

Abstract
Insulin receptor (IR) and class I major histocompatibility complex molecules associate with one another in cell membranes, but the functional consequences of this association are not defined. We found that IR and human class I molecules (HLA-I) associate in liposome membranes and that the affinity of IR for insulin and its tyrosine kinase activity increase as the HLA:IR ratio increases over the range 1:1 to 20:1. The same relationship between HLA:IR and IR function was found in a series of B-LCL cell lines. The association of HLA-I and IR depends upon the presence of free HLA heavy chains. All of the effects noted were reduced or abrogated if liposomes or cells were incubated with excess HLA-I light chain, β2-microglobulin. Increasing HLA:IR also enhanced phosphorylation of insulin receptor substrate-1 and the activation of phosphoinositide 3-kinase. HLA-I molecules themselves were phosphorylated on tyrosine and associated with phosphoinositide 3-kinase when B-LCL were stimulated with insulin.
INTRODUCTION
The manifold effects of insulin on cell physiology are mediated by a specific insulin receptor (IR). Insulin binding triggers a conformational change in IR, stimulating its tyrosine kinase activity and leading to its autophosphorylation (Rosen, 1987; White and Kahn, 1994). The insulin signal is propagated downstream of the IR kinase by the binding and tyrosine phosphorylation of docking proteins that connect IR to signaling pathways by mediating the binding of intracellular signaling proteins (Backer et al., 1992; Kuhne et al., 1993; Myers et al., 1994).
IR has been shown to associate with other proteins in the plane of the plasma membrane. Some of these associations, i.e., with PC-1, a transmembrane glycoprotein (Maddux et al., 1995) and with β adrenergic receptors (Karoor, et al., 1995; Baltensperger, et al., 1996), clearly have functional consequences. The first affects the kinase activity of IR, and the second affects the function of β adrenergic receptors. In contrast, no functional effects have been clearly shown for another lateral association of IR, i.e., that with the major histocompatibility complex (MHC) I molecules, mouse H2 and human HLA. Although the association of IR with these molecules has been demonstrated using biophysical and biochemical techniques (Fehlmann et al., 1985a; Phillips et al., 1986; Edidin and Reiland, 1990; Liegler et al., 1991), there is little evidence that this association has functional consequences for either IR or MHC I molecules. At best, some studies correlate MHC phenotype with insulin binding and signaling activity (Lafuse and Edidin, 1980; Kittur et al., 1987; Edidin, 1988), but others do not (Verland et al., 1989; Liegler et al., 1991). Thus the consequences, if any, of the association between IR and MHC I molecules remain unclear.
We have used a two-part strategy to detect the formation of molecular complexes between IR and human MHC I molecules, HLA, and to measure the functional consequences of this association for insulin-mediated signaling and the state of MHC phosphorylation. First, we studied the molecular proximity and biochemical activity of the purified IR and HLA proteins reconstituted together in artificial lipid bilayers, liposomes. This system addressed the specificity of the association between IR and HLA-I and its effect on the early events in insulin signaling, insulin binding, and receptor autophosphorylation. Second, we applied the conclusions drawn from experiments in liposomes to the association and function of IR and HLA in cells of the human B-LCL 721 and in the HLA loss mutants derived from it (Kavathas et al., 1980). We used these cells earlier to correlate HLA phenotype and IR affinity for insulin (Kittur et al., 1987). Here, we show that the effect of HLA-I on the affinity and the kinase activity of IR correlates with and depends upon the ratio of HLA:IR in both liposomes and cells. Increasing this ratio increased not only IR affinity for insulin, but also for a given concentration of insulin, receptor autophosphorylation, phosphorylation of HLA-I, phosphorylation of insulin receptor substrate-1 (IRS-1), and the activation of phosphoinositide 3-kinase (PI-3 kinase). Studies of the kinetics of phosphorylation in liposomes and of IR phosphopeptides suggest that HLA brings multiple IRs in proximity to one another, enhancing autophosphorylation without exposing new sites of phosphorylation on the IR. Increased phosphorylation of HLA and other substrates appears, therefore, to be a consequence of increased receptor activation. The physical and functional association of HLA and IR appeared to depend upon the presence of β2-microglobulin (β2m)-free HLA heavy chains because incubating either liposomes or cells with excess β2m abolished the effects associated with high HLA:IR ratio.
MATERIALS AND METHODS
Unless otherwise noted all reagents were were from Sigma (St. Louis, MO). 125I-labeled insulin (specific activity, 2200 mCi/mmol), γ- 32P-labeled ATP, and 32P-labeled orthophosphoric acid (H332PO4) were from Dupont (Wilmington, DE). Octyl-β-d glucopyranoside was from Calbiochem (La Jolla, CA). Fluorescein isothiocyanate (FITC) and sulforhodamine sulfonyl chloride (Texas red, TxR) were from Molecular Probes (Junction City, OR).
Antibodies
Hybridomas BB7.2, anti-HLA-A2, and aIR-1, anti-human IR were obtained from ATCC (Rockville, MD). Monoclonal antibodies (mAbs) GS0C142.1 and aB8 (anti-HLA-A1 and anti-HLA-B8) were the gifts of Dr. Paul Gladstone (Bristol-Myers Squibb Pharmaceutical Research Institute, Seattle, WA). mAb 4D12 (anti-HLA-B5) was the gift of Prof. Barton Haynes (Duke University, Durham, NC) and mAb Ke2 (against a monomorphic epitope of HLA heavy chain [Schreiber et al., 1984]) was the gift of Prof. Roger Kennett (University of Pennsylvania, Philadelphia, PA). mAbs were purified from spent culture medium, using a Protein-A Sepharose column (Ey et al., 1978).
Rabbit polyclonal anti-IR β-chain antibody LC 711 was from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phosphoserine monoclonal antibody was from Sigma. Mouse anti-phosphotyrosine mAb 4G10 and ab3 IR antibody were from Oncogene Sciences (Seattle, WA). Rabbit polyclonal antibody against the p85 a-subunit of P I-3 kinase and anti-IRS-1 mAb were from Transduction Laboratories (Lexington, KY). C4 anti-actin mAb was the gift of Dr. Trina Schroer (Department of Biology, Johns Hopkins University).
Proteins were conjugated to CNBr-activated Sepharose beads to a concentration of 2–3 mg of protein per gram of dry gel using our laboratory protocol (Chakrabarti et al., 1992).
Purification of IR
A Chinese hamster ovary (CHO) cell line transfected with human insulin proreceptor gene coupled to a gene for methotrexate resistance (Yoshimasa et al., 1990) was the gift of Dr. D. F. Steiner (University of Chicago, Chicago, IL). Cells were grown in Eagles’ minimum essential medium (without deoxynucleosides) + 10% fetal bovine serum supplemented with G418 (400 μg/ml) and methotrexate (50 nM). Under these conditions cells expressed >107 IR molecules per cell. Cells were lysed by homogenizing at 4°C in 1% Triton-X-100 in 50 mM HEPES, 0.15 M NaCl, pH 7.8, 1 mM in phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml aprotinin. The lysate was then centrifuged at 15,000 × g for 30 min, and the supernatant was incubated with wheat germ agglutinin (WGA)-Sepharose beads for 3 h. The beads were transferred to a column and washed 2× with 50 mM HEPES, 0.5 M NaCl, 0.5% Octyl-β-d-glucopyranoside, pH 7.8, 1 mM PMSF, 2 μg/ml aprotinin, and the adsorbed glycoproteins were eluted with steps of 0.1 M, 0.2 M, 0.3 M N-acetyl glucosamine in the above buffer. The peak fractions eluted with 0.3 M N-acetyl glucosamine were pooled, their proteins content measured using Bradford or BCA assays (Bio-Rad, Richmond, CA), and their purity assessed by SDS-PAGE. The identity of the band at 94 kDa was confirmed by probing as Western blot with anti-IR β-chain antibody. A typical preparation yielded 150–200 μg of IR.
Purification of HLA-A2
The MHC class I molecule HLA-A2 was purified from a Triton X-100 extract of JY cells as described elsewhere (Parham, 1983; Chakrabarti et al., 1993.). After SDS-PAGE, a single major band was observed at 45 kDa; the identity of the protein was confirmed by Western blotting with HC-10 mAb against denatured HLA I heavy chain (Stam et al., 1986, 1990)
Labeling of Purified Proteins
Affinity-purified HLA-A2 was labeled with fluorescein, Fl, or sulforhodamine (TxR), using our laboratory protocol (Chakrabarti et al., 1993).The labeled proteins were purified by affinity chromatography on a BB 7.1 Sepharose column. SDS-PAGE of fluorescently labeled HLA-A2 showed one major band at 45 kDa and a smear of weak fluorescence at lower molecular masses. IR was fluorescein-labeled after preincubating IR with excess insulin. The IR was then labeled with FITC for 2–3 h in 0.1 M HEPES, 0.1 M NaCl, pH 7.8. The labeled receptor was repurified from WGA-Sepharose affinity column after gel filtration on a Sephadex G-50 column. Glycophorin was labeled with FITC or TxR in 0.1 M sodium borate buffer, pH 9.0, at 4°C. Free dye was separated from the protein on a Sephadex-G-25 column.
The fluorescein:protein M ratios of tagged HLA-A2, glycophorin, and IR were 1.2, 2.1, and 1.4. TxR:protein ratios of tagged HLA-A2, glycophorin, and IR were 1.5, 1.4, and 2.1. The fluorescein concentration was estimated from absorbance at 495 nm (e = 63,000 M−1 cm−1) and TxR concentration from absorbance at 594 nm (e = 85,000 cm−1).
Liposomes
Our preparation of liposomes for flow cytometric energy transfer experiments has been described in detail (Chakrabarti et al., 1992). Briefly, proteoliposomes of 0.2–0.4 μm diameter formed when a mixture of lipid, dimyristoylphosphatidylcholine, and protein in octylglucoside was either diluted 10-fold in buffer or was dialyzed against buffer. More than 80% of native or fluorescently labeled HLA-A2 were present in the correct transmembrane orientation as estimated from their lateral diffusion and from the binding of Fl-Fab (Chakrabarti et al., 1992). Seventy percent of the functionally active IR was incorporated into liposomes as estimated by comparing the specific binding of 125I-insulin to IR in the liposome pellet with binding to the supernatant remaining after the liposomes were formed. Liposomes made with either lipid alone or with HLA-A2 but no IR bound <6% of the input insulin.
Flow Cytometric Energy Transfer
Fluorescence resonance energy transfer (FRET) between pairs of proteins labeled with fluorescein and TxR was detected following our published methods for FRET in liposomes (Chakrabarti et al., 1992). FRET was measured in terms of the quenching of donor fluorescence at 525 nm.
Insulin Binding to Liposomes
Proteoliposomes containing IR alone, IR + HLA, or IR + glycophorin were incubated with 125I-insulin (1 × 10−11 M) and unlabeled insulin (10 pM to 0.5 μM) at 37°C for 2 h in 0.2 ml of buffer, 50 mM HEPES, 0.15 M NaCl, 0.1% BSA, pH 7.9, and protease inhibitors. Bound insulin was separated from free insulin by adding 1 ml of prechilled ethanol followed by centrifugation in a Eppendorf centrifuge, model 5415C, for 5 min (Frandsen and Bacchus, 1987). The pellet was washed four times with 1.5 ml of prechilled ethanol and 125I was measured in a Beckman (Fullerton, CA) Biogamma counter. Duplicate counts were within 3% of one another. The data from different batches of the liposome preparations were pooled for each combination of reconstituted proteins. The dissociation constant (KD) for insulin binding and the number of IR in each liposome preparation were calculated using the curve-fitting program LIGAND (Munson and Rodbard, 1980).
IR Autophosphorylation in Liposomes
For autophosphorylation experiments, a constant amount of affinity-purified IR was incorporated into liposomes either alone or together with HLA-A2 or with glycophorin. γ-32P-ATP (15 mCi) was included in the incorporation buffer, 50 mM HEPES, 0.5% Octyl-β-d-glucoside, 0.15 M NaCl, 20 mM MgCl2, 20 mM MnCl2, pH 7.8, 2 mM + PMSF, and aprotinin. After the liposomes were formed, they were incubated with excess unlabeled ATP. The amount of γ-32P-ATP entrapped in the liposomes was 60–65% of the input radioactivity up to an HLA:IR ratio of 20:1. Liposomes with higher HLA:IR ratios were leaky, resulting in poor encapsulation of γ-32P-ATP. Autophosphorylation of IR was stimulated by adding insulin to a suspension of ATP-containing proteoliposomes in a volume of 0.2 ml and incubating the mixture at 37°C for 60 min. The reaction was terminated by the addition of 600 μl of a chilled (4°C) buffer containing 50 mM HEPES, 150 mM NaCl, 40 mM EDTA, 30 mM NaF + protease inhibitors followed by addition of Triton X-100 to the mixture to a final concentration of 0.2%. Phosphorylated receptors were isolated by incubating the liposome lysate mixed with WGA-Sepharose beads at 4°C for 2–3 h. After incubation, the beads were centrifuged and washed three times with Buffer A. Then they were boiled in 2× Laemmli buffer containing 0.3 M N-acetylglucosamine before electrophoresis in 7.5% SDS-PAGE gel. The gels were then dried and placed over x-ray film for 24–48 h at −80°C. Densities of the bands in the developed film were quantitated on a Molecular Dynamics (Sunnyvale, CA) PhosphoImager.
Kinetics of Autophosphorylation
Liposomes reconstituted with purified IR and HLA:IR (10:1) were prepared as described above. The autophosphorylation reaction was initiated by the addition of 1 nM insulin at 37°C. Aliquots of the reaction mixture were taken at intervals between 0 and 180 min and quenched by the addition of SDS-PAGE Laemmli buffer followed by electrophoresis. The extent of autophosphorylation was determined by the quantitation of a 94-kDa band corresponding to the β chain of IR in the autoradiograms.
The kinetic data were analyzed using the model developed by Kohanski (1993a) for the insulin-stimulated autophosphorylation of IR. According to this model autophosphorylation of IR can be described as a sum of two exponential decays:
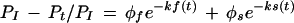 |
Where PI = phosphorylation of IR at infinite time and Pt = phosphorylation of IR at time t, φf and φs are the fractions of the autophosphorylation due to fast and slow phases, and ks and kf are the rate constants for the two phases.The initial rates of phosphorylation are plotted by assuming that the slow phase of reaction is negligible, φs = 0. Then log[PI − Pt/PI]= φf − kf(t).
Phosphopeptide Analysis
Phosphopeptides were prepared from 32P-labeled β chain isolated on SDS-PAGE gels, following a published protocol (Kohanski, 1993b). Gel regions corresponding to labeled β chain (94-kDa fragment) of IR from samples of HLA-A2/IR and IR alone liposomes were located by autoradiography of wet unfixed gel immediately after electrophoresis. These regions were excised from the gel, suspended in a 12 ml of water in a conical tube, and rocked for 15 min. This washing was repeated twice followed by the transfer of the gel fragment in a 1.5-ml Eppendorf tube. Gel was crushed with a wooden applicator stick, and 0.5 ml of 50 mM ammonium carbonate buffer was added. EndoLysC (1 mg) was added in a small volume, and the tubes were rocked at room temperature. After 14 h, 1 mg of proteinase K was added, and the digestion was continued for an additional 6 h. At the end of the digestion, the digest was frozen at −80°C. The frozen sample was dried under vacuum, reconstituted with distilled water, and redried. This cycle was repeated until no residue was seen after drying. Finally the sample was dissolved in 100 ml of water and frozen. The phosphopeptides were resolved by reverse phase HPLC on a 2×150 mm Hypersil-ODS column (3-μm beads, 120 Å pores) following the protocol described by Kohanski (1993b). Radioactivity in each fraction was detected as Cerenkov radiation, counting for 10 min per fraction.
Substrate Phosphorylation
We used poly(Glu,Tyr) (ratio of Glu:Tyr = 4:1) as an external substrate for IR kinase in the presence and absence of HLA-A2 in the lipid bilayer. Poly(Glu,Tyr) was added at a final concentration of 1 mg/ml during the reconstitution of IR and HLA:IR liposomes for phosphorylation. After stopping the reaction as described above, extracts were run on a 13% SDS-PAGE and visualized by autoradiography. For quantitative estimates, the entire lane from below the IR β-chain band to the 19-kDa marker was cut out and counted as described by Hansen et al. (1989).
B-LCL Cells
The B-lymphoblast cell line LCL-721 and HLA variant cell lines derived from it (Kavathas et al., 1980; for references to each cell line used here see Kittur et al., 1987; Reiland, 1990) were the generous gift of Dr. R. Demars (Department of Genetics, University of Wisconsin, Madison). Table 2 lists the HLA phenotypes of the LCL used in our experiments. Cells of all LCL-721 cell lines were grown in RPMI 1640 medium containing 15% heat-inactivated fetal calf serum (Intergen, Phoenix, AZ). Cell line 961 was grown in the presence of HAT, 20 mg/ml neomycin, and 1% antibiotic-antimycotic solution (10 μg streptomycin, 0.5 μg fungizone/ml; GIBCO, Grand Island NY).
Table 2.
HLA phenotypes and HLA/IR of B-LCL used in our studies
| Cell Line | HLA Phenotype | HLA/IRa |
|---|---|---|
| 721.1 | A2 B5 C | 1.5 |
| 721.45.1 | A2 B5 C | 3.3 |
| 961 | -B5- | 5 |
| 721.53 | A2-C | 16 |
| 721.13 | A1 B8 C | 23 |
HLA:IR was estimated from insulin binding to IR and Fl-β2m binding to HLA (Kittur et al., 1987; Hochman et al., 1991).
Insulin-Stimulated Phosphorylation of [32P] Phosphate-loaded Cells
Two 10-cm plates, each containing∼ 3 × 106 cells were serum-starved for 2 h; then the cells were washed twice with phosphate-free medium and incubated for 1 h at 37°C in the same medium containing 2 mCi of γ-32P-orthophosphate. Insulin was then added to one of the plates to a concentration of 1 nM, and both plates were incubated for 5 min at 37°C. After incubation at 37°C, cells in each plate (stimulated and control) were washed with ice-cold PBS and lysed in 1 ml of 50 mM HEPES, 0.15 M NaCl, 2 mM PMSF, aprotinin (2 μg/ml), leupeptin (3 μg/ml), 0.1 M NaF, 30 mM sodium pyrophosphate, 2 mM EDTA, 1 mM sodium orthovanandate, 1% Nonidet P-40, at pH 7.8 (RIPA buffer or lysis buffer) (Levy-Toledano et al., 1994; Hotamisligil et al., 1996) for 30 min at 4°C. The solution was centrifuged at 15,000 × g for 30 min. An aliquot of the supernatant was incubated with protein A-Sepharose for 1 h. Proteins of interest were immunoprecipitated from the cleared supernatant by incubating it at 4°C with specific antibody and precipitating the antigen–antibody complexes with protein A-Sepharose beads. The beads were washed five times in lysis buffer, resuspended in 20 μl of Laemmli buffer boiled for 5 min, and stored at −80°C.
Western Blotting of Immunoprecipitated Proteins
Immunoprecipitated proteins were resolved by SDS-PAGE and transferred by electroblotting to nitrocellulose paper. To reduce nonspecific binding of probe antibodies, the nitrocellulose papers were incubated for 2 h in PBS containing 5% BSA and 0.2% Tween-20. The papers were then incubated for 120 min with antibodies to the proteins of interest. The nitrocellulose papers were then washed six times with PBS containing 0.2% Tween-20 and incubated with peroxidase- or alkaline phosphatase-conjugated goat anti-mouse or goat anti-rabbit antibody. The bound anti-Ig was visualized by a chemiluminescence reaction using an ECL detection kit (Amersham Corp. Chicago, IL).
RESULTS
Association of HLA and IR in Liposomes
To establish the molecular proximity of IR and HLA-A2, we prepared small liposomes containing both Fl-IR and TxR-HLA-A2 and measured the fluorescence resonance energy transfer between these molecules in terms of donor fluorescence quenching (Table 1). In the presence of TxR-HLA-A2, fluorescence of Fl-IR was quenched 24%. In contrast, Fl-IR fluorescence was quenched <10% (our lower limit of reliability for detecting FRET) when TxR-glycophorin was included in the liposomes instead of TxR-HLA.
Table 1.
Quenching of fluorescent donor-labeled insulin receptor, HLA-A2 or glycophorin by fluorescent acceptor-labeled proteins
| Donor | Acceptor | % Quenching |
|---|---|---|
| Fl-IR | TxR-HLA-A2 | 24 ± 4a |
| Fl-IR + β2m | TxR-HLA-A2 | 12 ± 3a |
| Fl-IR | TxR-glycophorin | 9 ± 2b |
| Fl-HLA-A2 | TxR-HLA-A2 | 26 ± 1 |
| Fl-HLA-A2 + unlabeled IR | TxR-HLA-A2 | 23 ± 3c |
HLA:IR, 10:1.
Glycophorin:IR, 10:1.
HLA:IR, 10:1.
HLA-I molecules self-associate when incorporated into liposomes (Chakrabarti et al., 1992). We confirmed this point, detecting 26% quenching of Fl-HLA-A2 in the presence of TxR-HLA-A2 under the conditions employed in the energy transfer experiments. Incorporation of unlabeled IR together with the labeled HLA molecules (HLA:IR 10:1) did not affect the quenching of FL-HLA (23%). This suggests that IR, at the surface concentrations used here, does not significantly disrupt HLA/HLA associations. The self-association of HLA-I molecules depends upon the presence of nondenatured, but β2m-free HLA heavy chains. Incubation of HLA-containing liposomes or cells treated with excess human β2m disrupts clusters of HLA-I molecules (Chakrabarti et al., 1993; Matko et al., 1993). We found that excess human β2m also disrupted the association of IR and HLA. Addition of β2m reduced energy transfer, measured as the quenching of Fl-IR fluorescence, in Fl-IR/TxR-HLA-A2 liposomes to about 12%.
Insulin Binding to Liposomes
The association of HLA and IR increases the affinity of IR for insulin. IR reconstituted into liposomes bound insulin at a single high-affinity site with a KD of 1.3 ± 0.2 nM (mean of five independent fitting experiments ± SD). In the presence of HLA-A2 per IR, affinity of insulin for IR increased about 10-fold to 0.1 ± 0.05 nM, but the number of binding sites was 1/5 to 1/10 that of liposomes containing IR alone (three experiments), even though the amount of IR incoporated was the same whether or not HLA was present. Thus IR appears to aggregate in the presence of HLA. Binding was specific for IR. Liposomes made with either lipid alone or with HLA-A2, but no IR, bound <6% of the input insulin. Glycophorin also did not affect IR affinity for insulin; KD for insulin in glycophorin/IR liposomes was 1.4 nM.
When HLA:IR (10:1) liposomes were incubated with excess (6–10 μM) β2m before the addition of insulin, their KD for insulin was 1.8 ± 0.4 nM, close to that of liposomes containing only IR but incubated with β2m (KD, 1.6 ± 0.4 nM). Thus excess human β2m affects the functional association as well as the physical association of IR and HLA.
Insulin-stimulated Autophosphorylation of IR in Liposomes
Insulin binding activates IR kinase, which leads to the central event in the insulin-signaling cascade, autophosphorylation of the IR β-chain. We investigated the effect of HLA on IR phosphorylation using liposomes that had entrapped γ-32P ATP. The autophosphorylation of IR was enhanced in the presence of HLA-A2 (Figure 1A,C). At a constant HLA:IR ratio, the degree of IR phosphorylation depended on insulin concentration (Figure 1B). At a constant insulin concentration, IR autophosphorylation increased as HLA:IR increased over a range of 1:1 to 20:1 (Figure 1C). Phosphorylation of an exogenous substrate, poly(Glu,Tyr), was also increased, about twofold in HLA:IR 10:1 liposomes compared with that in IR-only liposomes, over insulin concentrations ranging from 10−10 to 10−7 M. The incorporation of another protein, glycophorin, with IR did not enhance IR autophosphorylation.
Figure 1.

Insulin-stimulated autophosphorylation of the β subunit of IR, reconstituted together with HLA-A2 into liposomes containing γ-32P ATP. (A) Effect of increasing HLA:IR ratio on autophosphorylation stimulated by 10−9 M insulin. (B) Insulin concentration and autophosphorylation of IR in HLA:IR 10:1 liposomes. (C) Effects of HLA/IR on the insulin (10−9 M)-stimulated autophosphorylation of IR. IR/Gly, glycophorin/IR 10:1. (D) Effect of excess β2m on the autophosphorylation of IR in IR/HLA-A2-reconstituted liposomes. Liposomes were preincubated with 6 μM β2m for 2 h at 37°C followed by treatment with 10−9 M insulin at 37°C. Control samples were taken through the same steps but without β2m.
Incubation of liposomes with β2m reduced the insulin-dependent autophosphorylation of IR, although not to the level measured in the absence of HLA (Figure 1, C and D). Incubation with β2m did not change the autophosphorylation level of IR in the absence of HLA (Figure 1D).
The presence of HLA molecules increased the initial rate of IR autophosphorylation as well as its maximum. Figure 2 compares the kinetics of IR phosphorylation in 10:1 HLA:IR liposomes with that in liposomes containing IR alone. It can be seen that the phosphorylation rate is increased about threefold in the presence of HLA. The same residues of IR β-chain were phosphorylated in the presence of 10:1 HLA:IR as in the absence of HLA. An HPLC peptide map of 32P-phosphorylated β-chain detected the same peaks of phosphorylation in molecules stimulated in the presence or absence of HLA (10:1 HLA:IR). The peaks corresponded to bis- and mono-phosphorylated peptides of the carboxy-terminal domain, tris-, bis-, and monophosphorylated peptides of the activation loop, and one peptide of the juxtamembrane domain (Kohanski, 1993b). One phosphopeptide whose source was not identified was also detected in each sample.
Figure 2.

Effect of HLA-A2 on the initial rate of IR autophosphorylation in liposomes. Rates are plotted as log[PI- Pt/PI] versus time, where PI is level of maximal phosphorylation and Pt is level of phosphorylation at time t. 32P-ATP-containing liposomes were made with or without 10:1 HLA-A2/IR. Phosphorylation was stimulated by adding insulin to a final concentration of 1 nm. ○, IR alone; ▪, HLA-A2/IR 10:1. Solid and dashed lines are best fits to the data. For IR alone y = −0.01 to 0.029x; for HLA/IR y = −0.11 to 0.075x. Correlation coefficients are 0.99 for IR alone and 0.98 for HLA/IR.
HLA:IR and IR Function in B-LCL Cells
IR responses to insulin in HLA:IR liposomes served as a guide for studying HLA and IR function in B-lymphoblasts bearing different amounts of IR and different amounts of HLA-I allomorphs. The phenotypes of the B-LCL cells used in this study are listed in Table 2. Earlier we measured the affinity of IR on these cells (Kittur et al., 1987) and concluded that the measured KD depended upon the HLA phenotype of a particular cell line; however, later work (Reiland and Edidin, 1993) showed that all four HLA-A and HLA-B allomorphs present on these cells could be coprecipitated with IR, requiring a complicated model for allomorph-specific affects on IR function.
In addition to differing in HLA phenotype, the B-LCL cell lines also differ in HLA:IR ratio when HLA is measured in terms of β2m binding. For example, cell lines LCL-721.1 and LCL- 721.45.1 express the same HLA phenotype, A2/B5/(C), but there is almost a fourfold difference in their HLA:IR ratio (Table 2). Given the results in HLA:IR liposomes, we replotted our data on insulin binding to B-LCL cells as a function of HLA:IR. It can be seen from Figure 3 that affinity of IR on these cells, expressed as KA, increases∼ 10-fold over a large range of HLA:IR, from 1:1 to >200:1. This trend suggests that in cells as in liposomes the HLA:IR ratio rather than HLA phenotype influences IR affinity and IR function as well.
Figure 3.

The affinity of B-LCL cell lines for insulin as a function of HLA/IR. Values for KA and number of IR per cell are taken from Kittur et al. (1987), Reiland (1990), and Reiland and Edidin (1993). Number of HLA per cell, measured in terms of binding of Fl-β2m are taken from Hochman et al. (1991) and Reiland (1990).
Autophosphorylation of IR and the Effect of β2m on IR Phosphorylation in B-LCL Cells
We examined insulin-stimulated autophosphorylation of IR in B-lymphoblast LCL cells 721.1, 721.45.1, 961, 721.13, and 721.53. Brief exposure to 10 nM insulin stimulated the autophosphorylation of the 96-kDa band of the β-chain in all of these cells (Figure 4A). However, the extent of the insulin-stimulated autophosphorylation (the increase over unstimulated controls) was different in each cell line. It did not correlate with HLA phenotype (compare the result for 721.1 and 721.45.1) but did correlate with HLA:IR. The differences in insulin-stimulated phosphorylation were not due solely to differences in affinity of IR for insulin. Over three decades of insulin concentration, insulin-stimulated phosphorylation of IR was∼ twofold higher in 961 cells (HLA:IR 5:1) than in 721.1 cells (HLA:IR 1.5:1) (Figure 4B). If the enhanced phosphorylation was due only to HLA effects on IR affinity, we would expect the same level of phosphorylation would be reached at 10−8 M insulin.
Figure 4.

(A) Insulin-stimulated autophosphorylation of the β subunit of IR in B-LCL cells. Cells were metabolically labeled with H332PO4 followed by treatment with 10−9 M insulin at 37°C as described in MATERIALS AND METHODS, IR was immunoprecipitated from cell extracts by anti-IR b chain antibody. Each value is the average of three experiments. (B) Autophosphorylation as a function of insulin concentration in 721.1 (HLA:IR 1.5:1) and 961 (HLA:IR 5:1) B-LCL cells. (C) β 2m and autophosphorylation of IR in the B-LCL line 721.13 (HLA/IR 15/1). Cells were preincubated with 6 μm β2m overnight followed by treatment with 10−9 M insulin.
Liposomes (HLA:IR) incubated with exogenous β2m bind and respond to insulin as if they contain only IR. This same effect of β2m on IR function was also seen in 721.13 cells (HLA:IR 15:1), incubated overnight with human β2m (6 μM). In these cells, insulin-stimulated IR phosphorylation was lower than that in cells incubated in control medium (Figure 4B).
Phosphorylation of HLA in B-LCL Cells
Our experiments with liposomes showed that the association of IR and HLA-I results in the insulin-stimulated phosphorylation of HLA. We examined this in B-LCL by using anti-phosphoserine, and anti-phosphotyrosine antibodies to probe Western blots of HLA immunoprecipitated from insulin-stimulated cells. There was little change in the serine phosphorylation of HLA after insulin stimulation (Figure 5A). In contrast, tyrosine phosphorylation of HLA greatly increased (Figure 5B). Probing the blots with antibody to HLA heavy chain (HC-10) and with anti-actin showed that the phosphorylated species was HLA and not contaminating actin (data not shown). HC-10 (Figure 5C) blots also showed that only a portion of HLA immunoprecipitated was phosphorylated. The tyrosine phosphorylation was also confirmed by phosphoaminoacid analysis (data not shown).
Figure 5.

Insulin-stimulated phosphorylation of HLA-A2 in 721.1 and 721.45.1 cells. Cells were serum starved and stimulated with 10−9 M insulin for 5 min at 37°C. Cells were lysed and HLA-I molecules immunoprecipitated with KE-2 antibody. The immunoprecipitates were analyzed by electrophoresis on 12% SDS-PAGE followed by Western blotting as described in MATERIALS AND METHODS. Immunoblots were probed with antiphosphoserine antibody (A), anti-phosphotyrosine antibody (B), or HC-10 antibody (C).
After establishing the site of phosphorylation of HLA, we next determined whether all the allomorphs of HLA were phosphorylated after insulin stimulation. When three different HLA-I molecules, HLA-A2 and HLA-B5 from LCL 721.45.1 and HLA-B8 from LCL 727.13 cells, were separately immunoprecipitated after insulin stimulation of the cells, all three were found to be labeled on tyrosine (Figure 6).
Figure 6.

Insulin-stimulated phosphorylation of three different allomorphs of HLA-I. Cells of lines 721.1 and 721.53 were serum starved and stimulated with 10−9 M insulin for 5 min at 37°C. (a) Cells were lysed and HLA-I molecules immunoprecipitated with mAb specific for individual allomorphs. Lysate from 721–1 cells was divided into two equal parts after protein estimation. One portion of the lysate was treated with BB7.2 mAb (anti-HLA-A2) and the other with 4D12 mAb (anti-HLA-B5). Lysate from the 721–53 cell was incubated with aB8 mAb (anti-HLA-B8). The immunoprecipitate was analyzed by electrophoresis on 12% SDS-PAGE followed by Western blotting as described in MATERIALS AND METHODS. Immunoblots were probed anti-phosphotyrosine antibody. (b) Same blot reprobed with HC-10 antibody.
Downstream Signaling by IRS-1 and PI-3 Kinase in B-LCL Cells
In most cell types insulin action leads to the tyrosine phosphorylation of a cytoplasmic protein with an apparent molecular mass between 160 and 185 kDa, IRS-1. Once phosphorylated, this protein serves as a docking protein, binding multiple SH2 domain-containing proteins (Sun et al., 1991; Myers et al., 1994). An anti-IRS-1 mAb detected a 165-kDa protein in immunoblots of the tyrosine-phosphorylated proteins immunoprecipitated from insulin-stimulated LCL 721.1 and 721.45 cells Figure 7a). As was the case for HLA, the phosphorylation of IRS-1 was higher in LCL 721.45.1 than in LCL 721.1 cells, correlating with the HLA:IR ratios of these two cell lines. IRS-1 was not associated with HLA; no HLA heavy chain was detected when immunoblots of immunoprecipitated IRS-1 were probed with mAb against free HLA heavy chain (data not shown).
Figure 7.

IRS-1 and PI-3 kinase in insulin- stimulated B-LCL cells. (a and b) Cells of lines 721.1 and 721.45.1 were stimulated with insulin and lysed as described in the legend of Figure 6. Phosphoproteins were immunoprecipitated from the lysate with anti-phosphotyrosine mAb or anti-IRS-1 mAb, resolved by SDS-PAGE, and analyzed in Western blots. (a) Blots of antiphosphotyrosine immunoprecipitates were probed with anti-IRS-1 mAb. (b) Blots of antiIRS-1 immunoprecipitates were probed with antibody against the p85 subunit of PI-3 kinase. (c) Cells were treated as described for panels a and b except that HLA-I molecules were precipitated from the lysate by KE-2 mAb. Blots were probed with antibody against the p85 subunit of PI-3 kinase.
Phosphorylation of IRS-1 promotes its association with several proteins, among them PI 3-kinase, a heterodimer consisting of 85-kDa regulatory and 110- kDa catalytic subunits (Carpenter et al., 1990). To determine whether IRS-1 associates with PI-3 kinase during the insulin stimulation, immunoblots of IRS-1 immunoprecipitated from control and insulin-stimulated LCL 721.1 and LCL 721.45.1 cells were probed with antibodies against the p85 subunit. PI-3 kinase was associated with IRS-1 in insulin-stimulated cells (Figure 7b). Surprisingly, PI-3 kinase was also coprecipitated with HLA molecules from these cells (Figure 7c). The intensity of the p85 band from LCL 721.45.1 was higher than from LCL 721.1. Thus it appears that extent of the association of PI-3-kinase with cell HLA correlates with HLA:IR. This is not due to the nonspecific binding of the PI-3 kinase to the precipitating antibody. Treatment of the cell lysate with an irrelevant antibody (against caveolin, a protein is not present in lymphocytes) did not precipitate PI-3 kinase either in the absence or presence of insulin (data not shown).
DISCUSSION
In this paper we show for the first time that the association of HLA-I molecules with IR has biological consequences. The receptor’s affinity for insulin and its tyrosine kinase activity are increased when HLA-I molecules are present and the increase depends upon HLA:IR. We also show that HLA-I molecules themselves are involved in the insulin-signaling cascade. They are phosphorylated on tyrosine after insulin binds IR and as a consequence bind at least one of the downstream molecules of the signaling cascade, PI-3 kinase.
We detected significant fluorescence resonance energy transfer between Fl-IR and TxR-HLA molecules in liposomes, showing that the molecules can directly associate with one another in the plane of the membranes. To the extent that no significant FRET was detected between Fl-IR and TxR-glycophorin, the association between IR and HLA-A2 is specific. This evidence for association of HLA and IR in membranes is consistent with our observation that the lateral diffusion of HLA molecules in liposomes is significantly reduced in the presence of IR (our unpublished results) and with our earlier finding that antibody-induced aggregation of IR in cell membranes reduced the lateral diffusion of HLA, and aggregation of HLA reduced the lateral diffusion of IR (Edidin and Reiland, 1990).
HLA-A2/IR association enhances receptor function, beginning with affinity for insulin. At the resolution of our curve-fitting program LIGAND (Munson and Rodbard, 1980), it appears that at HLA:IR 10:1 all IR are converted from KD ∼ 1 nM to KD ∼0.1 nM. The insulin-stimulated autophosphorylation of IR was also enhanced by association with HLA in liposome membranes. This enhancement does not appear to be due solely to the increased affinity of IR in the presence of HLA, since IR phosphorylation in high HLA:IR cells was greater than that in low HLA:IR cells even at very high concentrations of insulin.
Addition of HLA I molecules to IR liposomes increased the initial rate of phosphorylation of the IR β-chain, as well as the total amount of phosphorylation. Phosphopeptide maps of IR stimulated in the presence or absence of HLA were identical. This, together with the kinetic data, suggests that the autophosphorylation rate of IR aggregated in the presence of HLA is higher than the rate if they are dispersed in the membrane. This model is also consistent with an increase in apparent affinity of IR and reduction in number of binding sites in the presence of HLA. The apparent binding of insulin to clustered receptors should be higher than the binding to single receptors because clustering enhances the probability of a ligand rebinding to receptor. There is no evidence that sites of phosphorylation of β-chain are different in the presence or absence of HLA. All of the other consequences of HLA/IR association, phosphorylation of exogenous substrates in both liposomes and cells, are consistent with an initial enhancement of IR autophosphorylation due to clustering.
The model suggested by the results in liposomes proved to apply to B-LCL cells. The B-LCL lines used in this study bind insulin with different affinities (Kittur et al., 1987), and we had earlier ascribed the differences in affinity to allele-specific associations of HLA-I molecules with IR. This model was not supported by chemical cross-linking experiments in which all four HLA-A and HLA-B molecules of the B-LCL 721 lines were precipitated with IR (Reiland and Edidin, 1993). However, when we revisited our old data, in the light of the results on HLA:IR in liposomes, the cells’ affinity for insulin correlated well with HLA:IR. It appears that this ratio, and not the particular allomorphs of HLA-I present, determines IR affinity and subsequent signaling in B-lymphoblasts.
In B-lymphoblasts the extent of phosphorylation of IR itself, phosphorylation of IRS-1, the binding of PI-3 kinase, and the phosphorylation of HLA-I were all increased with increasing HLA:IR. Our finding that cell HLA-I is phosphorylated on a tyrosine residue that is conserved in the cytoplasmic domains of HLA-A and HLA-B (but not HLA-C) was prefigured by experiments in vitro (Pober et al., 1978; Braydon et al., 1983; Guild and Strominger, 1984; Keith and Said, 1994). Later work showed that both human (HLA) and mouse (H-2) MHC-I molecules are phosphorylated on tyrosine when cells are stimulated by phorbol esters (Feurstein et al., 1985; Peyron and Fehlmann, 1988); some reports suggested that tyrosine phosphorylation of mouse H-2 molecules is stimulated by insulin (Fehlmann et al., 1985b; Peyron and Fehlmann, 1988; Burke et al., 1989).
β2m-free HLA heavy chains are required for the lateral aggregation of HLA molecules and excess β2m disperses these aggregates (Matko et al., 1994; Chakrabarti et al., 1992). We found the same requirements and an effect of β2m on the physical as well as functional association of HLA and IR. In cells, affinity for insulin and insulin-stimulated phosphorylation correlated with HLA:IR when HLA was measured in terms of β2m binding to free HLA heavy chains. FRET studies indicate the HLA/IR association is disrupted by β2m. Incubation of HLA/IR liposomes with excess β2m reduced the affinity of IR to insulin to that of IR in the absence of HLA. Incubation of cells or liposomes with excess β2m reduced insulin-stimulated autophosphorylation of IR to levels found in membranes with low or no HLA. These incubations rescue a subset of free HLA-I heavy chains that have not denatured (Matko et al., 1994). The role of these chains in association of HLA/IR remains to be defined. It is possible that dissociation of β2m unmasks a site on the HLA heavy chain that can bind to IR; another possibility is that the site is created by conformational changes in the HLA heavy chain consequent to the loss of β2m. Mutants, particularly in the α3 domain of the HLA heavy chain, may help to locate the IR-binding regions in the chain.
The involvement of MHC I molecules with the IR-signaling cascade expands our view of MHC I molecules as signal transducers. Cross-linking of MHC I molecules triggers early (rises in free Ca++, tyrosine phosphorylation) and late (cytokine production, increased receptor display, and cell proliferation) responses by T-cells (Tscherning and Claësson, 1994; Bregenholt et al., 1996). The effects of cross-linking may require one or more signaling cascades that involve phosphorylation on the conserved cytoplasmic tyrosine of HLA. This is strongly suggested by our finding that HLA-I molecules are not only phosphorylated on tyrosine after insulin stimulation but are also associated with PI-3 kinase, a dual specificity lipid and serine kinase (Carpenter et al., 1990).
Activation of PI-3-kinase is believed to be initiated by association of its regulatory subunit with phosphotyrosine in activated receptors or in accessory proteins such as IRS-1 (Levy-Toledano et al., 1994; Songyang et al., 1995). Binding of PI-3 kinase to tyrosine phosphoproteins involves an SH2 domain of the enzyme and the motif YXXM on the phosphoprotein (Piccione et al., 1993; Nolte, et al., 1996). HLA molecules lack this SH2-binding domain, consistent with our failure to coprecipitate IRS-1 and HLA. Hence, binding of PI-3 kinase to HLA-I molecules may involve some intermediary proteins that themselves contain SH2 domains. The intermediary proteins could be parts of other signaling pathways. There is increasing evidence for cross-talk between such pathways (cf., Profrock and Schulz, 1991; Daub et al., 1996).
HLA molecules are associated with tyrosine kinase receptors other than IR, for example, epidermal growth factor-receptor (Schreiber et al., 1984). Epidermal growth factor receptors dimerize upon ligand binding, and this stimulates their kinase activity (Heldin, 1995). In contrast, IR appears to undergo an intramolecular dimerization between its constituent monomers upon ligand binding (White and Kahn, 1994). However, cross-linkers, polycations, lectins, or antibodies activate IR kinase and may do so by aggregating multiple IR molecules (O’Brien et al., 1987; Li et al., 1992, and references therein). Some experiments with mutant receptors also suggest that intermolecular dimerization can play a role in insulin activation of IR (Mynarcik and Whittaker, 1995). If MHC-I molecules enhance such dimerization, this could explain their effects on IR function. Indeed, we note that, in liposomes and cells with high HLA:IR, there is a increase in the level of IR kinase in the absence of insulin (for examples see Figures 1, 2, and 5). We also note that association of HLA with IR and other signal-transducing receptors could explain why HLA-I molecules lacking cytoplasmic domains can still transmit signals after cross-linking (Gur et al., 1990).
HLA/IR associate in membranes of lymphocytes, adipocytes, and hepatoma cells (Due et al., 1986; Samson et al., 1986; Cousin et al., 1987; Olsson et al., 1994; Shibata et al., 1995). Our studies have characterized the association and its consequences in B-lymphoblasts, cells that respond to physiological concentrations of insulin (Helderman, 1983; Snow, 1985; Valentine et al., 1993). The importance of HLA/IR associations in other IR-positive cells is not established. However, we note that early work on mouse liver plasma membranes correlated modest differences in affinity for insulin and responses to insulin with MHC haplotype and with the amount of MHC per unit membrane (Lafuse 1978; Lafuse and Edidin, 1980). Hepatocytes bear relatively high numbers of IR but lower levels of MHC-I molecules than lymphocytes (Klein, 1986). Hence, HLA/IR associations may be somewhat less important for IR function in hepatocytes than in lymphocytes. However, we expect that HLA has a significant effect on IR function in adipocytes (Hohlfeld and Engel 1994) because it appears that at least some mouse adipocytes (from fat pads) express very high levels of MHC-I molecules (Edidin, unpublished observations). On the other hand, HLA/IR associations are irrelevant to IR function in mature muscle myotubes because these cells lack MHC-I (Hohfeld and Engel, 1994).
We expect that some of the physiological questions about the importance of HLA/IR can be addressed in mice genetically engineered to lack β2m (Koller and Smithes, 1989; Koller et al., 1990; Zijlstra et al., 1989, 1990) or IR. Although plasma insulin was not significantly higher in diabetes-susceptible NOD-B2 mnull than in normoglycemic mice expressing class I molecules (Serreze et al., 1994), we predict that tissue-specific responses of β2m-knockout mice to insulin will prove different from those of their normal counterparts. In turn, the consequences of IR for HLA function may become evident from the study of cellular immune responses of IR null mice (Accili, et al., 1996).
ACKNOWLEDGMENTS
We thank Drs. Martha Zuniga, M. Daniel Lane, and Stephen Desiderio for critical comments. We thank Dr. R.A. Kohanski for advising us on phosphopeptide analysis of IR and for allowing us to use the HPLC column in his laboratory. This work was supported by National Institutes of Health grant R37-AI14584 to Michael Edidin.
REFERENCES
- Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- Backer JM, Myers MG, Jr, Shoelson SE, Chin DJ, Sun XJ, Mirapeix M, Hu P, Margolis B, Skolinik EY, Schlessinger J, White MF. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11:3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltensperger K, Karoor V, Paul H, Ruoho A, Czech M, Malbon CC. The beta-adrenergic receptor is a substrate for insulin receptor tyrosine kinase. J Biol Chem. 1996;271:1061–1064. doi: 10.1074/jbc.271.2.1061. [DOI] [PubMed] [Google Scholar]
- Braydon CG, Raymond LE, Strominger JL. HLA-A2 and HLA-B7 antigens are phosphorylated in vitro by Rous sarcoma virus kinase (pp60v-src) at a tyrosine residue encoded in a highly conserved exon of the intracellular domain. Proc Natl Acad Sci USA. 1983;80:2894–2898. doi: 10.1073/pnas.80.10.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregenholt S, Röpke M, Skov S, Claësson. MH. Ligation of MHC class I molecules on peripheral blood T lymphocytes induces new phenotypes and functions. J Immunol. 1996;157:993–999. [PubMed] [Google Scholar]
- Burke T, Pollock K, Cushley W, Charles SW. Phosphorylation of Class I but not Class II MHC molecules by membrane-localized protein kinase C. Mol Immunol. 1989;26:1095–1104. doi: 10.1016/0161-5890(89)90053-9. [DOI] [PubMed] [Google Scholar]
- Carpenter CL, Duckworth BC, Auger KR, Cohen P, Bischaffhausen BS, Cantley LC. Purification and characterization of Phosphoinositide 3-kinase from rat liver. J Biol Chem. 1990;265:19704–19711. [PubMed] [Google Scholar]
- Chakrabarti A, Makto J, Rahman NA, Barisas BG, Edidin M. Self-association of Class I major histocompatibility complex molecules on liposomes and cell surface membrane. Biochemistry. 1992;31:7182–7189. doi: 10.1021/bi00146a022. [DOI] [PubMed] [Google Scholar]
- Cousin J-L, Samson M, Pilch PF, Fehlman M. Internalization of insulin receptor and HLA antigens in human hepatoma cells. Biochem J. 1987;242:403–410. doi: 10.1042/bj2420403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Ullrich A, Wallasch C, Weiss FU. Role of transactivation of the EGF receptor in signaling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Due C, Simonsen M, Olsson L. The major histocompatibility complex class I heavy chain as a structural subunit of the human cell membrane insulin receptor: implication for the range of biological functions of histocompatibility antigens. Proc Natl Acad Sci USA. 1986;83:6007–6011. doi: 10.1073/pnas.83.16.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M. MHC antigens and membrane receptor complexes. Immunol Today. 1988;9:218–219. doi: 10.1016/0167-5699(88)91218-2. [DOI] [PubMed] [Google Scholar]
- Edidin M, Reiland. J. Dynamic measurements of the associations between class I MHC antigens and insulin receptors. Mol Immunol. 1990;27:1313–1317. doi: 10.1016/0161-5890(90)90036-y. [DOI] [PubMed] [Google Scholar]
- Ey PL, Prowse SJ, Jenkin CR. Isolation of pure IgG1, IgG2a and IgG2b immunoglobulins from mouse serum using protein A-Sepharose. Immunochemistry. 1978;15:429–436. doi: 10.1016/0161-5890(78)90070-6. [DOI] [PubMed] [Google Scholar]
- Fehlmann M, Chvatchko Y, Brandenburg D, van Obberghen E, Brossette N. The subunit structure of the insulin receptor and molecular interactions with the major histocompatibility complex antigens. Biochimie. 1985a;67:1155–1159. doi: 10.1016/s0300-9084(85)80114-0. [DOI] [PubMed] [Google Scholar]
- Fehlmann M, Peyron J-F, Samson MA, van Obberghen E, Brandenburg D, Brossette N. Molecular association between major histocompatibility complex class I antigens and insulin receptor in mouse liver membranes. Proc Natl Acd Sci USA. 1985b;82:8634–8637. doi: 10.1073/pnas.82.24.8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein N, Monos DS, Cooper HL. Phorbol ester effect in platelets: lymphocytes and leukemic cells (HL-60) is associated with enhanced phosphorylation of class I HLA antigens. Biochem Biophys Res Commun. 1985;126:206–213. doi: 10.1016/0006-291x(85)90592-3. [DOI] [PubMed] [Google Scholar]
- Frandsen EK, Bacchus RA. New, simple insulin-receptor assay with universal application to solubilized insulin receptors and receptors in broken and intact cells. Diabetes. 1987;36:335–340. doi: 10.2337/diab.36.3.335. [DOI] [PubMed] [Google Scholar]
- Guild BC, Strominger JL. Human and murine class I MHC antigens share conserved serine 335, the site of HLA phosphorylation in vivo. J Biol Chem. 1984;259:9235–9240. [PubMed] [Google Scholar]
- Gur H, El-Zaatari F, Geppert TD, Wacholtz MC, Taurog JD, Lipsky PE. Analysis of T-cell signaling by class I MHC molecules: the cytoplasmic domain is not required for signal transduction. J Exp Med. 1990;172:1267–1270. doi: 10.1084/jem.172.4.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T, Stagested J, Pedersen L, Roth RA, Goldstein A, Olsson L. Inhibition of insulin receptor phosphorylation by peptide derived from major histocompatibility complex class I antigen. Proc Natl Acad Sci USA. 1989;86:3123–3126. doi: 10.1073/pnas.86.9.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helderman JH. T cell cooperation for the genesis of B-cell insulin receptors. J Immunol. 1983;131:644–652. [PubMed] [Google Scholar]
- Heldin C-H. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Hochman JH, Jiang H, Matyus L, Edidin M, Pernis B. Specific association of fluorescent β2m with cell surfaces: the affinity of different H-2 and HLA antigens for β2m. J Immunol. 1991;146:1862–1867. [Google Scholar]
- Hohfeld R, Engel AG. The immunobiology of muscle. Immunol Today. 1994;15:269–274. doi: 10.1016/0167-5699(94)90006-X. [DOI] [PubMed] [Google Scholar]
- Hotamisligil SG, Peraldi P, Budvari A, Ellis R, White MF, Spigelman BM. IRS-1 mediated inhibition of insulin receptor tyrosine kinase activity in TNF-a-and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Karoor V, Baltensperger K, Paul H, Czech M, Malbon CC. Phosphorylation of tyrosyl residues 350/354 of the β-adrenergic receptor is obligatory for counter regulatory effects of insulin. J Biol Chem. 1995;270:25305–253. doi: 10.1074/jbc.270.43.25305. [DOI] [PubMed] [Google Scholar]
- Kavathas P, Bach FH, Demars R. Gamma ray induced loss of expression of HLA and glyoxalase. Proc Natl Acad Sci USA. 1980;77:4251–4255. doi: 10.1073/pnas.77.7.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith MS, Said AG. CD31 (PECAM-1), Cdw32(FcgRII), and anti-HLA class I monoclonal antibodies recognize phosphotyrosine containing proteins on the surface of human neutrophils. J Immunol. 1994;152:5902–5911. [PubMed] [Google Scholar]
- Kittur D, Shimzu Y, Demars R, Edidin M. Insulin binding to human B lymphoblast is a function of HLA haplotype. Proc Natl Acad Sci USA. 1987;84:1351–1355. doi: 10.1073/pnas.84.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J. Natural History of the Major Histocompatibility Complex. New York: John Wiley & Sons; 1986. pp. 152–174. [Google Scholar]
- Kohanski RA. Insulin receptor autophosphorylation I Autophosphorylation kinetics of the native receptor and its cytoplasmic kinase domain. Biochemistry. 1993a;32:5766–5772. doi: 10.1021/bi00073a007. [DOI] [PubMed] [Google Scholar]
- Kohanski RA. Insulin receptor autophosphorylation II. Determination of autophosphorylation sites by chemical sequence analysis and identification of juxtamembrane domain sites. Biochemistry. 1993b;32:5773–5780. doi: 10.1021/bi00073a008. [DOI] [PubMed] [Google Scholar]
- Koller BH, Marrack P, Kappler JW, Smithes O. Normal development of mice deficient in β2m, MHC class I proteins and CD8+ T-Cells. Science. 1990;248:1227–1230. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- Koller BH, Smithes O. Inactivating the β2-microglobulin locus in mouse embryonic stem cells by homologous recombination. Proc Natl Acad Sci USA. 1989;86:8932–8935. doi: 10.1073/pnas.86.22.8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne MR, Pawson T, Lienhard GE, Feng GS. The insulin receptor substrate-1 associates with the SH2-containing phosphotyrosine phosphatase Syp. J Biol Chem. 1993;268:11479–11481. [PubMed] [Google Scholar]
- Lafuse W. The role of histocompatibility-2 locus in modifying cyclic-AMP. Ph.D. Thesis. Baltimore, MD: Johns Hopkins University; 1978. [Google Scholar]
- Lafuse W, Edidin M. Influence of the mouse major histocompatibility complex H-2, on liver adenylate cyclase activity and on glucogen binding to liver cell membranes. Biochemistry. 1980;19:49–54. doi: 10.1021/bi00542a008. [DOI] [PubMed] [Google Scholar]
- Levy-Toledano R, Taouis M, Blaettler DH, Gorden P, Taylor SI. Insulin-induced activation of Phosphatidyl inositol 3-Kinase. J Biol Chem. 1994;269:31178–31182. [PubMed] [Google Scholar]
- Li S-l, Yan P-f, Paz B, Fujita-Yamaguchi Y. Human insulin receptor β-subunit transmembrane/cytoplasmic domain expressed in a baculovirus expression system: purification, characterization, and polylysine effects on the protein tyrosine kinase activity. Biochemistry. 1992;31:12455–12462. doi: 10.1021/bi00164a023. [DOI] [PubMed] [Google Scholar]
- Liegler T, Szollosi J, Hyun W, Goodenow RS. Proximity measurements between H-2 antigens and the insulin receptor by fluorescence energy transfer: evidence that a close association does not influence insulin binding. Proc Natl Acad Sci USA. 1991;88:6755–6759. doi: 10.1073/pnas.88.15.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddux BA, Sbraccia P, Kumakura S, Sasson S, Youngren J, Fisher A, Spener S, Grupe A, Henzel W, Stewart TA, Raven GM, Goldfine ID. Membrane glycoprotein PC-1 and insulin resistance in non-insulin dependent diabetes mellitus. Nature. 1995;373:448–451. doi: 10.1038/373448a0. [DOI] [PubMed] [Google Scholar]
- Makto J, Bushkin Y, Wei T, Edidin M. Clustering of Class I HLA molecules on the surface of activated and transformed human cells. J Immunol. 1994;52:3353–3360. [PubMed] [Google Scholar]
- Makto J, Ohki K, Edidin M. Luminescence quenching by nitroxide spin labels in aqueous solution: studies on the mechanism of quenching. Biochemistry. 1992;31:703–711. doi: 10.1021/bi00118a010. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. A versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–231. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Myers MG, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci. 1994;94:289–293. doi: 10.1016/0968-0004(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Mynarcik DC, Whittaker J. Insulin receptor transmembrane signaling: Evidence for an intramolecular oligomerization mechanism of activation. J Recept Signal Transduction Res. 1995;15:887–904. doi: 10.3109/10799899509049863. [DOI] [PubMed] [Google Scholar]
- Nolte R, Eck M, Schlessinger J, Shoelson SE, Harrison S. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phoshopeptide complexes. Nat Struct Biol. 1996;3:364–37. doi: 10.1038/nsb0496-364. [DOI] [PubMed] [Google Scholar]
- O’Brien RM, Soos MA, Siddle M. Monoclonal antibodies to the insulin receptor stimulate the intrinsic tyrosine kinase activity by cross-linking receptor molecules. EMBO J. 1987;6:4003–4010. doi: 10.1002/j.1460-2075.1987.tb02743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson L, Goldstein A, Stagsted J. Regulation of receptor internalization by the major histocompatibility complex class I molecule. Proc Natl Acad Sci USA. 1994;91:9086–9090. doi: 10.1073/pnas.91.19.9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P. Monoclonal antibodies against HLA products and their use in immunoaffinity purification. Methods Enzymol. 1983;92:110–146. doi: 10.1016/0076-6879(83)92012-8. [DOI] [PubMed] [Google Scholar]
- Peyron J-F, Fehlmann M. Phosphorylation of class I histocompatibility antigens in human B lymphocytes: regulation by phorbol esters and insulin. Biochem J. 1988;256:763–768. doi: 10.1042/bj2560763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Moule ML, Delovitch TL, Yip CC. Class I histocompatibility antigen and insulin receptors: evidence for interactions. Proc Natl Acad Sci USA. 1986;83:3474–3478. doi: 10.1073/pnas.83.10.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccione E, Case RD, Domchek SM, Hu P, Chaudhuri M, Backer JM, Schlessinger J, Shoelson SE. PI 3-kinase p85 SH2 domains specificity defined by direct phosphopeptide/SH2 domain binding. Biochemistry. 1993;32:3197–3202. doi: 10.1021/bi00064a001. [DOI] [PubMed] [Google Scholar]
- Pober JS, Guild BC, Strominger JL. Phosphorylation in vivo and in vitro of human major histocompatibility antigens (HLA-A & HLA-B) in the carboxyl-terminal intracellular region. Proc Natl Acad Sci USA. 1978;75:6002–6010. doi: 10.1073/pnas.75.12.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profrock A, Schulz I. Receptor for insulin interact with Gi-proteins and for epidermal growth factor with Gi- and Gs proteins in rat pancreatic Acinar cells. Biochem Biophys Res Commun. 1991;175:380–386. doi: 10.1016/0006-291x(91)91575-w. [DOI] [PubMed] [Google Scholar]
- Reiland J. Studies of the physical association of the Class I MHC antigens with insulin receptors on B-Lymphoblast HLA mutants. Ph.D. Thesis. Baltimore, MD.: The Johns Hopkins University; 1990. [Google Scholar]
- Reiland J, Edidin M. Chemical cross-linking detects association of insulin receptor with four different Class I leukocyte antigen molecules on the cell surface. Diabetes. 1993;42:619–625. doi: 10.2337/diab.42.4.619. [DOI] [PubMed] [Google Scholar]
- Rosen OM. After insulin binds. Science. 1987;23:1457–1465. doi: 10.1126/science.2442814. [DOI] [PubMed] [Google Scholar]
- Samson M, Cousin J-L, Fehlmann M. Crosslinking of insulin receptor to MHC antigens in human B-lymphocytes: evidence for selective molecular interactions. J Immunol. 1986;137:2293–2298. [PubMed] [Google Scholar]
- Schreiber AB, Schlessinger J, Edidin M. Interaction between major histocompatibility complex and epidermal growth factor receptors on human cells. J Cell Biol. 1984;98:725–730. doi: 10.1083/jcb.98.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serreze DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major histocompatibility complex class I-deficient NOD-β2m null mice are diabetes and insulitis resistant. Diabetes. 1994;43:505–509. doi: 10.2337/diab.43.3.505. [DOI] [PubMed] [Google Scholar]
- Shibata H, Suzuki Y, Omata E, Tanaka S, Kojima I. Dissection of GLUT4 recycling pathway into exocytosis and endocytosis in rat adipocytes. Evidence that GTP-binding proteins are involved in the processes. J Biol Chem. 1995;270:11489–11495. doi: 10.1074/jbc.270.19.11489. [DOI] [PubMed] [Google Scholar]
- Snow EC. Insulin and growth hormone function as minor growth factors that potentiate lymphocyte activation. J Immunol. 1985;135:776S. [PubMed] [Google Scholar]
- Songyang Z, Margolis B, Chaudhuri M, Shoelson SE, Cantley LC. The phosphotyrosine interaction domain of Shc recognizes the tyrosine-phosphorylated NPXY motif. J Biol Chem. 1995;270:14863–14866. doi: 10.1074/jbc.270.25.14863. [DOI] [PubMed] [Google Scholar]
- Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chain permit biochemical characterization of certain HLA-C products. J Immunol. 1986;137:2299–2307. [PubMed] [Google Scholar]
- Stam NJ, Vroom TM, Peters PJ, Pastoors EB, Ploegh HL. HLA-A- and HLA-B- specific monoclonal antibodies reactive with free heavy chains in western blots, in formalin-fixed paraffin embedded tissue sections and in cryo-immunoelectron microscopy. Int Immunol. 1990;2:113–120. doi: 10.1093/intimm/2.2.113. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF. The structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- Tscherning T, Claësson MH. Signal transduction via MHC class-I molecules in T cells. Scand J Immunol. 1994;39:117–121. doi: 10.1111/j.1365-3083.1994.tb03349.x. [DOI] [PubMed] [Google Scholar]
- Valentine MA, Licciardi KA, Clark EA, Krebs EG, Meier KE. Insulin regulates serine/threonine phosphorylation in activated human B-lymphocytes. J Immunol. 1993;150:96–105. [PubMed] [Google Scholar]
- Verland S, Simonsen M, Gammeltoft, Allen S, H, Flavell RA, Olsson L. Specific molecular interaction between the insulin receptor and a D product of MHC class I. J Immunol. 1989;143:945–951. [PubMed] [Google Scholar]
- White MF, Kahn CR. Insulin signaling. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- Yoshimasa Y, Paul JL, Whittaker J, Steiner DF. Effects of amino acid replacement within the tetrabasic cleavage site on processing of human insulin receptor precursor expressed in chinese hamster ovary cells. J Biol Chem. 1990;265:17230–17237. [PubMed] [Google Scholar]
- Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin-deficient mice lack CD4−8+ cytolytic T-cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- Zijlstra M, Li E, Sajjadi F, Subramani S, Jaenisch R. Germ-line transmission of a disrupted β2m gene produced by homologous recombination in embryonic stem cells. Nature. 1989;342:435–438. doi: 10.1038/342435a0. [DOI] [PubMed] [Google Scholar]