

Abstract
For decades, the binding of prostaglandin H2 (PGH2) to multiple target proteins of unrelated protein structures which mediate diverse biological functions has remained a real mystery in the field of eicosanoid biology. Here, we report that the structure of a PGH2 mimic, U46619, bound to the purified human TP, was determined and compared with that of its conformation bound to the COX-downstream synthases, prostacyclin synthase (PGIS) and thromboxane A2 synthase (TXAS). Active human TP protein, glycosylated and in full length, was expressed in Sf-9 cells using a baculovirus (BV) expression system and then purified to near homogeneity. The binding of U46619 to the purified receptor in a nonionic detergent-mimicked lipid environment was characterized by high-resolution NMR spectroscopy. The conformational change of U46619, upon binding to the active TP, was evidenced by the significant perturbation of the chemical shifts of its protons at H3 and H4 in a concentration-dependent manner. The detailed conformational changes and 3D structure of U46619 from the free form to the TP-bound form were further solved by 2D 1H NMR experiments using a transferred NOE (trNOE) technique. The distances between the protons of H11 and H18, H11 and H19, H15 and H18, and H15 and H19 in U46619 were shorter following their binding to the TP in solution – down to within 5 Å, which were different than that of the U46619 bound to PGIS and U44069 (another PGH2 mimic) bound to TXAS. These shorter distances led to further separation of the U46619 α and ω chains, forming a unique “rectangular” shape. This enabled the molecule to fit into the ligand-binding site pocket of a TP model, in which homology modeling was used for the transmembrane (TM) domain, and NMR structures were used for the extramembrane loops. The proton perturbations and 3D conformations in the TP-bound U46619 were different with that of the PGH2 mimics bound to PGIS and TXAS. The studies indicated that PGH2 can adopt multiple conformations in solution to satisfy the specific and unique shapes to fit the different binding pockets in the TP receptor and COX-downstream enzymes. The results also provided sufficient information for speculating the molecular basis of how PGH2 binds to multiple target proteins even though unrelated in their protein sequences.
INTRODUCTION
Prostaglandin H2 (PGH2)1 is an endogenous lipid molecule produced by cyclooxygenase from arachidonic acid (AA). The unstable PGH2 is a potent ligand that activates the thromboxane A2 (TXA2) receptor (TP) and therefore mediates the receptor signaling that causes thrombosis and vasoconstriction, which can lead to strokes and heart attacks [1-2]. On the other hand, PGH2 also serves as a common substrate and can be isomerized to the biologically active prostaglandins (PG) E2 (PGE2), PGD2, PGF2, PGI2, and thromboxane A2 (TXA2) by their corresponding COX-downstream synthases, PGE2 synthase (PGES), PGD2 synthase (PGDS), PGF2 synthase (PGFS), PGI2 synthase (PGIS) and TXA2 synthase (TXAS) (Figure 1, [1-10]). The synthesized prostanoids mediate diverse and opposite pathophysiological processes within the vascular [1-2, 11-14], nervous [15], and reproductive [16-18] systems as well as cancers [19-20]. The primary structures of the TP receptor and the COX-downstream synthases are unrelated. The TP is a G protein-coupling receptor located on the cell membrane with seven transmembrane domains, while the COX-downstream synthases belong to P450 and glutathione-related proteins and other enzymes [15].
Figure 1.

Panel I. The multiple target proteins of PGH2 synthesized from arachidonic acid (AA) through the COX pathway. The COX-1 and COX-2 enzymes convert AA to PGH2 (A), which serves as a common substrate for COX-downstream synthases including PGDS, PGES, PGFS, PGIS and TXAS, and also serves as an agonist for the TP. The chemical structures of the PGH2 mimics, U46619 (A), and U44069 (B) are also shown. Panel II. The specific ligand binding activity of the purified TP from Sf9 cells. The purified TP protein (■) in PBS was incubated with increasing amounts (0 to 60 nM) of the ligand, [3H]U46619. Unlabeled U46619 (1μM) was added to the Sf9 cells as a negative control (●) before the radio-labeled ligand was added. The results are representative data from three assays (n=3) and are shown as means ± the standard error.
The molecular mechanisms of the PGH2 and its multiple protein-binding properties have been addressed for decades. However, very little information had been made available until the very recent successful purification of a large amount of recombinant TXAS [21] and PGIS [22]. Characterization of the PGH2 mimics’ interaction with the purified PGIS and TXAS in solution, using high-resolution NMR spectroscopic techniques, has revealed that the PGH2 mimics’ (U44069 and U46619) structures are highly flexible in solution. The PGH2 mimics underwent different conformational changes when bound to the active and purified TXAS and PGIS proteins in solution [21-22]. The altered conformations were favorable for the docking of PGH2 into the pockets of the enzymes’ substrate-binding sites with different configurations. For example, the free form of PGH2 in solution adopts a relaxed conformation with wide open α and ω chains. However, the conformation changes to a compact, oval shape with shorter distances between the chains when bound to PGIS and TXAS [21-22]. These observations have led us to hypothesize that PGH2 could adopt multiple conformations to specifically recognize the different target proteins. The PGH2 conformation that was bound to the TP-ligand pocket mediates signaling, which is different and specific in comparison to its binding with other COX-downstream synthases.
However, there is minimal structural information available for the molecular basis of PGH2 bound to TP. One of the major obstacles is the lack of an approach for preparing a large amount of the purified TP protein for structural characterization of the interaction between the receptor and its ligand. Recently, we reported the characterization of a solution structure of a TP ligand interacting with a constrained peptide that mimics the receptor-ligand recognition site by high-resolution NMR spectroscopy using the trNOE technique. This revealed useful information for understanding the molecular mechanism of the ligand-receptor interaction. In this paper, an attempt has been made to obtain purified TP protein and allow it to interact with PGH2. The conformation of PGH2, as a ligand bound to TP, was characterized by the binding of a PGH2 mimic, U46619, to the purified TP using a previously established trNOE technique. The detailed solution structure of U46619 bound to TP was solved, then utilized to reveal the specific conformation, and compared with the other PGH2 mimics bound to PGIS and TXAS. Thus, the conformation-dependent activation of the different target proteins by PGH2 was demonstrated.
EXPERIMENTAL PROCEDURES
Materials
2H2O, ethanol-d6 and DSS, (sodium 2, 2-dimethyl-2-silapentane-5-sulfonate) were purchased from Cambridge Isotope Laboratories (Andover, MA). U46619, a stable analog of PGH2, was obtained from Cayman Chemicals (Ann Arbor, MI). The detergent, n-Dodecyl-β-d-Maltoside (DM) was purchased from Calbiochem (San Diego, CA). All other chemicals were purchased from Sigma (St. Louis MO).
Expression and Purification of the Human TP
The expression and purification of the recombinant human TP protein are described as follows. Briefly, human TP cDNA was subcloned into the pVL1392 vector, which is suitable for the Baculovirus (BV) expression. A 6His-tag DNA sequence was also added to the isolated TP cDNA at the C-terminal position of the receptor protein to generate the pVL1392-TP-6His construct using a PCR approach. The pVL1392-TP-6His was packed into BV with Orbigen Sapphire DNA and the recombinant BV was then prepared and used for over-expressing the full length TP protein in Sf-9 cells. The TP protein was extracted from the Sf-9 cells using 1% DM, separated twice by ultracentrifugation for 1hr at 4°C and 250000 g, and purified by Fast Protein Liquid Chromatography (FPLC) on a Superdex-75 column (1 × 45 cm) with a flow rate of 0.2 mL/min using PBS with 0.2mM DM. The fractions containing the active TP protein were identified by binding assays, polyacrylamide gel electrophoresis (PAGE) analysis, and Western blot.
Electrophoresis, Western Blot and Ligand Binding Assay
The purified TP receptor was separated by 10% PAGE under denaturing conditions and then stained by protein staining or transferred to a nitrocellulose membrane for Western blot, in which a band recognized by anti-human TP receptor antibody was visualized by a second antibody linked to horseradish peroxidase, as previously described [23]. The ligand binding assay for the TP receptor was performed using a method described in Tai’s group [24-26]. The protein sample in 25mM Tris-HCl buffer, pH 7.4, containing 5mM CaCl2, was incubated with [3H]U46619 (Dupont/NEN) in a final reaction volume of 0.1 mL with vigorous shaking at room temperature for 60 min. The reaction was then terminated by adding 1 mL of ice-cold washing buffer (25mM Tris-HCl, pH 7.4). The unbound ligand was filtered through a Whatman GF/B glass fiber filter presoaked in ice-cold washing buffer using a vacuum system. The radioactivity of the TP -bound [3H]U46619 remaining on the filter was counted in 3 mL of scintillation cocktail using a Beckman β-Counter. For the competitive inhibition assay, unlabeled U46619 was added.
NMR Experiments
For the NMR experiment of the free form ligand, U46619 (final concentration 5.4 mM) was dissolved in 0.55 mL of 20 mM sodium phosphate buffer, pH 6.0, containing 0.2mM and 10% ethanol-d6 and 2H2O (to provide a lock signal), and then used for determination of the 1D and 2D (DQF-COSY, TOCSY and NOESY) 1H spectra. For the NMR titration experiment, the FPLC-purified, active TP protein was dissolved in the same buffer, and the spectrum was acquired. Later, different amounts (5, 10, 20 and 30μL) of U46619 in ethanol-d6 were added into the NMR sample (1mg/30μL) and incubated at 298K for 20 min and the 1D spectra were acquired again. The 2D DQF-COSY, TOCSY, and NOESY spectra for the final mixture of the titration (5.4 mM U46619 and approximately 80μM TP) were then recorded. All NMR experiments were carried out on a Bruker Avance 600MHz NMR spectrometer with a 5 mm triple-resonance probe at 298K. The water peak was suppressed by the Excitation Sculpting method [27]. All 1D spectra contain 16K data points. NOESY and TOCSY spectra contain 2048 × 512 data points, and the DQF-COSY spectra contain 4096 × 512 data points. The NOESY spectra were recorded with a mixing time of 200 ms. The TOCSY spectra were carried out with an MLEV-17 spin-lock pulse sequence with a total mixing time of 70 ms. Quadrature detection was achieved in the F1 by the states-TPPI method. The NMR data were processed using the Felix program (Accelrys, San Diego, CA). Shifted Sine-bell window functions of either 0° (for DQF-COSY), 60° (for TOCSY), or 90° (for NOESY) were used in both dimensions. Chemical shifts were referenced to the internal standard, DSS (contained in the 2H2O), which was set to 0 ppm.
NMR Structural Calculations
The overall conformation of the free U46619 or the TP-bound U46619 was determined through the inter-proton distances derived from the NOESY cross-peaks using a distance bound method [28]. The NOE cross-peaks were first assigned, and the volumes were measured and converted into upper bounds of the inter-proton distance constraints using the Felix 2000 software (Accelrys). The strong, medium, and weak peaks were set to correspond to the upper bound distances of 2.7, 3.5, and 5.0 Å, respectively. Distance constraints were input manually into the Discover program within the Insight II package (Accelrys, San Diego, CA), and restrained energy minimizations consisting of 1000 steps of conjugate gradient algorithm were then carried out to generate the structures of U46619.
Docking of PGH2 Mimics Into the Putative Ligand-Binding Site of the TP Model
The 3D structures of the TM domain of the human TP were constructed by homology modeling using the corresponding regions of the β2 adrenergic receptor’s X-ray structure (2RH1). The solution NMR structures of the TP extra- [29] and intra- [30] cellular loop segments were used to link the TM segments together to form a 3D TP model by using the Insight II protein modeling software. The PGH2 mimics were docked into the ligand pocket that was previously predicted by NMR spectroscopic and mutagenesis studies [29, 31], and by taking into account the ligand pocket in the β2 adrenergic receptor [32-33]. The docking study was performed by a 3D-Quantitative Structure Activity [34] using SYBYL Surflex-Dock software (Tripos, St. Louis, MO).
RESULTS
Multiple Binding of the PGH2 Mimic, U46619, to Purified TP, PGIS, and TXAS
The binding of the purified TP to PGH2 (Figure 1, panel IA) was inferred from an assay using [3H]-U46619 (Figure 1, panel II) as a ligand, since the PGH2 is not sufficiently stable in solution to be used for this assay. The glycosylated, full length human TP protein was expressed in Sf-9 cells using the BV expression system, and then purified by a high-yield purification procedure consisting of optimized detergent extraction, ultracentrifugation, and FPLC-separation. The purified TP showed roughly a single band in the protein staining (Figure 2A) and Western blot analysis (Figure 2B). A titration experiment was performed, in which increasing amounts of [3H]-U46619 were added to the purified TP in a DM detergent-mimicked lipid environment, and the Kd value (approximately 20 nM (Figure 1, panel II)) for the ligand binding was determined. On the other hand, it was previously reported that when the PGH2 served as substrate for PGIS and TXAS, it had Km values of approximately 10–30 μM [21, 35]. It was clear that the PGH2 had a much stronger affinity for binding to the TP than that of PGIS and TXAS. This information became very interesting for understanding the molecular basis behind the multiple binding activities and biological functions of the endogenous PGH2 produced by the COX.
Figure 2.
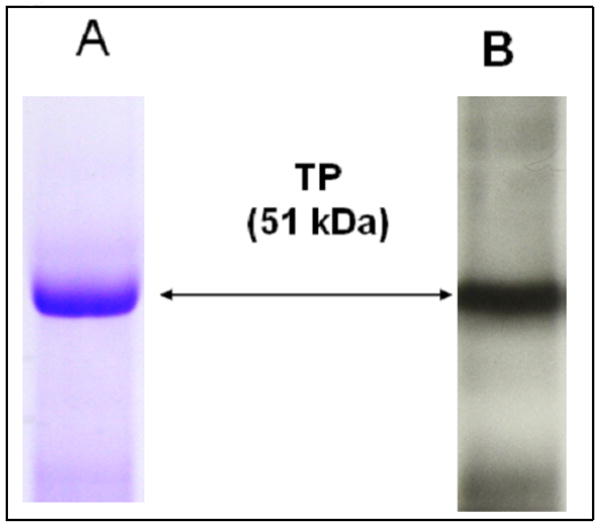
Purified TP protein analyzed by SDS-PAGE. The FPLC-purified TP protein (approximately 5 μg) was separated by 10% SDS-PAGE, and then stained with Coomacie blue (A) or transferred to nitrocellulose membrane and analyzed by western blot using TP peptide antibody (B). The bands of the TP protein with a molecular mass of approximately 51 kD are shown with arrows.
Identification of the Structural Changes of the PGH2 Mimic Upon Binding to the Purified TP, Using 1D 1H NMR Titration Experiments
Based on previous NMR structural studies for the interaction of U44069 with TXAS [21], and U46619 with PGIS [22], we discovered a conformational change in which an open hairpin structure changed to a more compact oval-like structure [21-22]. In addition, the oval shape of U46619 bound to PGIS was also different to that of U44069 bound to TXAS [22], which had a round shape. The experiments implied that the structure of U46619 could also become altered during its binding to the TP. To see the binding of the U46619 to the purified and active TP in solution, 1D 1H NMR titration experiments were performed as described in the Experimental Procedures. Upon binding of U46619 to TP, the chemical shifts of the proton resonance signals of both the ligand and protein were affected. Since the TP has a molecular mass of about 51 kDa and its NMR signal has shown difficulty in being observed, we focused primarily on the changes in the chemical shifts of U46619. Thus, the 1D 1H NMR titration experiments were performed in the presence of a large amount of U46619 and a very small amount of TP protein to adequately exclude the signals of the protein. The molar ratios of U46619 to TP protein in experiments B, C, D, and E in Figure 3 were 67.4:1, 45.7:1, 23.3:1, and 11.8:1, respectively. Significant perturbations of the proton chemical shifts at H3 and H4 of U46619 were observed following the addition of the purified TP in a concentration-dependent manner during the titration experiments (Figure 3). In contrast, shifts of the other U46619 protons (such as H12) were not significantly changed. The perturbed protons of the U46619 bound to TP (Figure 4B) are different in comparison to those from the U46619 bound to PGIS, and U44069 bound to TXAS (Table 1). These results provided evidence that U46619 was undergoing conformational changes following its binding to the TP in solution, and the U46619 protons, specifically H3 and H4, were likely involved in the direct and/or indirect contact with the active site residues of the TP protein. The experiment also indicated that the conformational changes of U46619 induced by the binding to the TP were different with that of PGIS and TXAS.
Figure 3.
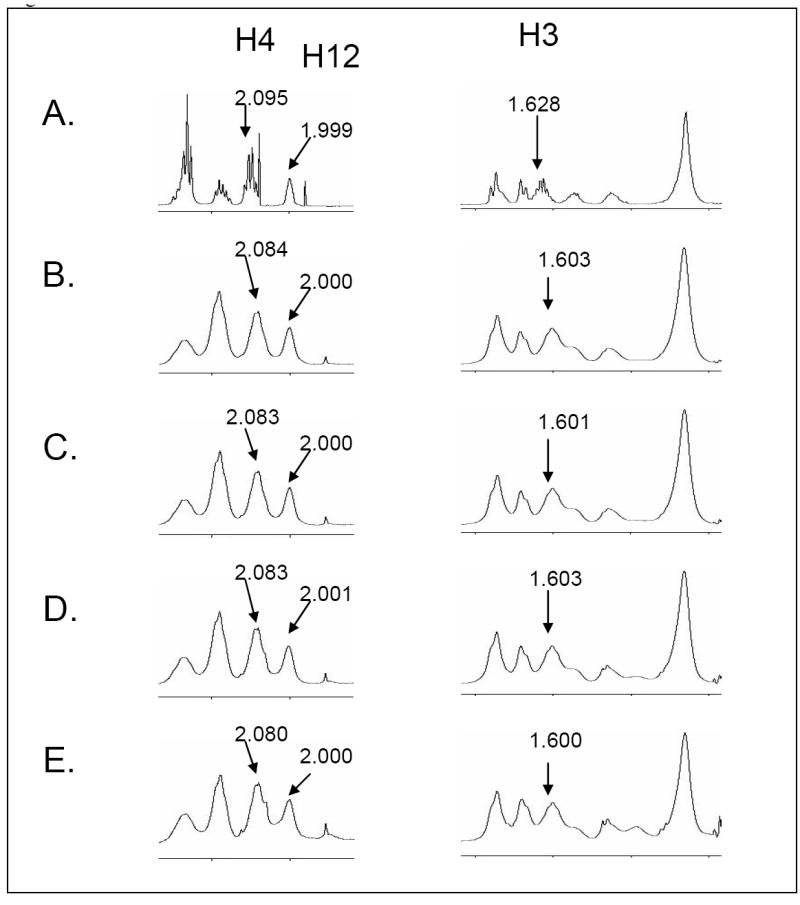
1D NMR titration of U46619 bound to the purified TP. Free, unbound U46619 is shown (A). The chemical shift signals of H3 and H4 or non-chemical shift signals (H12) from the U46619 are shown, following the addition of TP protein at ratios of 67.4:1 (B), 45.7:1 (C), 23.3:1 (D), and 11.8:1 (E).
Figure 4.

Intramolecular NOEs of U46619 bound to the TP protein. The new intramolecular NOEs of U46619 appeared after interaction with active TP (B) in comparison to that of its free state (A). The new NOE cross-peaks are boxed and labeled with the corresponding resonance assignments in the first and second dimensions.
Table 1.
Comparison of the perturbed protons in PGH2 mimics bound to different targeted proteins
| Target proteins | PGH2 mimic | Perturbed protons |
|---|---|---|
| TP | U46619 | H3, H4 |
| PGIS | U46619 | H11, H2C-2, H20 |
| TXAS | U44069 | H9, H13, H15 |
Determination and Characterization of the PGH2 Mimic’s Binding to the Purified TP by High-Resolution 2D 1H NMR, Using the Transferred NOE Effect Technique
In 2D NMR spectroscopy, the exchange-transferred nuclear Overhauser effect (transferred NOE, tr-NOE or et-NOE) has proven to be a useful technique for studying the bound conformation of ligands to proteins. Small U46619 molecules have a fast tumbling rate in their free states and slowly build up to appear as negative peaks in the NOESY spectrum. In contrast, the bound states of the U46619 have a slow tumbling rate and quickly build up to appear as positive peaks in NOESY spectrum, due to the large molecular weight of the ligand–protein complex [36-37]. In trNOE experiments, the NOESY spectra of U46619 were recorded in the presence of a very small amount of the purified TP protein. The cross-peak intensities of the NOESY spectra could be predominantly determined by the bound state NOEs. Therefore, by measuring the intensities of the NOE cross-peaks, the inter-proton distances in the bound state can be estimated and used for calculating the bound-state conformation. Also, the ligand’s binding to the protein can be indicated by a change in the signal of the cross-peaks from a negative (free ligand) to a positive (bound ligand) signal.
In these studies, the 2D 1H NMR spectra (NOESY, DQF-COSY and TOCSY) were recorded for U46619 in the presence and absence of the purified, active TP, using 200 ms mixing time. For the experiments, the ratio of the ligand to TP protein was approximately 67:1, which showed a very strong signal for the ligand and only minimal interference from the broad and weak protein peaks. The proton resonance assignments for the U46619 alone were completed on the basis of the proton chemical shifts and the through-bond coupling detected by DQF-COSY and TOCSY. The resonance assignments of U46619, in the presence of TP, were obtained by comparing the spectra of the mixture with that of the free U46619. The binding of the ligand to the active TP protein was clearly established by the identification of two major changes in the NOESY spectra: (a) The intramolecular cross-peaks of U46619 changed from the negative phase (Figure 4A, small molecule showing a negative phase) to the positive phase (Figure 4B; U46619 bound to the active TP became a large complex molecule with a positive phase). (b) New cross-peaks were observed in the NOESY spectrum of U46619 with the active TP protein (Figure 4B), which accounted for the conformational change upon interaction with the TP. For example, the NOE cross-peaks of H11/H18, H11/H19, H15/H18, and H15/H19, identified in the NOESY spectrum of U46619 with the TP protein (Figure 4B), were not found in the NOESY spectrum of the free U46619 (Figure 4A). This indicates that the distances between H11 and H18, H11 and H19, H15 and H18, and H15 and H19 became shorter upon binding to the receptor – down to within 5 Å. In addition, the new NOEs of the U46619 that resulted from its binding to the TP were different with those resulting from its binding to TXAS [21] and to PGIS [22]. The data indicated that the conformation of U46619 bound to TP was unique.
3D Solution Structure of the TP-Bound U46619
The complete assignments for the 2D spectra (NOESY (Figure 4B), and TOCSY (Figure 5)) with the proton resonances of U46619, in the presence of the TP receptor, provide information that generated 44 NOE constraints from the NOESY spectra for U46619 (Table 2). Ten structures of the TP-bound U46619 were obtained by initial structure calculations using the experimental distance constraints (Figure 6A). A refined average solution structure, shown in a rectangle shape, of the TP-bound U46619 is displayed in Figure 6B. The free U46619, shown in a triangle shape (Figure 6C) was also generated by a similar approach. The rectangle conformation of the TP-bound U46619 is clearly different with that of the PGIS-bound U46619, which is better shown with a round shape (Figure 6D) and the TXAS-bound U44069, which is illustrated with an oval shape (Figure 6E). These results first revealed that: (1) The conformation of U46619 underwent changes upon binding to the purified active TP in the lipid environment. (2) In the free form of U46619, the two arms (α and ω chains) were separated with a distance of approximately 10 Å. In contrast, the two arms were further separated in the TP-bound form of U46619, and formed a rectangle, which indicated that the topological arrangement of the PGH2-binding pocket in TP is likely a rectangular shape. (3) The conformation of the TP-bound form of U46619 is clearly different with the conformations of the PGH2 mimics (U46619 and U44069) bound to PGIS and TXAS, in which the two arms became closer forming an oval and round shape, respectively (Figure 6).
Figure 5.
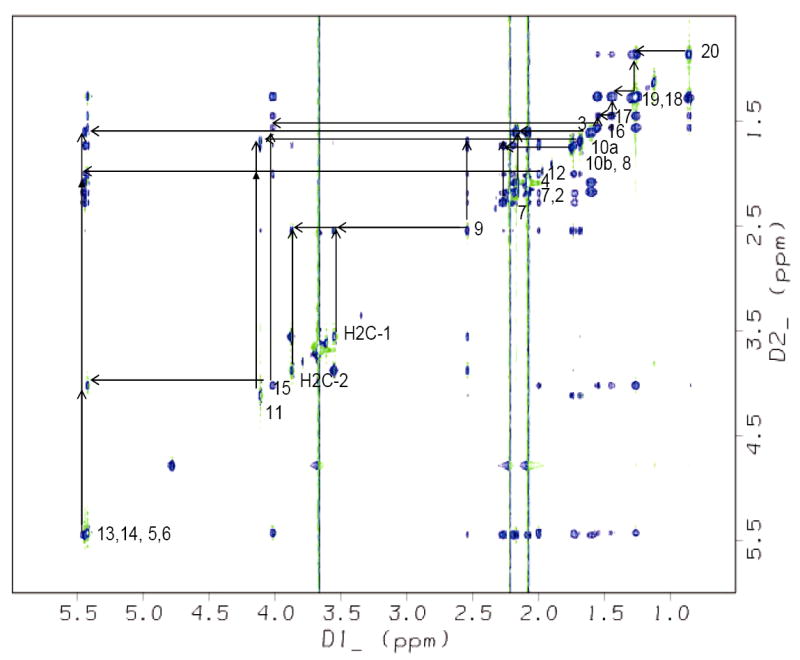
The TOCSY spectrum showing the resonance assignments of the TP-bound U46619 in solution. The coupling network between protons on adjacent carbons is indicated.
Table 2.
NOE constrains based on the upper limit distance of the bound form U46619 used to generate 3D conformation.
| Proton pairs | Distance (Å) | Proton pairs | Distance (Å) |
|---|---|---|---|
| H19 / H20 | 2.7 | H18 / H20 | 3.5 |
| H17 / H18 | 2.7 | H17 / H19 | 2.7 |
| H16 / H18 | 2.7 | H16 / H19 | 2.7 |
| H16 / H17 | 2.7 | H12 / H8 | 2.7 |
| H4 / H3 | 2.7 | H2 / H3 | 2.7 |
| H7-2 / H8 | 3.5 | H10b / H9 | 3.5 |
| H8 / H9 | 2.7 | H10a / H9 | 5.0 |
| H9 / H2C-1 | 2.7 | H10b / H2C-1 | 3.5 |
| H10a / H2C-1 | 2.7 | H9 / H2C-2 | 3.5 |
| H7-2 / H2C-2 | 3.5 | H2C-2 / H12 | 3.5 |
| H15 / H18 | 3.5 | H15 / H19 | 3.5 |
| H15 / H16 | 5.0 | H11 / H2C-1 | 5.0 |
| H11 / H9 | 5.0 | H11 / H8 | 3.5 |
| H11 / H4 | 5.0 | H11 / H18 | 5.0 |
| H11 / H19 | 5.0 | H11 / H10a | 5.0 |
| H11 / H10b | 3.5 | H5,H6 / H7-2 | 3.5 |
| H13,H14 / H20 | 5.0 | H13,H14 / H18 | 3.5 |
| H13,H14 / H19 | 3.5 | H13,H14 / H8 | 2.7 |
| H13,H14 / H12 | 2.7 | H13,H14 / H9 | 5.0 |
| H13,H14 /H2C-1 | 5.0 | H13,H14/H2C-2 | 5.0 |
| H13,H14 / H7-1 | 5.0 | H13,H14 / H7-2 | 3.5 |
| H13,H14 / H11 | 5.0 | H13,H14 / H15 | 2.7 |
Figure 6.
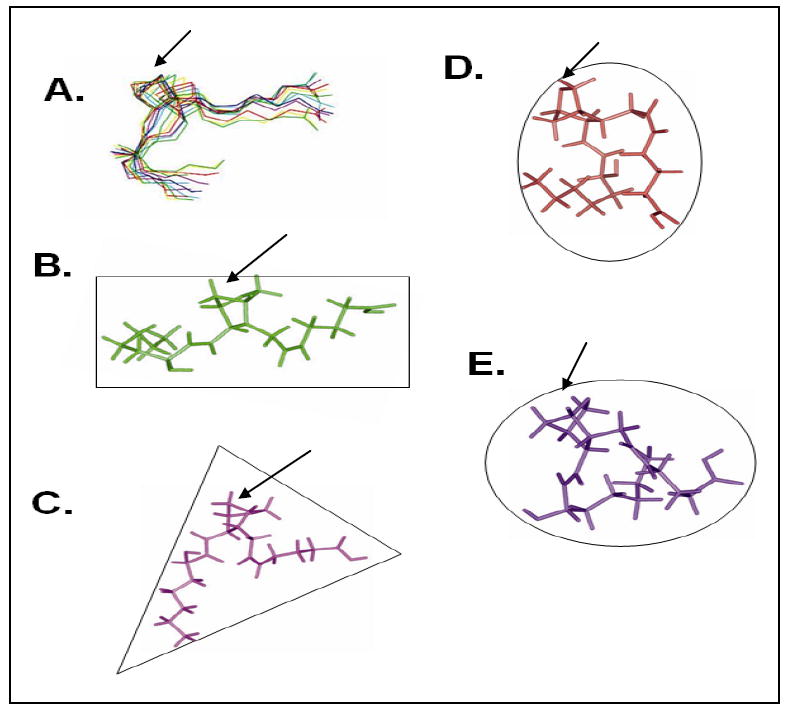
Comparison of the solution structures of the PGH2 mimics determined by NMR spectroscopy. The solution structures of the U46619 (bound to the purified TP) showing the 10 superimposed NMR structures (A) and an average NMR structure in the rectangle shape (B). The U46619 is also shown alone (C) in the triangle shape, and bound to the purified PGIS [22] (D, round shape). The structure of the U44069 bound to the purified TXAS [21] (E, oval shape) is also shown.
Docking of the TP-Bound U46619 With the Ligand-Binding Pocket of the TP Structural Model
A structural model of the human TP receptor, based on the extracellular loops’ structures determined by NMR spectroscopy and the conserved TM domains generated from homology modeling using bovine rhodopsin, has been characterized and reported [29]. Also, the crystal structure of the human β2 adrenergic receptor, a member of the G protein-coupled receptor (GPCR) family, was very recently reported [32-33]. This particular GPCR shares many homology characteristics with that of TP, especially in the TM domains. A TP seven-TM domain model (Figure 7, green colors) was generated by homology modeling using the x-ray structure of the TM domains from the β2 adrenergic receptor as templates. The TP extracellular loop structure was constructed by linking the NMR structures of the three loops to the corresponding TM domains and then optimized by energy minimization as described [29]. The TM ligand-binding site of the TP was localized by using previous mutagenesis information [31], which includes the interaction of the TP extracellular loop with its ligand, and the ligand pocket in the crystal structure of the β2 adrenergic receptor [32-33]. The NMR structure of the TP-bound U46619 conformation (Figure 6B, rectangle shape) could fit into the putative ligand pocket very well (Figure 7A). In contrast, docking of the free form of U46619 (Figure 6C, triangle shape) into the putative active site of the TP model caused severe steric clashes that resulted from the unreasonable energy increases by the overlapping atoms (circled in Figure 7B) between the ligand and the side chains of the protein (Figure 7B).
Figure 7.
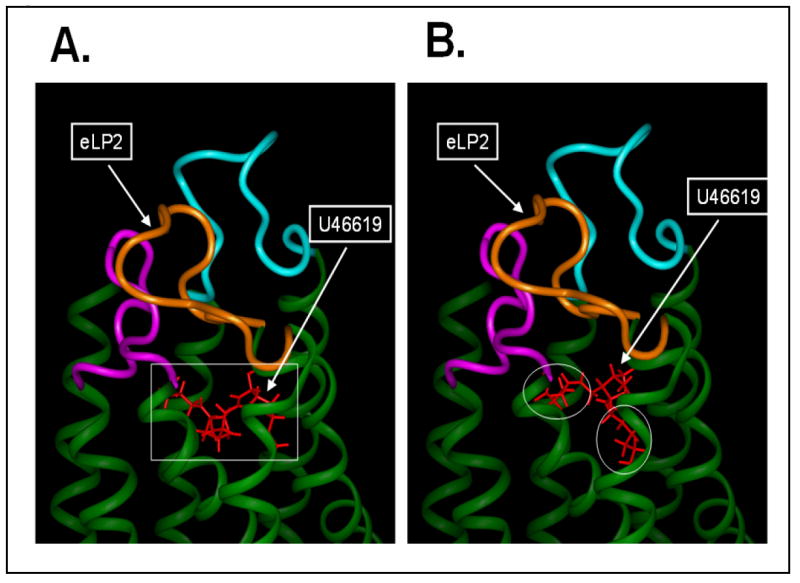
Comparison of the bound and free U46619 in the TP ligand pocket. A comparison of the U46619 in bound (rectangle shape, A) and free (triangle shape, B) forms were docked into the ligand pocket of the human TP model. The human TP model was constructed by linking the NMR structures of the extracellular loops [29] to the corresponding helical structures of the TM domains generated from homology modeling using the TM domain x-ray structures of the β2 adrenergic receptor [32-33]. The region of the free form U46619 overlapped with the TM domain in an unfit position to the pocket, as indicated with the circles in the panel B.
DISCUSSION
For decades, the PGH2 molecule’s binding to multiple target proteins with unrelated protein structures was a real mystery in the fields of protein chemistry and eicosanoid biology. One former explanation is that all of the proteins that share the PGH2 molecule as their common ligand might have similar ligand-binding pockets in their 3D conformations that fit PGH2. With the successful molecular cloning for all of the PGH2-binding target proteins in the past couple of decades, this hypothesis could possibly be applied for PGIS and TXAS because both belong to the microsomal P450 enzymes. However, the hypothesis is not supported by the structural information of the unrelated protein sequences between the TP (a GPCR) and PGES and PGDS (glutathione related enzymes) in comparison to the P450 enzymes, PGIS and TXAS. This has led us to hypothesize that the PGH2 has great flexibility and can adopt multiple conformations to fit the diverse ligand pockets with different topological arrangements and shapes, according to the different target proteins. To prove this concept, it is necessary to determine the PGH2 mimic structures bound to the purified target proteins. Thus, it is essential to have purified and active PGH2-target proteins available for structural studies, which were a major challenge until recently, when PGIS [21], TXAS [22], and the TP protein were successfully expressed in large-scale and purified.
Previously, by using the purified PGIS and TXAS proteins, we have successfully determined the solution structures of the PGH2 mimics bound to the active enzymes and demonstrated that the conformation of PGH2 bound to PGIS is significantly different with that of PGH2 bound to TXAS [22, Figure 6], even though both enzymes are included in the P450 family. This became more interesting in understanding the solution structure of PGH2 bound to the active TP protein, since it represents an unrelated protein to the P450 enzymes – PGIS and TXAS, in terms of protein sequences and biological functions.
High-resolution NMR spectroscopy is a very useful approach for determining the detailed changes in the PGH2 mimic once interacted with its targeted proteins in solution. Two types of NMR experimental approaches are generally used for studying the ligand and receptor interactions. One type of NMR approach focuses on the changes in NMR signals from the small ligand molecule produced upon binding to the receptor, in which the trNOE and saturation transferred difference (STD) techniques are preferred. The other type observes the chemical-shift changes in the targeted receptor protein (typically the amide 1H-15N single bond correlations) produced upon binding with its ligand. This approach is called “SAR by NMR” because structure-activity relationships (SAR) are obtained from NMR. The trNOE NMR technique is particularly useful in the identification of a moderate, ligand-binding protein as well as the determination of a bound conformation of the ligand through quantitative analysis of the intramolecular trNOEs (U46619-PGIS, [38-40]). The current study is the first proven experiment, which used advanced NMR techniques for the characterization of TP binding to the PGH2 mimic (Figures 3-6), all of which has been made possible through the successful large-scale expression and purification of the full length and active TP protein (Figure 2).
In these current studies, the 1D and 2D trNOE NMR experiments have provided clear results for the identification of the interaction between U46619 and the purified TP (Figure 3-5). The 3D structural determination of the TP-bound U46619, where the cross-peaks changed from a strong negative to positive in the NOESY spectrum and the intramolecular trNOEs of the bound U46619 are shown (Figure 4), has become important experimental evidence that supports the second hypothesis, in which PGH2 adopts a unique structure, when bound to the TP, that can be distinguished from that of its conformation bound to the COX-downstream synthases, PGIS and TXAS (Figure 6).
To control the spin diffusion which might affect the trNOEs resulting from the binding of U46619 to the TP protein, we have used the following strategies: (1) Using less TP protein. Less protein gives less spin diffusion, as identified by previous investigators [41]. Since the bound U46619 is a very small amount, when a small amount of TP protein is also used, a relatively longer mixing time (300 ms) can be applied to the experiment to get the necessary intensity in the NOESY spectrum. In other words, the spin diffusion effect of using a longer mixing time is partially cancelled by using a lower concentration of protein. (2) Using different mixing times for the NMR experiments. After comparison of the bound structures of U46619 obtained with the 200 and 300 ms mixing times, no significant differences were found, which indicated that little spin diffusion affects the experiments even when using the 300 ms mixing time. In addition, since we were using the distance-bound methods and the NOEs were only converted to the upper-bound limit of the distance, instead of an accurate interproton distance, the error introduced by spin-diffusion should be very minimal. In fact, several experimental results have proven that the intermolecular spin diffusion is not a general problem for bound ligand determination in the designed trNOE experiments [42].
It should also be noted that U46619 is not an endogenous compound, but it can perform similar activities to those of PGH2 when serving as an agonist for TP, such as triggering signaling by increasing intracellular calcium concentration through Gq protein coupling. In addition, the chemical structure of U46619 is identical to that of PGH2 except for a single atom at the O-9 position, where a carbon atom replaces the oxygen atom of PGH2. Thus, understanding the conformation of U46619’s binding to TP helps to characterize the endogenous PGH2 as it binds to and activates TP. However, it should be noted that minor differences may be present in the U46619’s binding compared to that of the endogenous PGH2.
In conclusion, the binding of a PGH2 mimic (U46619) to the purified TP adopts a unique conformation that fits into the ligand-binding pocket of the TP, which is different in comparison to that of U46619’s binding to PGIS and U44069’s binding to TXAS. The results have proven that PGH2 could adopt multiple conformations in solution upon interaction with its different targeted proteins.
Acknowledgments
This work was supported by NIH Grants (HL056712 and HL079389 (to KHR)). We thank Dr. Xiaolian Gao in the Biology & Biochemistry Department and Dr. Youlin Xia in the KECK/IMD NMR Center of the University of Houston, for access to the NMR facility and for providing valuable advice on taking the NMR spectra. In addition, we would like to thank the Baculovirus/Monoclonal Antibody Facility in Baylor College of Medicine (Houston, TX) for the large-scale expression of the TP protein.
Footnotes
This work was supported by NIH Grants (HL56712 and HL79389 (to KHR))
Abbreviations used: AA, arachidonic acid; TXA2, thromboxane A2; TXAS, TXA2 synthase; PGI2, prostacyclin or prostaglandin I2; PGIS, PGI2 or prostacyclin synthase; PGH2, prostaglandin H2; TP, TXA2 receptor; ER, endoplasmic reticulum; NMR, nuclear magnetic resonance; DQF-COSY, double-quantum-filtered correlation spectroscopy; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser enhancement spectroscopy; TOCSY, total correlation spectroscopy; 1D, one dimensional; 2D, two dimensional; 3D, three dimensional; DSS, sodium 2,2-dimethyl-2-silapentane-5-sulfonate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Needleman P, Turk J, Jackschik BA, Morrison AR, Lefkowith JB. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 2.Granstrom E, Diczfalusy U, Hamberg M, Hansson G, Malmsten C, Samuelsson B. Prostaglandins and the Cardiovascular System. Raven Press; New York: 1982. pp. 15–58. [PubMed] [Google Scholar]
- 3.Majerus PW. J Clin Invest. 1983;72:1521–1525. doi: 10.1172/JCI111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller DK, Sadowski S, Soderman DD, Kuehl FA., Jr J Biol Chem. 1985;260:1006–1014. [PubMed] [Google Scholar]
- 5.Smith WL. Annu Rev Physiol. 1986;48:251–262. doi: 10.1146/annurev.ph.48.030186.001343. [DOI] [PubMed] [Google Scholar]
- 6.Picot D, Loll PJ, Garavito RM. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 7.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashir JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 8.Ren Y, Walker C, Loose-Mitchell DS, Deng J, Ruan KH, Kulmacz RJ. Arch Biochem Biophys. 1995;323:205–214. doi: 10.1006/abbi.1995.0027. [DOI] [PubMed] [Google Scholar]
- 9.Ruan KH, Wang LH, Wu KK, Kulmacz RJ. J Biol Chem. 1993;268:19483–19490. [PubMed] [Google Scholar]
- 10.Ruan KH, Li P, Kulmacz RJ, Wu KK. J Biol Chem. 1994;269:20938–20942. [PubMed] [Google Scholar]
- 11.Moncada S, Herman AG, Higgs EA, Vane JR. Thromb Res. 1977;11:323–344. doi: 10.1016/0049-3848(77)90185-2. [DOI] [PubMed] [Google Scholar]
- 12.Weksler BB, Ley CW, Jaffe EA. J Clin Invest. 1978;62:923–930. doi: 10.1172/JCI109220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingerman-Wojenski C, Silver MJ, Smith JB, Macarak E. J Clin Invest. 1991;67:1292–1296. doi: 10.1172/JCI110157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith WL, DeWitt DL, Allen ML. J Biol Chem. 1983;258:5922–5926. [PubMed] [Google Scholar]
- 15.Funk CD. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 16.Huang JC, Goldsby JS, Wun WS. Hum Reprod. 2004;19:1856–1860. doi: 10.1093/humrep/deh352. [DOI] [PubMed] [Google Scholar]
- 17.Huang JC, Wun WS, Goldsby JS, Matijevic-Aleksic N, Wu KK. Hum Reprod. 2004;19:2900–2906. doi: 10.1093/humrep/deh524. [DOI] [PubMed] [Google Scholar]
- 18.Huang JC, Wun WS, Goldsby JS, Wun IC, Falconi SM, Wu KK. Hum Reprod. 2003;18:2582–2589. doi: 10.1093/humrep/deg490. [DOI] [PubMed] [Google Scholar]
- 19.Sakai H, Suzuki T, Takahashi Y, Ukai M, Tauchi K, Fujii T, Horikawa N, Minamimura T, Tabuchi Y, Morii M, Tsukada K, Takeguchi N. FEBS Lett. 2006;580:3368–3374. doi: 10.1016/j.febslet.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Nie D, Che M, Zacharek A, Qiao Y, Li L, Li X, Lamberti M, Tang K, Cai Y, Guo Y, Grignon D, Honn KV. Am J Pathol. 2004;164:429–439. doi: 10.1016/S0002-9440(10)63133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruan KH, Wu J, Wang LH. Arch Biochem Biophys. 2005;444:165–173. doi: 10.1016/j.abb.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Ruan KH, Wu J, Cervantes V. Biochemistry. 2008;47:680–688. doi: 10.1021/bi701671q. [DOI] [PubMed] [Google Scholar]
- 23.Geng L, Wu J, So SP, Huang G, Ruan KH. Arch Biochem Biophys. 2004;423:253–265. doi: 10.1016/j.abb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Chiang N, Tai HH. Arch Biochem Biophy. 1998;352:207–213. doi: 10.1006/abbi.1998.0620. [DOI] [PubMed] [Google Scholar]
- 25.Chiang N, Kan WM, Tai HH. Arch Biochem Biophys. 1996;334:9–17. doi: 10.1006/abbi.1996.0423. [DOI] [PubMed] [Google Scholar]
- 26.Zhou H, Yan F, Tai HH. J Pharmacol Exp Ther. 2001;298:1243–1251. [PubMed] [Google Scholar]
- 27.Callihan D, West J, Kumar S, Schweitzer BI, Logan TM. J Magn Reson B. 1996;112:82–85. doi: 10.1006/jmrb.1996.0114. [DOI] [PubMed] [Google Scholar]
- 28.Wüthrich K. NMR of protein and nucleic acids. John Wiley & Sons; New York: 1986. [Google Scholar]
- 29.Ruan KH, Wu J, So SP, Jenkins LA, Ruan CH. Eur J Biochem. 2004;271:3006–3016. doi: 10.1111/j.1432-1033.2004.04232.x. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Feng M, Ruan KH. Arch Biochem Biophys. 2008;470:73–82. doi: 10.1016/j.abb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.So SP, Wu J, Huang G, Huang A, Li D, Ruan KH. J Biol Chem. 2003;278:10922–10927. doi: 10.1074/jbc.M209337200. [DOI] [PubMed] [Google Scholar]
- 32.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 34.AbdulHameed MD, Hamza A, Liu J, Huang X, Zhan CGJ. J Chem Inf Model. 2008;48:179–185. doi: 10.1021/ci700315c. [DOI] [PubMed] [Google Scholar]
- 35.Yeh HC, Hsu PY, Wang JS, Tsai AL, Wang LH. Biochim Biophys Acta. 2005;1738:121–132. doi: 10.1016/j.bbalip.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landy SB, Rao BDN. J Magn Reson. 1989;81:371–377. [Google Scholar]
- 37.Lippens RM, Cerf C, Hallenga KJ. J Magn Reson. 1992;99:268–281. [Google Scholar]
- 38.Leone M, Freeze HH, Chan CS, Pellecchia M. Curr Drug Discovery Technol. 2006;3:91–100. doi: 10.2174/157016306778108884. [DOI] [PubMed] [Google Scholar]
- 39.Angulo J, Rademacher C, Biet T, Benie AJ, Blume A, Peters H, Palcic M, Parra F, Peters T. Methods Enzymol. 2006;416:12–30. doi: 10.1016/S0076-6879(06)16002-4. [DOI] [PubMed] [Google Scholar]
- 40.Megy S, Bertho G, Gharbi-Benarous J, Baleux F, Benarous R, Girault JP. FEBS Lett. 2006;580:5411–5422. doi: 10.1016/j.febslet.2006.08.084. [DOI] [PubMed] [Google Scholar]
- 41.Campbell PA, Sykes BJ. J Magnetic Resonance. 1991;93:77–92. [Google Scholar]
- 42.Post CB. Curr Opin Struct Biol. 2003;13:581–588. doi: 10.1016/j.sbi.2003.09.012. [DOI] [PubMed] [Google Scholar]