

The non-ribosomal peptides (NRP) feature an enormous chemical diversity and have given rise to a multitude of therapeutics.1 However, their microbial synthesis by large multifunctional synthetases complicates the construction and sampling of large libraries of novel NRPs.2 In contrast, peptides synthesized by ribosomal translation are amenable to ultra-high throughput screening schemes such as mRNA-display,3 a combinatorial method that allows the sampling of more than 1013 individual molecules.4 In the past, however, these peptide libraries had to be constructed from the 20 canonical L-amino acids – a rather uniform set of building blocks compared to the variety of L-, D-, β-, N-methyl, and α,β-unsaturated amino acids from which NRPs are tailored. In an effort to reduce this limitation, we and others have shown that the in vitro translation of non-biological polymers from unnatural L-amino acids is indeed possible with a reconstituted Escherichia coli translation system (PURE-system).5-7 However, the ribosomal production of polymers with additional alternative backbone residues remains a challenging,8-10 yet indispensable step towards the production of NRP-like structures in an mRNA templated fashion.
One backbone alteration found in many natural products such as lantibiotics,11 microcystin,12 and thiopeptide antibiotics13 stems from the incorporation of dehydroalanine (ΔAla, Scheme 1). α,β-unsaturated amino acids exhibit rigidifying effects on the peptide backbone, which stabilizes secondary structures and increases the proteolytic stability of the parent molecule.14 Due to its moderate electrophilicity, ΔAla may also serve as warhead in protein reactive compounds15 and has proven a valuable intermediate for the preparation of cyclized, glycosylated or prenylated peptides through intra- or intermolecular Michael addition.16
Scheme 1.
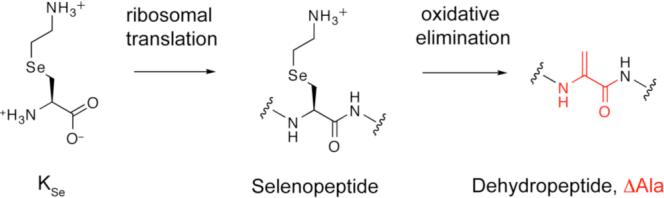
To harness this versatility for the in vitro selection of peptides we devised a method for constructing dehydropeptides through mRNA templated, ribosomal peptide synthesis. Van der Donk et al. reported the use of Se-phenylselenocysteine (SecPh) as a building block for solid phase peptide synthesis that can be converted to ΔAla via oxidative elimination.17 The chemical conditions for this process are compatible with biomacromolecules, however, ScPh is not a reported substrate for any of the E. coli aminoacyl tRNA synthetases and thus cannot be used for all-enzymatic peptide synthesis. To circumvent this problem, we investigated the possibility that selenalysine18 (KSe, Scheme 1) might be an efficient substrate for lysine aminoacyl-tRNA-synthetase and thus be incorporated into peptides as directed by lysine codons on translated mRNAs. We further surmised that such selenopeptides might undergo oxidative elimination to expose ΔAla in a posttranslational manner (Scheme 1).
To test these ideas, we assembled a reconstituted translation system from E. coli6 as previously described and tested the efficiency of KSe19,20 incorporation in place of lysine into a short peptide (Figure 1, peptide 1). 50 μl translation reactions containing the corresponding mRNA were incubated for 1h at 37° C. Product peptide yields were determined by purification on NTA-agarose beads and 35S-Met scintillation counting (Figure 1, left). Reactions supplemented with 0.2 mM lysine typically yielded 30 − 40 pmols of peptide. Reactions where lysine was replaced by 0.16 mM KSe produced similar amounts of product, which was subsequently identified as the desired selenopeptide by MALDI-TOF MS (mobs. = 1494.8 Da vs. mcalc. = 1494.6 Da). As judged by this analysis, the KSe containing reaction did not incorporate lysine. This is notable since endogenous traces of lysine in the translation mixture allow considerable synthesis of the lysine containing peptide (mobs. = 1428.6 Da vs. mcalc. = 1428.7 Da) (Figure 1, left).
Figure 1.
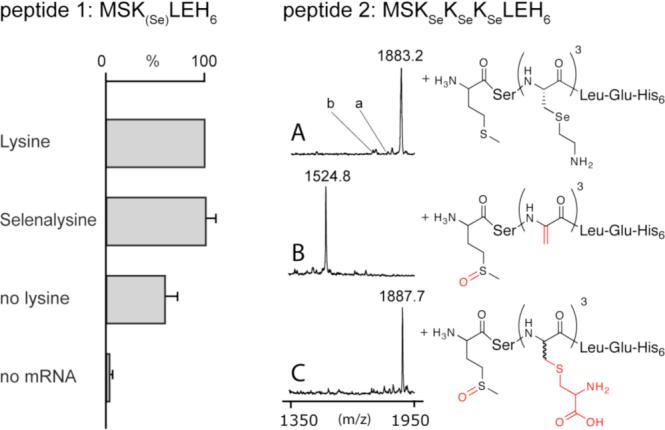
Selenalysine incorporation assay. Left: The efficiency of peptide 1 production was determined by scintillation counting of specific 35S-Met activity. The bars represent the average of three independent measurements and the averaged value for the lysine containing reactions (35 ± 3 pmol) was set to 100 %. Control translation reactions without supplemented lysine (no lysine) show a significant background due to the presence of contaminating lysine in the translation mixture. Right: MALDI-TOF analysis of the peptide 2 after translation (A, mcalc. = 1883.6 Da), oxidative elimination (B, mcalc. = 1524.6 Da), and Michael addition of L-cysteine (C, mcalc. = 1887.7 Da). Signals consistent with the production of peptides containing one (a) or two (b) lysines are indicated. To improve detectability by MALDI-TOF, peptides 1 and 2 were produced as the free N-terminal amine by omission of the formyl donor (10-formyl-5,6,7,8-tetrahydrofolic acid ) necessary for formylation of methionyl-tRNA.
As a more rigorous test of the efficiency and fidelity of lysine replacement by KSe, we prepared an mRNA containing three consecutive lysine codons. This template was also efficiently translated into selenopeptide (30 pmol/50 μl) with an observed mass of mobs. = 1883.2 Da (mcalc. = 1883.6 Da) indicating that incorporation of KSe does not significantly inhibit the translational machinery and that lysine incorporation is effectively outcompeted (Figure 1, A).
Translated and purified selenopeptide (Figure 1, peptide 2) was then converted into the corresponding dehydropeptide by incubation with 200 mM H2O2 at pH 5 − 6 on ice for 1 h.17 The mass difference between starting material and the oxidized peptide as determined by MALDI-TOF is consistent with elimination of three formal equivalents of 2-amino ethylselenol (−3 × 125 Da) accompanied by sulfoxidation of the N-terminal methionine (+16) (Figure 1, B). The formation of three electrophilic functions was further confirmed by intermolecular thiol-conjugation by incubating the oxidized peptide with 100 mM L-cysteine (Figure 1, C). Indeed, a large variety of nucleophiles may be reacted with ΔAla-containing peptides and thus our approach presents a general method for the site directed incorporation of small molecules into translated peptides or proteins.16
Finally, we aimed at introducing ΔAla into cyclic peptides so as to enhance our mimicry of ΔAla containing natural products and ultimately set the stage for the in vitro selection of novel, drug-like molecules. Biologically active peptides are very often cyclic, because cyclization improves proteolytic stability,21 membrane solubility22 and target affinity/specificity.23 We exploited the recent discovery that α,α'-dibromo-m-xylene can cross-link and therefore cyclize peptides that contain two cysteine residues.24 The two resulting thioether bonds are stable, apolar, and may improve bioavailability.
A model peptide (Figure 2, peptide 3) was produced by in vitro translation and adsorbed onto NTA-agarose beads. These beads were then treated with 5 mM α,α'-dibromo-m-xylene and 0.2 mM tris(carboxyethyl)-phosphine in a 1:4 acetonitrile:50 mM Tris-HCl buffer, pH 8.0 for 1 h at room temperature. The peptides were then eluted with 0.2 % TFA and oxidized with 20 mM H2O2 at pH 5.0 for 1 h and then analyzed by MALDI-TOF (Figure 2, E). The observed mass is consistent with the desired structure with a minor signal consistent with sulfoxidation of one of the two thioethers. Treatment of the same peptide with 200 mM H2O2 led to a mixture of non-, mono- and disulfoxidized species. Reduction of the oxidant concentration, however, led to satisfactory homogeneity. For the same reason we chose to substitute methionine with norleucine (MNor), an efficient analog with an oxidation-resistant side chain.25
Figure 2.
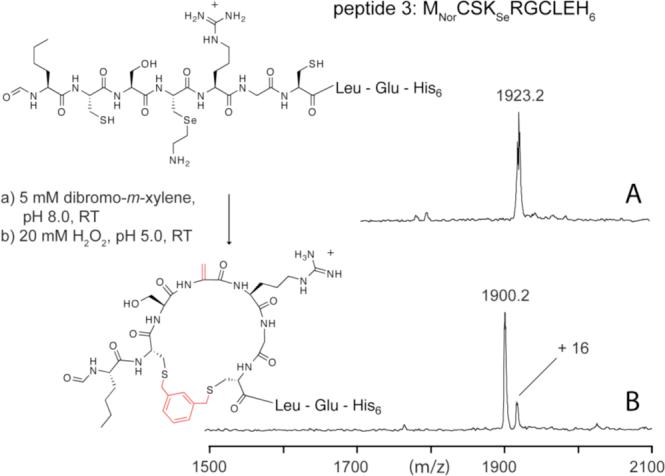
Incorporation of ΔAla into a cyclic scaffold. Two concomitant SN2 reactions between two peptide-borne thiols (A, mcalc. = 1923.7 Da) and one equivalent of α,α'-dibromo-m-xylene leads to peptide cyclization. Possible side-products such as the linear adduct of two equivalents of xylene and one peptide are not observed (B, mcalc. = 1900.8 Da). A minor signal 16 Da higher than the expected mass indicates partial sulfoxidation of the newly formed thioethers by the conditions used for ΔAla formation.
In summary, we have demonstrated the production of genetically encoded dehydropeptides. Our approach employs unmodified components from the translation machinery of E. coli, and mild but robust conditions to install multiple electrophilic functions in linear or cyclic peptides. The simplicity of this methodology should allow wide applicability, making highly decorated peptides available independently of solid phase peptide synthesis. Furthermore, having introduced ΔAla to the toolbox of mRNA-templated peptide synthesis, we may now embark on ultrahigh throughput selections for specific protein-reactive or catalytically active compounds.
Supplementary Material
Acknowledgment
We thank Drs. M.C.T. Hartman, K. Josephson and B. Seelig for their insightful advice. J.W.S. is an Investigator of the Howard Hughes Medical institute and F.P.S is supported by the National Institutes of Health (GM 074505-02).
Footnotes
Supporting Information Available: Experimental details are available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Walsh CT. Antibiotics, Actions, Origins, Resistance. ASM Press; Washington, D.C.: 2003. [Google Scholar]
- 2.Finking R, Marahiel MA. Annu. Rev. Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 3.Roberts RW, Szostak JW. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keefe AD, Szostak JW. Nature. 2001;410:715–718. doi: 10.1038/35070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 6.Josephson K, Hartman MCT, Szostak JW. J. Am. Chem. Soc. 2005;127:11727–11735. doi: 10.1021/ja0515809. [DOI] [PubMed] [Google Scholar]
- 7.Forster AC, Tan Z, Nalam MNL, Lin H, Qu H, Cornish VW, Blacklow SC. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6353–6357. doi: 10.1073/pnas.1132122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan Z, Forster AC, Blacklow SC, Cornish VW. J. Am. Chem. Soc. 2004;126:12752–12753. doi: 10.1021/ja0472174. [DOI] [PubMed] [Google Scholar]
- 9.Dedkova LM, Fahmi NE, Golovine SY, Hecht SM. J. Am. Chem. Soc. 2003;125:6616–6617. doi: 10.1021/ja035141q. [DOI] [PubMed] [Google Scholar]
- 10.Eisenhauer BM, Hecht SM. Biochemistry. 2002;41:11472–11478. doi: 10.1021/bi020352d. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee C, Paul M, Xie LL, van der Donk WA. Chem. Rev. 2005;105:633–683. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 12.MacKintosh RW, Dalby KN, Campbell DG, Cohen PTW, Cohen P, MacKintosh C. FEBS Lett. 1995;371:236–240. doi: 10.1016/0014-5793(95)00888-g. [DOI] [PubMed] [Google Scholar]
- 13.Bagley MC, Dale JW, Merritt EA, Xiong A. Chem. Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 14.Humphrey JM, Chamberlin AR. Chem. Rev. 1997;97:2243–2266. doi: 10.1021/cr950005s. [DOI] [PubMed] [Google Scholar]
- 15.Drahl C, Cravatt BF, Sorensen EJ. Angew. Chem. Int. Ed. 2005;44:5788–5809. doi: 10.1002/anie.200500900. [DOI] [PubMed] [Google Scholar]
- 16.Gieselman MD, Zhu YT, Zhou H, Galonic D, van der Donk WA. Chembiochem. 2002;3:709–716. doi: 10.1002/1439-7633(20020802)3:8<709::AID-CBIC709>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Okeley NM, Zhu YT, van der Donk WA. Org. Lett. 2000;2:3603–3606. doi: 10.1021/ol006485d. [DOI] [PubMed] [Google Scholar]
- 18.De Marco C, Busiello V, Digirolamo M, Cavallini D. Biochim. Biophys. Acta. 1976;454:298–308. doi: 10.1016/0005-2787(76)90232-x. [DOI] [PubMed] [Google Scholar]
- 19. The synthesis of KSe has been described before (Ref. 20). KSe synthesized from sodium borohydride reduced L-selenocysteine and 2-bromoethylamine was characterized by MS (EI) m/z (M +H)+ 213.0.
- 20.De Marco C, Rinaldi A, Dernini S, Cavallini D. Gazz. Chim. 1975;105:1113–1115. [Google Scholar]
- 21.March DR, Abbenante G, Bergman DA, Brinkworth RI, Wickramasinghe W, Begun J, Martine JL, Fairlie DP. J. Am. Chem. Soc. 1996;118:3375–3379. [Google Scholar]
- 22.Burton PS, Conradi RA, Ho NFH, Hilgers AR, Borchardt RT. J. Pharm. Sci. 1996;85:1336–1340. doi: 10.1021/js960067d. [DOI] [PubMed] [Google Scholar]
- 23.Khan AR, Parrish JC, Fraser ME, Smith WW, Bartlett PA, James MN. Biochemistry. 1998;37:16839–16845. doi: 10.1021/bi9821364. [DOI] [PubMed] [Google Scholar]
- 24.Timmerman P, Beld J, WPuijk WC, Meloen RH. Chembiochem. 2005;6:821–824. doi: 10.1002/cbic.200400374. [DOI] [PubMed] [Google Scholar]
- 25.Kiick KL, Weberskirch R, Tirrell DA. FEBS Lett. 2001;502:25–30. doi: 10.1016/s0014-5793(01)02657-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.