, H.M., A. Tanaka, G. Bozek, D. Nicolae, and U. Storb. 2006. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J. Immunol. 177:5386–5392. [DOI] [PubMed] [Google Scholar]
- 20.Guikema, J.E., E.K. Linehan, D. Tsuchimoto, Y. Nakabeppu, P.R. Strauss, J. Stavnezer, and C.E. Schrader. 2007. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J. Exp. Med. 204:3017–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan-Hammarstrom, Q., A.M. Jones, A. Lahdesmaki, W. Zhou, R.A. Gatti, L. Hammarstrom, A.R. Gennery, and M.R. Ehrenstein. 2005. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J. Exp. Med. 201:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martomo, S.A., W.W. Yang, and P.J. Gearhart. 2004. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J. Exp. Med. 200:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whiteside, D., R. McLeod, G. Graham, J.L. Steckley, K. Booth, M.J. Somerville, and S.E. Andrew. 2002. A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple cafe-au-lait spots. Cancer Res. 62:359–362. [PubMed] [Google Scholar]
- 25.Kadyrov, F.A., L. Dzantiev, N. Constantin, and P. Modrich. 2006. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 126:297–308. [DOI] [PubMed] [Google Scholar]
- 26.Jiricny, J. 2006. MutLalpha: at the cutting edge of mismatch repair. Cell. 126:239–241. [DOI] [PubMed] [Google Scholar]
- 27.Erdeniz, N., M. Nguyen, S.M. Deschenes, and R.M. Liskay. 2007. Mutations affecting a putative MutLalpha endonuclease motif impact multiple mismatch repair functions. DNA Repair (Amst.). 6:1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, Y., F. Yuan, S.R. Presnell, K. Tian, Y. Gao, A.E. Tomkinson, L. Gu, and G.M. Li. 2005. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 122:693–705. [DOI] [PubMed] [Google Scholar]
- 29.Clendenning, M., H. Hampel, J. LaJeunesse, A. Lindblom, J. Lockman, M. Nilbert, L. Senter, K. Sotamaa, and A. de la Chapelle. 2006. Long-range PCR facilitates the identification of PMS2-specific mutations. Hum. Mutat. 27:490–495. [DOI] [PubMed] [Google Scholar]
- 30.Catalan, N., F. Selz, K. Imai, P. Revy, A. Fischer, and A. Durandy. 2003. The block in immunoglobulin class switch recombination caused by activation-induced cytidine deaminase deficiency occurs prior to the generation of DNA double strand breaks in switch mu region. J. Immunol. 171:2504–2509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}