

Abstract
Myosin binding protein C (MyBP-C) is a thick-filament protein that limits cross-bridge cycling rates and reduces myocyte power output. To investigate mechanisms by which MyBP-C affects contraction, we assessed effects of recombinant N-terminal domains of cardiac MyBP-C (cMyBP-C) on contractile properties of permeabilized rat cardiac trabeculae. Here, we show that N-terminal fragments of cMyBP-C that contained the first three immunoglobulin domains of cMyBP-C (i.e., C0, C1, and C2) plus the unique linker sequence termed the MyBP-C “motif” or “m-domain” increased Ca2+ sensitivity of tension and increased rates of tension redevelopment (i.e., ktr) at submaximal levels of Ca2+. At concentrations ≥20 μM, recombinant proteins also activated force in the absence of Ca2+ and inhibited maximum Ca2+-activated force. Recombinant proteins that lacked the combination of C1 and the motif did not affect contractile properties. These results suggest that the C1 domain plus the motif constitute a functional unit of MyBP-C that can activate the thin filament.
INTRODUCTION
Myosin binding protein C (MyBP-C) is a sarcomeric protein comprised of repeating domains that are homologous to either Ig or fibronectin type III domains (Einheber and Fischman, 1990; Furst et al., 1992; Gautel et al., 1995). Skeletal isoforms of MyBP-C contain 10 such domains, termed C1 through C10, whereas cardiac MyBP-C (cMyBP-C) contains an additional Ig domain at its N terminus, referred to as C0 (see Fig. 1). Between domains C1 and C2 is a linker sequence, termed the MyBP-C “motif,” that is highly conserved among the different MyBP-C isoforms (Gautel et al., 1995).
Figure 1.

Schematic diagram showing the domain organization of cMyBP-C. (Top) Full-length cMyBP-C. (Bottom) Recombinant proteins used in this study.
Sequence analysis of the motif shows that it is unique to MyBP-C proteins, but its functional significance is not well understood. A regulatory role for the region has been proposed based on observations that the cMyBP-C motif is phosphorylated by protein kinase A in response to inotropic stimuli (Gautel et al., 1995; Gruen et al., 1999). The motif also binds myosin S2 (Gruen and Gautel, 1999), and phosphorylation by protein kinase A abolishes binding, suggesting that the motif–S2 interaction is important for the regulatory effects of cMyBP-C. Consistent with this idea, recombinant proteins containing the motif increased Ca2+ sensitivity of tension in permeabilized cardiac myocytes and skeletal muscle fibers (Kunst et al., 2000; Harris et al., 2004) and biphasically activated and inhibited actomyosin interactions in in vitro motility assays (Razumova et al., 2006; Shaffer et al., 2007). However, in motility assays the functional effects of either full-length cMyBP-C (Saber et al., 2008) or its N-terminal domains (Shaffer et al., 2007) did not depend on binding myosin S2, suggesting that interactions of the motif with ligands other than S2 are important in mediating the functional effects of cMyBP-C. Alternatively, sequences outside the motif could mediate observed functional effects of the cMyBP-C N terminus. Consistent with the latter idea, the cardiac-specific C0 domain has been reported to bind actin and increase Ca2+ sensitivity of tension (Kulikovskaya et al., 2003). Herron et al. (2006) also found that a proline-alanine–rich region preceding C1 activated contraction independent of Ca2+ in permeabilized cardiac preparations. The MyBP-C motif was not required for the latter activating effects. However, effects were only observed at high protein concentrations, and it was not determined whether activating effects persisted in the presence of Ca2+.
The purpose of the current study was to assess the importance of the MyBP-C motif in affecting actomyosin contractile properties, either alone or in combination with other N-terminal domains of MyBP-C. Results demonstrate that the MyBP-C motif together with the C1 domain is necessary for effects of the N terminus of cMyBP-C to increase Ca2+ sensitivity of tension, increase rates of tension redevelopment (ktr) at submaximal [Ca2+], and increase tension development in the absence of Ca2+.
MATERIALS AND METHODS
Expression and Purification of Recombinant MyBP-C N-terminal Proteins
N-terminal cMyBP-C proteins containing the C0C1, C1C2, or C3C4 domains were cloned with and without the MyBP-C motif (Fig. 1) by PCR amplification of desired sequences from the full-length mouse cMyBP-C cDNA sequence (GenBank accession no. AF097333). C1C2(-m) lacked 82 amino acids of the motif sequence beginning at residues LRSAFRR and ending at DLRGMLK, whereas C3mC4 included 100 amino acids of the motif beginning at DLDLRSA and ending at DEKKSTAF. Motif sequences of C0C1m, C1m, and mC2 included 5 additional amino acids and ended at AFQKKLE. Recombinant constructs were subcloned into the pQE-2 expression vector (QIAGEN) in frame with a 6X His-tag at the N-terminus of the clone sequence to facilitate protein purification. Protein expression in M15 cells (QIAGEN) was induced by IPTG, and expressed proteins were purified under native conditions using a Ni-NTA affinity column according to the manufacturer's directions. After column elution, fractions containing the expressed protein were pooled and dialyzed into AB buffer (in mmoles/L): 25 KCl, 25 imidazole, 4 MgCl2, 1 EGTA, and 1 DTT. Protein concentration in AB buffer was determined by protein assay (Thermo Fisher Scientific) using bovine gamma globulin as a standard or by UV absorbance at 280 nm using theoretical extinction coefficients calculated from protein primary sequences.
Preparation of Permeabilized Cardiac Trabeculae
Treatment of all animals was in strict accordance with the guidelines and protocols established by the Institutional Animal Care and Use Committees at the University of Washington and the University of California, Davis. Male Sprague-Dawley rats (200–250 g) were killed by intraperitoneal injection of sodium pentabarbitol. Hearts were then rapidly excised, and right ventricles were dissected in a Ringer's solution containing (in mM): 100 NaCl, 24 NaHCO3, 2.5 KCl, 1 MgSO4*7H2O, 1 Na2HPO4, and 1 CaCl2. Trabeculae were permeabilized in situ by incubation of splayed ventricles overnight in a relaxing solution (in mM: 100 KCl, 10 imidazole, 2 EGTA, 5 MgCl2, and 4 ATP) containing 50% glycerol and 0.4% Triton X-100 (Sigma-Aldrich) at 4°C. Individual trabeculae were then dissected from ventricle free walls, pinned to the bottom of a sylgar-coated Petri dish, and stored for up to 1 wk in glycerinated relaxing solution at 4°C. All skinning and storage solutions contained protease inhibitor cocktail (P8340; Sigma-Aldrich).
Experimental Apparatus
The apparatus for force measurements is shown schematically in Fig. 2. Permeabilized trabeculae were transferred from a glass Petri dish to a temperature-controlled stainless steel plate containing six experimental chambers (Aurora Scientific Inc.). The first chamber contained relaxing solution and was large enough to facilitate mounting of a trabecula between a force transducer (model 400A; Aurora Scientific Inc.) and a torque motor (model 312C; Aurora Scientific Inc.) for rapid adjustments of fiber length. Trabeculae were secured to the force transducer and motor as shown in Fig. 2 by placing each end of a trabecula into a trough constructed from a bisected 25-gauge needle. The ends of the trabecula were then overlayed with a segment (∼1 mm in length) of 4.0 suture material. The suture and trabecula were next secured to the trough by tying with a strand of unwaxed dental floss (approximately equivalent to 10.0 suture thread). After securing each end of the trabecula, the entire stainless steel plate containing the experimental wells and the trabecula was placed on the stage of an inverted microscope (IX71; Olympus) fitted with a 12–mega pixel digital camera (DP70; Olympus) for visualization of the trabecula and measurement of fiber dimensions and sarcomere length. Sarcomere length was adjusted to ∼2.3 μm in relaxing solution and monitored throughout the course of an experiment.
Figure 2.

Schematic diagram of the apparatus used to obtain force measurements and original chart recordings. (Top, inset) Attachment method for securing a permeabilized trabecula to metal troughs. One trough was attached to the active end of a force transducer and the other to a motor for rapid changes in length. (Bottom) Original chart records showing Ca2+-activated force in the presence and absence of 5 μM C1C2 or 10 μM C0C1. The pCa values are shown above each activation, and the presence of C1C2 or C0C1 in bath solutions is indicated by bars.
Experimental Protocols
pCa Solutions.
Relaxing and activating solutions were prepared using a custom software package as described previously (Martyn et al., 1994; Regnier et al., 2002). Solutions were maintained at 0.18 M ionic strength and pH 7.0 at 15°C and contained (in mM): 15 phosphocreatine, 15 EGTA, 80 MOPS, 1 free Mg2+, at least 38 Na+ plus K+, 1 dithiothreitol, and 5 ATP. Ca2+ concentration (reported as pCa = −log[Ca2+]) was set by varying amounts of CaCl2.
Steady-state Force
The amount of force generated in a given experimental solution was measured as the difference in calibrated output voltage of the force transducer recorded before and after rapid slackening of a trabecula to 80% of its resting length (where resting length was set at sarcomere length of ∼2.3 μm). Input commands to the motor for length control were initiated via a PC using the 600A software package (Aurora Scientific Inc.) and delivered to the motor via a D/A converter. Length and force output signals were digitized via an A/D converter and were displayed and stored on a PC. All force measurements were acquired at 15°C.
The protocol to determine Ca2+-activated force was as follows: force was first measured in a pCa 9.0 solution followed by transfer of the trabecula to a pre-activating solution (reduced Ca2+ buffering) for ∼2 min before transfer into an activating solution containing Ca2+ and measurement of developed force. Ca2+-activated force was then calculated as the difference between resting force measured at pCa 9.0 and the force measured in Ca2+-activating solutions with free Ca2+ concentrations of pCa < 9.0. Final values for Ca2+-activated force were normalized to the width of the preparation by assuming a 2:1 width/height cross-sectional area.
Force–pCa relationships were constructed from force measurements as described above for a series of activations at submaximal [Ca2+] (pCa > 4.5). The order of activating solutions was chosen randomly for each trabecula. After each Ca2+ activation at a submaximal pCa, the trabecula was returned to relaxing solution (pCa 9.0). Maximal force measurements were obtained at saturating Ca2+ (i.e., at pCa 4.5) at the beginning and the end of each series of submaximal force measurements. Typically, the decline in force from the first measurement in pCa 4.5 to the last pCa 4.5 measurement was <10%. Data from experiments in which the total decline was >15% were discarded. This included experiments in which multiple force–pCa relationships were measured in a single trabecula.
Force-pCa data were plotted by expressing submaximal force (P) at each pCa as a fraction of maximal force measured at pCa 4.5 (Po), that is P/Po, where P/Po = Prel. The pCa50 and slope, nH, for each curve were determined by fitting the normalized data to the Hill equation:
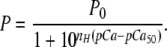 |
Rate of Tension Redevelopment
The rate of force redevelopment (ktr) after a release and restretch maneuver (Brenner and Eisenberg, 1986) was measured during each activation after steady-state force had reached a plateau value in a given pCa solution. Each trabecula was released by 20% of its resting length for a brief (20-ms) duration followed by a rapid underdamped restretch to achieve ∼105% of the original fiber length before returning to the original length. The slack-restretch maneuver is intended to mechanically detach cross-bridges bound to the thin filament while the thin filament is activated in the presence of Ca2+. The subsequent redevelopment of force after the slack and restretch step reflects the rate of cross-bridge transition from weakly bound to strongly bound force-generating states (Brenner and Eisenberg, 1986). The resulting force transient is used to calculate ktr according to the relation:
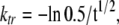 |
where t1/2 is the half-time of force redevelopment and ktr is expressed in reciprocal seconds (s−1) as described previously (Regnier et al., 1998).
Force Measurements after Incubation with Recombinant MyBP-C Proteins
Control force-pCa or ktr-pCa curves were first measured for each trabecula as described above in the absence of added protein. Each trabecula thereby served as its own control. Individual trabeculae were then incubated for 15 min in relaxing solution (pCa 9.0) containing added recombinant protein followed by transfer into a pre-activating solution that also contained added protein. Trabeculae were then transferred to Ca2+-activating solutions that typically lacked added protein to avoid potentially confounding effects of changes in [Ca2+]. After each force measurement in a given Ca2+-activating solution, trabeculae were rapidly returned into relaxing solution with added protein before the next Ca2+ activation. Recombinant proteins were added to the pCa 9.0 and pre-activating solutions as concentrated stock solutions in AB buffer to achieve the desired final concentration. Dilution of pCa 9.0 relaxing solution or pre-activating solutions with proteins in AB was typically ∼5% (vol/vol) but not >10%. Control experiments showed that addition of AB buffer alone (without added proteins) to achieve 10% dilution of pCa 9.0 or pre-activating solutions did not affect steady-state force or ktr measurements (not depicted). The magnitude of force measured upon reaching steady-state in Ca2+-activating solutions with or without added proteins was not different (Fig. 2).
Statistical Analysis
All values are reported as average ± SEM. Data were compared using ANOVA followed by Bonferroni post-hoc comparisons. Significance was considered at P < 0.05.
RESULTS
The MyBP-C Motif Is Necessary But Not Sufficient to Increase Ca2+ Sensitivity of Tension in Permeabilized Trabeculae
We had previously shown that a recombinant protein containing the C1 through C2 domains of cMyBP-C (i.e., C1C2; Fig. 1) increased Ca2+ sensitivity of tension in permeabilized mouse myocytes (Harris et al., 2004). To determine whether the MyBP-C motif was required for the effects of C1C2, we created a mutant C1C2 protein that lacked the motif sequence between the C1 and C2 domains (i.e., C1C2[-m]) and compared its effects to those of C1C2 (replete with motif) on the Ca2+ sensitivity of force in permeabilized rat cardiac trabeculae. As shown in Fig. 3 A, 10 μM C1C2 added to relaxing (pCa 9.0) and pre-activating solutions resulted in significantly greater Ca2+-activated force developed in submaximal pCa solutions, as indicated by the leftward shift of the force–pCa relationship compared with control curves generated before the addition of protein. These data thus agree with previous observations that C1C2 increased Ca2+ sensitivity of tension in permeabilized mouse myocytes (Harris et al., 2004). However, in marked contrast to the effects of C1C2, 20 μM C1C2(-m), which lacked the motif, did not increase Ca2+ sensitivity of tension, suggesting that the motif is required for the effects of C1C2 on force at submaximal pCa concentrations.
Figure 3.

Effects of the MyBP-C motif on Ca2+ sensitivity of tension. (A) Incubation of trabeculae with 10 μM C1C2 (squares; n = 6) resulted in a leftward shift of the tension–pCa relationship relative to control (circles), indicating an increase in Ca2+ sensitivity of tension (ΔpCa50 = 0.30 ± 0.05). 20 μM C1C2(-m) (triangles; n = 3) that lacks the MyBP-C motif was without effect on the tension–pCa relationship. (B) 20 μM C2C4 without the motif (triangles; n = 4) and 20 μM C3mC4 with the motif (squares; n = 3) had no effect on Ca2+ sensitivity of tension.
To determine whether the motif was sufficient to induce the leftward shift of the tension–pCa relationship, we next investigated whether the motif could confer the ability to increase Ca2+ sensitivity to other segments of cMyBP-C. Because single Ig domains may be unstable when expressed alone (Politou et al., 1994a, 1994b), we expressed the motif as a linker between domains C3 and C4. As shown in Fig. 3 B, the three-domain segment C2C4 (inclusive of C3) did not significantly affect force–pCa relationships. However, the addition of the motif between domains C3 and C4 (i.e., C3mC4) also failed to induce a leftward shift of the tension–pCa relationship. These results suggest that Ig domains C2, C3, and C4 do not affect force and that the addition of the motif to these domains is not sufficient to increase Ca2+ sensitivity of tension.
We next investigated whether the Ca2+-sensitizing effects of the motif required the presence of the flanking regions C1 or C2. We therefore generated protein constructs that combined the motif with one or the other of the flanking domains, i.e., C1m or mC2. As shown in Fig. 4 A, the C1m construct was effective at inducing a leftward shift of the force–pCa relationship, whereas mC2 at a comparable concentration failed to induce a leftward shift. Collectively, these results suggest that the motif plus C1, but not the motif plus C2, are necessary for the Ca2+-sensitizing effects of C1C2.
Figure 4.

The combination of C1 and the MyBP-C motif is required to increase Ca2+ sensitivity of tension. (A) 10 μM mC2 (triangles; n = 4) had no effect on tension–pCa relationships relative to control (circles), whereas 5 μM C1m (squares; n = 6) significantly increased Ca2+ sensitivity of tension (ΔpCa50 = 0.12 ± 0.04). (B) 10 μM C0C1 (triangles; n = 3) had no effect on tension–pCa relationships relative to control (circles), whereas 5 μM C0C1m (squares; n = 4) significantly increased Ca2+ sensitivity of tension (ΔpCa50 = 0.27 ± 0.05).
To determine whether C1 without the motif was effective at increasing Ca2+ sensitivity of tension, effects of C0C1 were also assessed. As shown in Fig. 4 B, C0C1 did not significantly affect Ca2+ sensitivity of force. This result is consistent with observations that C1C2(-m), which lacks the motif but includes C1, also did not affect force–pCa relationships (Fig. 3). However, the addition of the motif to C0C1 (i.e., C0C1m) again conferred a marked leftward shift to the force–pCa relationship. These results thus suggest that a combination of both the motif and C1 are minimal requirements for the Ca2+-sensitizing effects of C1C2.
Fig. 5 shows a comparison of the force–pCa relationships for proteins containing the C1 and m-domains. The relative efficacy of the different proteins as assessed by the midpoint shift in force–pCa relations (i.e., ΔpCa50) was C0C2 > C0C1m ≥ C1C2 > C1m. Thus, although C1m was the minimal subunit capable of inducing a leftward shift of the force–pCa relationship, it was not as effective as proteins containing either the C0 or C2 domains in addition to C1m. Summary data for effects of the different proteins are shown in Table I.
Figure 5.

Relative effects of proteins containing C1 and the MyBP-C motif on Ca2+ sensitivity of tension. Tension–pCa relationships were determined after incubation of trabeculae with 5 μM C1m (upward triangles; n = 6), 5 μM C1C2 (diamonds; n = 4), 5 μM C0C1m (squares; n = 4), and 5 μM C0C2 (downward triangles; n = 6). Data for C1m and C0C1m were redrawn from Fig. 4. Controls curves (circles) were obtained in each trabecula before the addition of recombinant protein.
TABLE I.
Summary Data of Effects of Recombinant N-terminal Proteins on Tension–pCa Relationships
| Protein (n) | [ ] (μM) | pCa50 (Control) |
pCa50 (After protein) |
ΔpCa50 | Hill coefficient (nH) (Control) |
Hill coefficient (nH) (After protein) |
|---|---|---|---|---|---|---|
| C0C2 (6) | 5 | 5.32 ± 0.02 | 5.73 ± 0.05a | 0.42 ± 0.04 | 5.43 ± 0.32 | 2.19 ± 0.14a |
| C1C2 (4) | 5 | 5.30 ± 0.02 | 5.54 ± 0.03a | 0.25 ± 0.05 | 6.45 ± 0.86 | 2.65 ± 0.25a |
| C0C1m (4) | 5 | 5.32 ± 0.01 | 5.59 ± 0.05a | 0.27 ± 0.05 | 7.66 ± 0.72 | 2.71 ± 0.34a |
| C1m (6) | 5 | 5.34 ± 0.02 | 5.46 ± 0.04a | 0.12 ± 0.04 | 6.83 ± 0.89 | 3.72 ± 0.34a |
| C1C2 (5) | 10 | 5.29 ± 0.02 | 5.58 ± 0.05a | 0.30 ± 0.05 | 5.02 ± 0.33 | 2.11 ± 0.14a |
| mC2 (4) | 10 | 5.30 ± 0.01 | 5.32 ± 0.03 | 0.02 ± 0.03 | 6.63 ± 0.44 | 5.90 ± 0.67 |
| C0C1 (3) | 10 | 5.34 ± 0.01 | 5.32 ± 0.01 | −0.02 ± 0.01 | 6.61 ± 0.70 | 7.50 ± 0.83 |
| C2C4 (4) | 20 | 5.32 ± 0.01 | 5.32 ± 0.01 | 0.00 ± 0.01 | 6.73 ± 0.21 | 6.15 ± 0.71 |
| C1C2(-m) (3) | 20 | 5.33 ± 0.02 | 5.34 ± 0.01 | 0.01 ± 0.01 | 9.62 ± 2.078 | 8.67 ± 2.04 |
| C3mC4 (3) | 20 | 5.30 ± 0.02 | 5.29 ± 0.02 | −0.01 ± 0.01 | 4.96 ± 0.62 | 5.56 ± 0.42 |
Significance, P < 0.05 versus control.
C1 Plus the MyBP-C Motif Increase Rates of Force Redevelopment (ktr) at Submaximal Ca2+ Activation
To assess whether the motif or C1 affected rates of force activation in addition to their effects on steady-state force–pCa relationships, the rate of force redevelopment after a release and restretch maneuver (i.e., ktr) was measured during each Ca2+ activation. Fig. 6 A shows representative force recordings after a release and restretch protocol for control conditions in the absence of added protein and after incubation of a trabecula with 5 μM C0C2. Incubation with C0C2 significantly accelerated ktr, such that values were near maximal in all pCa solutions (Fig. 6 B). Thus, the Ca2+ dependence of ktr was virtually eliminated by incubation in 5 μM C0C2. Because the rate of tension redevelopment varies as a function of the total level of thin-filament activation (i.e., ktr is increased as a result of both Ca2+ and strongly bound cross-bridges; Gordon et al. [2000]), ktr values were also plotted as a function of the relative force obtained at each pCa. As shown in Fig. 7 A, ktr values measured after incubation with C0C2 were near maximal even at low levels of force. Thus, C0C2 increased ktr to near maximum levels independent of the amount of force generated or, presumably, the number of cycling cross-bridges.
Figure 6.

Effects of C0C2 on rates of tension redevelopment (ktr). (A) Smoothed force traces showing steady-state force measurements after a release and restretch maneuver (ktr) during maximal Ca2+ activation (pCa 4.5) and during submaximal Ca2+ activation (pCa 5.4). Incubation with 5 μM C0C2 accelerated ktr at low Ca2+ (pCa 5.8) to near maximal rates. (B) 5 μM C0C2 accelerated ktr at all levels of submaximal Ca2+ activation.
Figure 7.

Effects of recombinant N-terminal proteins on rates of tension redevelopment (ktr) relative to changes in force. (A) 5 μM C0C2 (triangles; n = 6) increased ktr to near maximal at all levels of submaximal Ca2+ activation. (B) 5 μM C0C1m (triangles; n = 4) and 5 μM C1C2 (squares; n = 4) increased ktr to near maximal at all levels of submaximal Ca2+ activation, whereas 5 μM C1m (diamonds; n = 6) increased ktr to near maximal only at values at or below the pCa50 for force generation.
As shown in Fig. 7 B, C0C1m and C1C2 were also effective at increasing ktr at submaximal Ca2+. C1m appeared to represent the minimal domains required for these effects. However, increases in ktr induced by C1m were evident only for a limited range of pCa and force values below the pCa50 of force generation (Fig. 7 B). Thus, the order of efficacy for protein effects on ktr was similar to that observed for effects on pCa–force relationships, i.e., C0C2 > C0C1m ≥ C1C2 > C1m. Proteins that did not contain the combination of C1 plus the motif did not affect ktr (not depicted).
Effects of C1 and the MyBP-C Motif on Ca2+-independent and Ca2−-activated Force
In all of the preceding experiments, the concentration of recombinant proteins used were chosen such that they did not significantly affect either resting force in the absence of Ca2+ (pCa 9.0) or maximum force in the presence of saturating Ca2+ (pCa 4.5). However, we frequently observed that C0C2 concentrations ≥10 μM increased force measured in pCa 9.0 solutions. Activation of Ca2+-independent force by N-terminal cMyBP-C proteins has been reported and was attributed to a proline-alanine–rich linker sequence between C0 and C1 (Herron et al., 2006). We therefore investigated whether proteins containing this linker and/or the MyBP-C motif increased force in the absence of Ca2+ (pCa 9.0), and whether maximal force in the presence of saturating Ca2+ (pCa 4.5) was affected by the recombinant proteins.
Fig. 8 shows effects of increases in [C0C2] on Ca2+-independent force developed in relaxing (pCa 9.0) solutions, total force measured in pCa 4.5 solutions, and maximum Ca2+-activated force (i.e., active force developed in a pCa 4.5 solution minus force measured at pCa 9.0). The data show a trend for concentrations of C0C2 ≥ 10 μM to increase force independent of Ca2+ with activating effects in pCa 9.0 significant at 20 μM C0C2. Total force (Ca2+ dependent and independent) measured at pCa 4.5 was reduced at 30 μM C0C2, and there was a significant decrease in the Ca2+-activated component of force generated at saturating Ca2+ (pCa 4.5). Similar effects were observed for C1C2, C0C1m, and C1m (Fig. 8). All activating and inhibitory effects were reversible after a 20-min washout of proteins from pCa 9.0 and pre-activating solutions. Table II shows summary data for effects of recombinant proteins on force at pCa 9.0 and at pCa 4.5.
Figure 8.

Effects of C0C2, C1C2, C0C1m, and C1m on total Ca2+-independent and -dependent force. Effects of 10, 20, and 30 μM protein are shown on total force at pCa 4.5 (black bars) and Ca2+-independent force at pCa 9.0 (gray bars). Ca2+-activated force (white bars) is force at pCa 4.5 minus force at pCa 9.0. Washout (Wash) shows force values after a 20-min incubation in relaxing solutions without added proteins.
TABLE II.
Summary Data of Effects of Recombinant N-terminal Proteins on Ca2+-independent and -dependent Force
| Protein (n) | Concentration (μM) |
Total force (pCa 4.5) P/P0 |
Ca2+-independent force (pCa 9.0) P/P0 |
Ca2+-dependent force P/P0 |
|---|---|---|---|---|
| Control (13) | — | 1.0 ± 0.0 | 0.11 ± 0.01 | 0.88 ± 0.01 |
| C0C2 (5) | 30 | 0.80 ± 0.06a | 0.44 ± 0.03a | 0.36 ± 0.06a |
| C1C2 (4) | 30 | 0.60 ± 0.10a | 0.43 ± 0.06a | 0.21 ± 0.12a |
| C0C1m (5) | 30 | 0.79 ± 0.07 | 0.42 ± 0.10a | 0.30 ± 0.12a |
| C1m (3) | 30 | 0.88 ± 0.11 | 0.57 ± 0.29 | 0.31 ± 0.17a |
| mC2 (4) | 30 | 0.95 ± 0.08 | 0.09 ± 0.02 | 0.85 ± 0.06 |
| C0C1 (3) | 30 | 1.00 ± 0.03 | 0.07 ± 0.01 | 0.93 ± 0.04 |
| C1C2(-m) (3) | 30 | 1.07 ± 0.09 | 0.14 ± 0.01 | 0.94 ± 0.08 |
| C3mC4 (3) | 30 | 0.91 ± 0.05 | 0.08 ± 0.02 | 0.83 ± 0.04 |
Significance, P < 0.05 versus control.
DISCUSSION
The purpose of this study was to investigate the functional significance of the unique motif or “m-domain” of MyBP-C in mediating contractile effects induced by the N terminus of MyBP-C. Results show that the motif is necessary but not sufficient by itself to induce the functional effects elicited by the N terminus of MyBP-C in permeabilized trabeculae. Instead, results demonstrate that a combination of the C1 domain and the motif is necessary and suggest that together these domains constitute a functional unit of MyBP-C.
The effects on contraction reported here for the C1 domain and the motif have not been reported previously for the C1 domain or for the C1 and motif domain in combination with one another. However, the results confirm and extend previous observations for contractile effects of C1C2 (which contains C1 and the motif) and localize the Ca2+-sensitizing effects of C1C2 to the C1 and m-domains. This conclusion was evident because constructs that lacked motif, such as C1C2(-m), C0C1, and C2C4, were all without effect on contractile properties at the concentrations tested (Figs. 3 and 4 and Table I). However, the motif by itself was not sufficient to increase Ca2+ sensitivity because neither C3mC4 nor mC2 induced leftward shifts of the tension–pCa relationship. Instead, only proteins that included the combination of the C1 domain and the motif, such as C0C1m, C0C2 (inclusive of C1m), and C1m, were able to increase Ca2+ sensitivity of tension (Table I).
The finding that the combination of C1 and the motif is required to elicit contractile effects suggests that these two domains together constitute a functional subunit of cMyBP-C. Consistent with this idea, recent structural studies of the cMyBP-C N terminus showed that C1 and the motif adopt a stable spatial arrangement with respect to one another, and that the arrangement is maintained in the presence or absence of C0 (Jeffries et al., 2008). Thus, C1 and the motif together may form an extended structural surface that mediates functional effects of the cMyBP-C N terminus. The idea that multiple Ig domains can create an extended interaction surface is further supported by observations that three titin domains (i.e., A168-A170) constitute the binding site for the ubiquitin ligase, MuRF-1 (Mrosek et al., 2007; Muller et al., 2007). Alternatively, the requirement for both the C1 and m-domains could indicate that one domain is required for stabilization of the other. For instance, Ig domains may be unstable when expressed alone, but may be stabilized by additional N-terminal amino acids (Politou et al., 1994a, 1994b). The requirement for C1 may then reflect structural stabilization of the m-domain. In this case, effects may still be specifically related either to C1 or the linker between C1 and the motif (Muller et al., 2007) because C3 was unable to substitute for C1 when the motif was placed between C3 and C4.
Although we did not find that other N-terminal cMyBP-C domains elicited independent effects on force generation, the two domains flanking C1m, C0 and C2, significantly enhanced effects of C1m (Fig. 5). Of these, the cardiac-specific C0 domain was the most effective, and proteins containing the combination of C0 and C1m (e.g., C0C2 and C0C1m) induced the greatest increases in Ca2+ sensitivity of tension. Proteins that contained C0 along with C1m also increased ktr at all submaximal activation levels and virtually eliminated the activation dependence of ktr (Fig. 7). However, the effects of C0 to potentiate effects of C1m reported here contrast with another report where the C0 domain alone was sufficient to increase Ca2+ sensitivity of tension and inhibit maximal Ca2+-activated force (Kulikovskaya et al., 2003). In that study, the effects of C0 correlated with the phosphorylation state of endogenous cMyBP-C, such that effects of C0 were diminished when endogenous cMyBP-C was dephosphorylated. Because it is likely that trabeculae used in the current study were substantially dephosphorylated (McClellan et al., 2001), independent functional effects of C0 may not have been readily apparent.
The C1 and m-domains were also effective at activating force in the absence of Ca2+ (Fig. 8 and Table II). This result is similar to observations made by Herron et al. (2006) who reported that the N terminus of cMyBP-C activated force independent of Ca2+. However, in those experiments the effect was attributed to a linker sequence rich in proline and alanine between the C0 and C1 domains. In contrast, in the current experiments we found that the C0C1 protein that includes the pro-ala sequence was without effect on Ca2+ sensitivity of tension and did not activate tension under relaxing conditions at pCa 9.0, even at concentrations up to 50 μM. Although a precise explanation for the apparent discrepancy is unclear, species-specific differences may be relevant because mouse sequences were used in the current study, whereas human proteins were used in the previous work (Herron et al., 2006). Notably, the proline-alanine–rich region differs significantly (∼52% identity) between mouse and human and so may contribute to the functional differences observed in the two studies. Muscle type differences may also exist because Kunst et al. (Kunst et al., 2000) using human C0C1 in permeabilized skeletal muscle obtained results similar to those reported here and found that 30 μM C0C1 failed to affect Ca2+ sensitivity of tension and did not activate tension at pCa 9.0.
The findings that C1 and the motif increased Ca2+ sensitivity of tension, increased ktr at submaximal [Ca2+], and activated tension in the absence of Ca2+ (pCa 9.0) suggest that together these two domains are potent activators of the thin filament. Although the precise mechanism(s) by which the C1 domain and motif exert these effects is not apparent from these studies, several explanations are possible. The first is that the C1 domain and the motif bind directly to the thin filament and thereby promote its activation. According to this scenario, effects of C1m might be analogous to those of strongly bound cross-bridges. Consistent with this idea, the effects of C1m are similar to those of NEM-S1, a chemical derivative of myosin S1 that lacks enzymatic activity but binds strongly to and activates the thin filament (Nagashima and Asakura, 1982; Swartz and Moss, 1992; Fitzsimons et al., 2001a, 2001b). Notably, in cardiac muscle both C1m and NEM-S1 activate force in the absence of Ca2+ (pCa 9.0), induce a leftward shift in the tension–pCa relationship, and accelerate ktr at submaximal [Ca2+] (Fitzsimons et al., 2001b). Both C1m and NEM-S1 also inhibit maximal Ca2+-activated force at pCa 4.5. Because the latter effect of NEM-S1 is attributed to competitive inhibition of endogenous (cycling) cross-bridges by high concentrations of NEM-S1 (Swartz and Moss, 1992), the similarities may suggest that C1m similarly limits S1 binding. However, although C1C2 has been shown to bind to actin (Razumova et al., 2006), competitive binding of C1m with myosin S1 has not yet been established.
A second possibility is that instead of binding to the thin filament, C1m interacts with the thick filament and promotes strong cross-bridge formation. According to this scenario, C1m might enhance formation of strongly bound cross-bridges and thereby indirectly activate the thin filament. Consistent with this idea, there is evidence that MyBP-C stabilizes a strongly bound post-power stroke state (Palmer et al., 2004). Furthermore, interactions with the thick filament are plausible because C1C2 binds to myosin S2 (Gruen and Gautel, 1999). However, arguing against this scenario, S2 binding was not required for activating effects of C1C2 on actomyosin interactions in an in vitro motility assay because C1C2 affected motility even when S1 catalytic domains (without S2) were used to drive actin filament sliding (Shaffer et al., 2007).
Other possibilities include the idea that the different functional effects of C1m (e.g., to increase Ca2+ sensitivity of tension, increase ktr at submaximal [Ca2+], and to activate tension independent of Ca2+) are mediated through separate, independent interactions with one or both filaments. In addition, the possibility that the observed effects of C1m are due to competition with endogenous cMyBP-C for binding to sites normally occupied by endogenous cMyBP-C cannot be excluded. This possibility seems unlikely because C1C2 also increased Ca2+ sensitivity of tension in myocytes from cMyBP-C knockout mice that lack cMyBP-C (Harris et al., 2004). The latter result suggests that the observed effects are not dependent on the presence of endogenous cMyBP-C and therefore cannot be due to binding competition with endogenous cMyBP-C. Furthermore, because cMyBP-C is present at a limited molar ratio with respect to myosin (Offer et al., 1973; Craig and Offer, 1976), it is likely that even when endogenous cMyBP-C is present (as in the current experiments), a majority of available binding sites are open and accessible for interactions with exogenously added protein.
The idea that exogenously added proteins can bind to all available binding sites and not just to those ordinarily occupied by endogenous cMyBP-C might explain the present observations that added recombinant proteins elicit a significant increase in the Ca2+ sensitivity of tension, whereas knockout of endogenous cMyBP-C had comparatively modest effects on Ca2+ sensitivity of tension (Harris et al., 2002; Stelzer et al., 2006). In contrast, knockout of cMyBP-C accelerated rates of tension redevelopment (ktr) at submaximal Ca2+ (Korte et al., 2003; Stelzer et al., 2006), similar to the effects of adding exogenous proteins described here. The similarity of effects despite opposite perturbations (protein knockout vs. protein addition) suggests that cMyBP-C may affect actomyosin interaction kinetics through multiple, distinct mechanisms. For instance, cMyBP-C has been proposed to tether myosin heads to the thick filament (Calaghan et al., 2000). Release of cross-bridge heads by disrupting the tether, either by knockout of cMyBP-C or phosphorylation of the motif, may thereby increase the spatial extent of myosin heads and hence accelerate interactions with actin (Colson et al., 2008). On the other hand, Nagayama et al. (2007) found evidence for both phosphorylation-dependent and -independent effects of cMyBP-C on cardiac contraction and relaxation and suggested that binding interactions with sarcomeric proteins other than S2 may be important for effects of cMyBP-C. In this regard, we and others have shown that the N terminus of cMyBP-C interacts with actin (Kulikovskaya et al., 2003; Razumova et al., 2006). It will therefore be of interest to determine whether interactions with actin, perhaps via the C1 and m-domains, mediate functional effects of cMyBP-C.
In summary, we have shown that the combination of C1 and the unique m-domain of cMyBP-C function together to increase Ca2+ sensitivity of tension, increase ktr at submaximal [Ca2+], and activate tension in the absence of Ca2+ (pCa 9.0). These results suggest that these two domains together constitute a functional subunit of cMyBP-C that promotes thin filament activation. Future studies will be aimed at identifying the binding partner(s) for C1 and the MyBP-C motif and determining the mechanisms by which they affect actomyosin interactions.
Acknowledgments
These studies were supported by National Institutes of Health (NIH) grant HL080367 to S.P. Harris and NIH grant HL65497 to M. Regnier.
Lawrence G. Palmer served as editor.
K.L. Bezold and S.P. Harris' present address is Dept. of Neurobiology, Physiology, and Behavior, University of California, Davis, Davis, CA 95616.
Abbreviations used in this paper: cMyBP-C, cardiac myosin binding protein C; MyBP-C, myosin binding protein C.
References
- Brenner, B., and E. Eisenberg. 1986. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. USA. 83:3542–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calaghan, S.C., J. Trinick, P.J. Knight, and E. White. 2000. A role for C-protein in the regulation of contraction and intracellular Ca2+ in intact rat ventricular myocytes. J. Physiol. 528:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson, B.A., T. Bekyarova, M.R. Locher, D.P. Fitzsimons, T.C. Irving, and R.L. Moss. 2008. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 103:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, R., and G. Offer. 1976. The localization of C-protein in rabbit skeletal muscle. Proc. R. Soc. Lond. B. Biol. Sci. 192:451–461. [DOI] [PubMed] [Google Scholar]
- Einheber, S., and D.A. Fischman. 1990. Isolation and characterization of a cDNA clone encoding avian skeletal muscle C-protein: an intracellular member of the immunoglobulin superfamily. Proc. Natl. Acad. Sci. USA. 87:2157–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons, D.P., J.R. Patel, K.S. Campbell, and R.L. Moss. 2001. a. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 117:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons, D.P., J.R. Patel, and R.L. Moss. 2001. b. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J. Physiol. 530:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst, D.O., U. Vinkemeier, and K. Weber. 1992. Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J. Cell Sci. 102:769–778. [DOI] [PubMed] [Google Scholar]
- Gautel, M., O. Zuffardi, A. Freiburg, and S. Labeit. 1995. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 14:1952–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A.M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- Gruen, M., and M. Gautel. 1999. Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 286:933–949. [DOI] [PubMed] [Google Scholar]
- Gruen, M., H. Prinz, and M. Gautel. 1999. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 453:254–259. [DOI] [PubMed] [Google Scholar]
- Harris, S.P., C.R. Bartley, T.A. Hacker, K.S. McDonald, P.S. Douglas, M.L. Greaser, P.A. Powers, and R.L. Moss. 2002. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 90:594–601. [DOI] [PubMed] [Google Scholar]
- Harris, S.P., E. Rostkova, M. Gautel, and R.L. Moss. 2004. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ. Res. 95:930–936. [DOI] [PubMed] [Google Scholar]
- Herron, T.J., E. Rostkova, G. Kunst, R. Chaturvedi, M. Gautel, and J.C. Kentish. 2006. Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ. Res. 98:1290–1298. [DOI] [PubMed] [Google Scholar]
- Jeffries, C.M., A.E. Whitten, S.P. Harris, and J. Trewhella. 2008. Small-angle X-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J. Mol. Biol. 377:1186–1199. [DOI] [PubMed] [Google Scholar]
- Korte, F.S., K.S. McDonald, S.P. Harris, and R.L. Moss. 2003. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 93:752–758. [DOI] [PubMed] [Google Scholar]
- Kulikovskaya, I., G. McClellan, J. Flavigny, L. Carrier, and S. Winegrad. 2003. Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 122:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst, G., K.R. Kress, M. Gruen, D. Uttenweiler, M. Gautel, and R.H. Fink. 2000. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 86:51–58. [DOI] [PubMed] [Google Scholar]
- Martyn, D.A., P.B. Chase, J.D. Hannon, L.L. Huntsman, M.J. Kushmerick, and A.M. Gordon. 1994. Unloaded shortening of skinned muscle fibers from rabbit activated with and without Ca2+. Biophys. J. 67:1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan, G., I. Kulikovskaya, and S. Winegrad. 2001. Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys. J. 81:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrosek, M., D. Labeit, S. Witt, H. Heerklotz, E. von Castelmur, S. Labeit, and O. Mayans. 2007. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 21:1383–1392. [DOI] [PubMed] [Google Scholar]
- Muller, S., S. Lange, M. Gautel, and M. Wilmanns. 2007. Rigid conformation of an immunoglobulin domain tandem repeat in the A-band of the elastic muscle protein titin. J. Mol. Biol. 371:469–480. [DOI] [PubMed] [Google Scholar]
- Nagashima, H., and S. Asakura. 1982. Studies on co-operative properties of tropomyosin-actin and tropomyosin-troponin-actin complexes by the use of N-ethylmaleimide-treated and untreated species of myosin subfragment 1. J. Mol. Biol. 155:409–428. [DOI] [PubMed] [Google Scholar]
- Nagayama, T., E. Takimoto, S. Sadayappan, J.O. Mudd, J.G. Seidman, J. Robbins, and D.A. Kass. 2007. Control of in vivo contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 116:2399–2408. [DOI] [PubMed] [Google Scholar]
- Offer, G., C. Moos, and R. Starr. 1973. A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification, and characterization. J. Mol. Biol. 74:653–676. [DOI] [PubMed] [Google Scholar]
- Palmer, B.M., T. Noguchi, Y. Wang, J.R. Heim, N.R. Alpert, P.G. Burgon, C.E. Seidman, J.G. Seidman, D.W. Maughan, and M.M. LeWinter. 2004. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ. Res. 94:1615–1622. [DOI] [PubMed] [Google Scholar]
- Politou, A.S., M. Gautel, C. Joseph, and A. Pastore. 1994. a. Immunoglobulin-type domains of titin are stabilized by amino-terminal extension. FEBS Lett. 352:27–31. [DOI] [PubMed] [Google Scholar]
- Politou, A.S., M. Gautel, M. Pfuhl, S. Labeit, and A. Pastore. 1994. b. Immunoglobulin-type domains of titin: same fold, different stability? Biochemistry. 33:4730–4737. [DOI] [PubMed] [Google Scholar]
- Razumova, M.V., J.F. Shaffer, A.Y. Tu, G.V. Flint, M. Regnier, and S.P. Harris. 2006. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: evidence for long-lived cross-bridges. J. Biol. Chem. 281:35846–35854. [DOI] [PubMed] [Google Scholar]
- Regnier, M., D.M. Lee, and E. Homsher. 1998. ATP analogs and muscle contraction: mechanics and kinetics of nucleoside triphosphate binding and hydrolysis. Biophys. J. 74:3044–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier, M., A.J. Rivera, C.K. Wang, M.A. Bates, P.B. Chase, and A.M. Gordon. 2002. Thin filament near-neighbour regulatory unit interactions affect rabbit skeletal muscle steady-state force-Ca(2+) relations. J. Physiol. 540:485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saber, W., K.J. Begin, D.M. Warshaw, and P. VanBuren. 2008. Cardiac myosin binding protein-C modulates actomyosin binding and kinetics in the in vitro motility assay. J. Mol. Cell. Cardiol. 44:1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer, J.F., M.V. Razumova, A.Y. Tu, M. Regnier, and S.P. Harris. 2007. Myosin S2 is not required for effects of myosin binding protein-C on motility. FEBS Lett. 581:1501–1504. [DOI] [PubMed] [Google Scholar]
- Stelzer, J.E., D.P. Fitzsimons, and R.L. Moss. 2006. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys. J. 90:4119–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, D.R., and R.L. Moss. 1992. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 267:20497–20506. [PubMed] [Google Scholar]