

Summary
We examined the leukemic stem cell potential of blasts at different stages of maturation in childhood acute lymphoblastic leukemia. Human leukemic bone marrow was transplanted intrafemorally into NOD/scid mice. Cells sorted using the B precursor differentiation markers CD19, CD20 and CD34 were isolated from patient samples and engrafted mice before serial transplantation into primary or subsequent (up to quaternary) recipients. Surprisingly, blasts representative of all the different maturational stages were able to reconstitute and re-establish the complete leukemic phenotype in vivo. Sorted blast populations mirrored normal B precursor cells with transcription of a number of stage-appropriate genes. These observations have informed a model for leukemia-propagating stem cells in childhood ALL.
Significance
There is an ongoing debate whether all malignancies are maintained by a small population of immature cancer stem cells (as for myeloid leukemias) or whether the majority of malignant cells possess some stemness. This question is of key clinical relevance as in vivo clonogenic stem cells constitute the primary therapeutic target. We have developed an in vivo assay permitting the analysis of the stem cell potential of human ALL blasts at different stages of maturation. Using this approach we demonstrate that B precursor blasts mirroring different stages of maturation display self-renewal and possess malleability within their altered B cell developmental programme. Thus, leukemic lymphoid progenitors may not lose their self-renewal capability with maturation.
Introduction
Acute lymphoblastic leukemia (ALL) is the most frequent malignancy of childhood. Despite its overall good response to current treatment protocols and an approximately 80% long-term event-free survival rate (Pui, 2000), there is a big demand for new therapies for patients with high-risk and relapsed ALL with a cure rate below 50%.
There is an ongoing debate as to the existence of a rare cancer stem cell population in childhood ALL, (Clarke et al., 2006; Kelly et al. 2007; Kennedy et al. 2007; Adams et al. 2007) which, if it exists, provides a key target for novel curative therapies. The presence of leukemic stem cells has been clearly defined in AML by xeno-transplantation studies. It has been demonstrated that cells with the ability to reestablish the human leukemia in immune-deficient NOD/scid mice were exclusively present within the CD34+CD38- stem cell fraction (Lapidot et al., 1994; Bonnet et al., 1997). Like the normal hematopoietic stem cell compartment, the LSC compartment in AML is heterogeneous and organized as a hierarchy with distinct subclasses that differ in their proliferative and self-renewal capacities. With a clonal tracking approach and serial transplantation of candidate LSC it has been possible to define short-term, long-term and quiescent long-term LSCs within this hierarchy (Hope et al., 2004). These profound similarities between normal and leukemic hematopoietic stem cells support the hypothesis that AML arises within the normal HSC compartment and retains a hierarchy similar to normal hematopoiesis.
For ALL, the picture is less clear. Our understanding of the hierarchy of childhood B-precursor ALL has been limited by the lack of appropriate in vitro and in vivo models. The original hypothesis, as proposed by Mel Greaves, suggested that the success of treatment for childhood ALL is linked to the transformation of a B-cell progenitor prone to undergo apoptosis (Greaves, 1993). In contrast, adult and certain types of high-risk ALL may originate in a more primitive stem cell equipped with multiple protective mechanisms to resist chemotherapy. This hypothesis is supported by studies showing that in the most common subtype of childhood ALL, ALL/t(12;21), blasts and pre-leukemic stem cells harboring t(12;21) are found exclusively in the more mature CD19+ population (Hotfilder et al., 2002; Castor et al., 2005; Hong et al., 2008). Similarly, in high hyperdiploid ALL, the hyperdiploidy is restricted to lymphoid cells (Kasprzyk et al., 1999). However, the identification of leukemic subclones with unrelated DJ rearrangements (Stankovic et al., 2000), and of diagnostic cytogenetic abnormalities in lineage marker negative cells (Quijano et al., 1997) argues for involvement of more primitive cells in certain ALL patients. Moreover involvement of immature CD34+CD19- cells in two types of high-risk ALL, namely infant ALL with a translocation t(4;11) and Philadelphia chromosome-positive ALL (Hotfilder et al., 2005; Castor et al., 2005), highlights the heterogeneity of ALL.
Most importantly, there is a paucity of functional in vivo studies showing successful engraftment of ALL subpopulations in immune-deficient mice. The studies published to date have presented heterogeneous results. In two studies, only cells with an immature stem cell-like immunophenotype (either CD34+CD38- or CD34+CD19-) were able to engraft and re-initiate the leukemia in immune-deficient mice following intravenous injection (Cobaleda et al., 2000; Cox et al., 2004), while two more recent studies demonstrated engraftment of more mature CD19+ lymphoid blasts rather than immature CD19- cells (Castor et al., 2005; Hong et al., 2008).
These conflicting results indicate that key questions regarding leukemic stem cells in ALL remain unresolved. In which cell does childhood ALL arise (cell of origin)? Is there heterogeneity of stem cell involvement in ALL? What is the phenotype of the in vivo propagating leukemic stem cells? Is it a rare cell with a primitive immunophenotype, or do the majority of blasts retain some stemness? The aim of this study was to develop a more sensitive and consistent functional assay for self-renewing candidate ALL stem cell populations. Sorted blasts mirroring different stages of B cell maturation were able to fully re-constitute and maintain the human leukemia through serial transplantations. These populations also express a number of stage appropriate B cell developmental genes.
Results
Flow sorted ALL cells serially engraft NOD/scid mice following intrafemoral injection
To establish a sensitive in vivo assay for self-renewing leukemic stem cell populations, unmanipulated and flow sorted bone marrow cells from 13 children with ALL were transplanted via intrafemoral injection into NOD/scid mice and engrafting leukemias were serially transplanted into secondary, tertiary and quaternary recipients. Seven leukemias were classified as high-risk ALL and the remaining 6 leukemias were from standard-risk patients (see supplemental experimental procedures). Overall, cells from 6 out of 7 high-risk (86%) and 2 out of 6 standard-risk (33%) leukemias engrafted. The 8 engrafting leukemias were serially transplanted in 199 mice. Of the 185 mice which received at least the minimally engrafting cell dose of 2 × 103 cells (see below), 115 (62%) showed engraftment with more than 5% human leukemic blasts in the bone marrow and 112 mice (61%) developed overt leukemia with 20 - 100% blasts (mean = 89%, SD = 18%).
The leukemia in the mice recapitulates the original disease within the patient and remains stable through four serial transplantations
To confirm that our mouse model recapitulates the original disease a thorough morphological, flow cytometric and molecular analysis of the human leukemias in the mice was performed. Human leukemic engraftment was assessed and quantified by flow cytometric analysis of cells within the lymphocyte gate (scatter profile) expressing CD19, CD34 and CD45. A representative comparison of the initial and post transplantation blasts demonstrates a conserved immunophenotype (Figure S1). In all mice analyzed, the human cells had a B-cell precursor phenotype consistent with the original diagnosis. Consistent with the flow cytometry data, bone marrow from leukemic mice showed a massive infiltration of mainly L1 lymphoblasts. Both of these findings remained stable from primary to quaternary transplants (Figure 1). Engraftment of human cells also correlated with the development of splenomegaly, indicating that transplanted mice developed overt leukemia (Figure S2).
Figure 1. The morphology and immunophenotype of the leukemic blasts remain stable for four passages in the mice.

(A) Original patient #14 blast cell morphology and immunophenotype. (B-E) Bone marrow samples derived from mice serially transplanted with CD34+CD19+ cells from the patient shown in A. Blast cell morphology and immunophenotype resemble the original patient sample in all four consecutive transplantations. (scale bar = 10μm)
Engrafted human cells demonstrate high levels of leukemia-specific karyotypic or immunogenetic changes, indicating engraftment of leukemia and not normal human hematopoiesis
As engraftment of normal human myeloid cells is dependent on pre-transplant conditioning of the mice by irradiation (except for the transplantation of very high cell doses), co-engraftment of normal human hematopoietic cells was not expected in our non-irradiated recipient mice (Spiegel et al., 2004). This was confirmed by flow cytometry that failed to detect human B-lineage negative cells expressing the myeloid markers CD33 or CD15 (Figures S1). To molecularly confirm engraftment of leukemic rather than normal lymphoid progenitor cells, the proportion of leukemic cells within the different human subpopulations in the mice was quantified with FISH probes that detect the leukemia-specific aberrations. Twenty-one leukemic mice transplanted with samples containing the following chromosomal aberrations were selected for this analysis: t(4;11), t(11;19), dup(21q) and t(12;21). Human CD34+CD19-, CD34+CD19+ and CD34-CD19+ cell populations were purified from the marrow of engrafted mice by flow sorting these cells directly onto glass slides. The leukemia-specific chromosomal aberration could be detected in the majority of human cells at a mean frequency of 85% (SD = 12.0%) in each cell fraction (Table1 & Figure S3A). As the cells sorted directly onto the slides were not always nicely spread and sometimes slightly damaged, FISH analysis was likely to underestimate the percentage of aberrant cells.
Table 1. All analyzed engrafted subpopulations are leukemic.
High proportions of cells with leukemia-specific chromosomal aberrations were detected by FISH analysis in every engrafted subpopulation (mean = 85.4%, SD = 12.2%). Whenever possible, two specimens per engrafted mouse bone marrow were examined and shown data represent the mean values. (x = no data available)
| Patient | Mouse No. | Subpopulation and Dosage | Transplantation (Prim.-Quart.) | Final Engraftment Level (%) | FISH-positive human cells in the mouse (%) | ||
|---|---|---|---|---|---|---|---|
| CD34+CD19- | CD34+CD19+ | CD34-CD19+ | |||||
| #14 dup(21q) | 153 | 6.0 × 106 unsorted | primary | 96 | 74.1 (unsorted) | ||
| 177 | 2.0 × 105 CD34+CD19+ | secondary | 98 | 60 | 84 | x | |
| 179 | 2.0 × 105 CD34+CD19+ | secondary | 96 | 91 | 74 | 84 | |
| 229 | 2.0 × 104 CD34+CD19+ | tertiary | 97 | x | 86 | x | |
| 232 | 2.0 × 104 CD34+CD19+ | tertiary | 97 | x | 87 | x | |
| 249 | 1.5 × 104 CD34+CD19- | tertiary | 98 | x | 80 | x | |
| 213 | 3.0 × 104 CD34-CD19+ | tertiary | 97 | 77 | 43 | 72 | |
| 219 | 3.0 × 104 CD34-CD19+ | tertiary | 99 | 90 | 80 | x | |
| #1 t(4;11) | 51 | 1.0 × 105 CD34+CD19- | primary | 76 | 69 | 69 | x |
| 65 | 6.0 × 104 CD34+CD19+ | secondary | 97 | x | 75 | 65 | |
| #9 t(4;11) | x | Patient Sample | x | x | 100 | 98 | 99 |
| 235 | 3.0 × 104 CD34+CD19- | primary | 99 | 89 | 78 | 80 | |
| 236 | 2.0 × 105 CD34+CD19+ | primary | 99 | 88 | 88 | 90 | |
| 239 | 3.0 × 104 CD34+CD19- | primary | 93 | 99 | 97 | x | |
| 240 | 2.0 × 105 CD34+CD19+ | primary | 97 | x | 94 | 98 | |
| #15 t(11;19) | 162 | 11.5 × 106 unsorted | primary | 93 | 89 | 95 | 98 |
| 202 | 2.0 × 106 CD34+CD19+ | secondary | 100 | 98 | 94 | 99 | |
| 206 | 4.0 × 104 CD34-CD19+ | secondary | 96 | x | 87 | 97 | |
| 243
244 |
2.0 × 106 CD34+CD19+
2.0 × 104 CD34+CD19+ |
tertiary
tertiary |
98
98 |
x
x |
92
93 |
90
x |
|
| #866/06 t(12;21) | 167 | 2.0 × 105 CD34-CD19+ | secondary | 70 | x | 99 | 93 |
| 104 | 7.0 × 106 unsorted | primary | 34 | x | 98 | 93 | |
To confirm the leukemic nature of human engraftments from patient samples without a known chromosomal marker, we analyzed unsorted bone marrow of 26 leukemic mice by PCR to detect and quantify patient-specific clonal immunoglobulin heavy chain gene rearrangements. These rearrangements had already been established as a feasible minimal residual disease (MRD) marker within the patient bone marrow after initial diagnosis (#1075/04 - Vd2-Dd3, VH1.2-JH4b; #1002/05 - VH1.2-JH4b, DH2.15-JH4b). We detected high MRD loads in 26 leukemic mouse bone marrows transplanted with cells from the two patients. Therefore, consistent with the flow cytometric data, the molecular analysis detects human leukemic engraftment in the mice.
Sorted CD34+CD19-, CD34+CD19+ and CD34-CD19+ populations all contain leukemia-initiating cells
CD34+CD19-, CD34+CD19+ and CD34-CD19+ cells were purified from the primary patient sample or most often from engrafted primary or subsequent mice and serially transplanted into primary, secondary, tertiary and quaternary recipients to identify which population would contain leukemic stem cell activity. To our surprise, all three leukemic subpopulations had the capacity to engraft and reconstitute the leukemia in NOD/scid mice after intrafemoral transplantation. First, we looked at the engraftment of sorted subpopulations from the 2 patients with standard-risk ALL. One patient was diagnosed with ALL/t(12;21), an ALL subtype that is thought not to involve the CD34+CD19- compartment (Hotfilder et al., 2002; Castor et al., 2005). In concordance with these previous studies, CD19+ cells from the two standard-risk ALL patients were able to transfer the leukemia into secondary, tertiary and quaternary mice (Table 2A). Interestingly, this was independent of the expression of CD34. In contrast, 2 mice transplanted with CD34+CD19- cells showed no engraftment. A similar picture was seen in mice transplanted with cells from high-risk patients (Table 2B): CD19+ cells, irrespective of whether they expressed CD34, were able to transfer the leukemia into recipient mice. In addition, the more immature CD34+CD19-population was also shown to contain cells with leukemia-initiating capability at high frequency. Leukemia development after transplantation of the different subpopulations was a consistent and reproducible finding across the 115 mice which engrafted. The mean time-to-leukemia development was 11.7 weeks (SD 5.4 weeks), regardless of which population was transplanted (Figure S4A). At the point of sacrifice, all three populations had produced mean engraftment levels of >80% human cells (Figure S4B).
Table 2. Engraftment of populations from high-risk and standard-risk ALL, sorted using CD19/34 or CD19/20.
This table summarizes all 196 mice that were transplanted with purified ALL subpopulations from the 10 engrafting leukemias (patients #1, 9, 12, 14, 15, 862/02, 1075/04, 1002/05, L736 and L754) and with a minimal cell dose of 2 × 103 cells.
| Standard Risk ALL (n=3)
|
High Risk ALL (n=7)
|
||||
|---|---|---|---|---|---|
| Transplanted subpopulation | Transplanted mice | Engrafted mice | Transplanted subpopulation | Transplanted mice | Engrafted mice |
| CD34+CD19- | 2 | 0 (0%) | CD34+CD19- | 37 | 15 (40.5%) |
| CD34+CD19+ | 5 | 4 (80.0%) | CD34+CD19+ | 80 | 58 (72.5%) |
| CD34-CD19+ | 18 | 11 (61.1%) | CD34-CD19+ | 41 | 27 (65.9%) |
| CD19+CD20-/low | 3 | 1 (33.3%) | CD19+CD20-/low | 4 | 3 (75.0%) |
| CD19+CD20high | 2 | 1 (50.0%) | CD19+CD20high | 4 | 2 (50.0%) |
To confirm these surprising results, independent xenograft transplantation experiments were initiated at the Northern Institute for Cancer Research in Newcastle using ALL blasts sorted to be CD19+CD20-/low or CD19+CD20high. Eight primary and five secondary NOD/scid IL2γnull mice were transplanted with sorted blasts from two ALL patients (#L754 and #L736) and then assessed using diagnostic bone marrow punctures taken between 12 and 20 weeks, without being sacrificed (Table 2). Of 7 mice receiving 8 × 104 - 1 × 105 CD19+CD20-/low cells, 4 mice showed full engraftment with 7-34% human leukemic infiltrates, 2 showed low levels of human cells (4-5%) and one did not engraft. One engrafted mouse has been harvested and transplanted into secondary mice. Of 4 primary and 2 secondary mice transplanted with 8 × 104 - 1 × 105 CD19+CD20high cells, 3 have shown good engraftment (8-19% blasts), one shows low levels of engraftment (2.5% blasts) and two mice died at 10 weeks post transplant before engrafting (< 0.1% blasts). The engraftment of sorted populations again demonstrates recapitulation of the original patient immunophenotype (Figure S5). Ten of these 13 mice are still alive and appear in good health at the time of manuscript submission and we continue to monitor their engraftment. As both CD19+CD20-/low and the more mature CD19+CD20high blasts engraft, these data confirm our observation that leukemic blasts, mirroring different stages of B cell maturation, display leukemic stem cell activity.
Of the 185 mice in the original experiments, 21 were transplanted with cells directly sorted from the original diagnostic bone marrow sample (pts # 1, 9 and 12) (Table S1). Primary mice engrafted with CD34+CD19- (n = 2), CD34+CD19+ (n = 4) and CD34-CD19+ blasts (n = 3). Moreover, 8 additional NOD/scid IL2Rγnull mice were transplanted in Newcastle with 1 × 105 CD19+CD20-/low or CD19+CD20high blasts directly sorted from the original patient sample (patient #L754). Five of those primary mice had human leukemic blasts in the murine bone marrow at 12 weeks after transplantation (Figure S5). We are therefore confident that there was no difference in engraftment, whether the cells were sorted directly from the original patient sample or from the leukemia recovered from engrafted mice.
The intrafemoral NOD/scid assay is a highly sensitive and specific model for engraftment of sorted leukemic populations
To evaluate the sensitivity of our mouse model we performed a limiting dilution assay by transplanting CD34+CD19-, CD34+CD19+ and CD34-CD19+ cells from patient #12 in descending concentrations into NOD/scid mice. CD34+CD19- cells: 2 mice each transplanted with 1.0 × 103, 1.0 × 102 and 1.0 × 101 cells; CD34+CD19+ and CD34-CD19+ cells: 2 mice each transplanted with 1.0 × 104, 1.0 × 103 and 1.0 × 102 cells. None of the mice transplanted with CD34+CD19- cells and one mouse each transplanted with 1.0 × 104 CD34+CD19+ or CD34-CD19+ cells engrafted. Overall, 4 out of 7 mice (57%) transplanted with 2 − 7 × 103 CD34+CD19- cells and 3 out of 6 mice (50%) transplanted with 1.0 × 104 CD19+ cells engrafted. Thus a minimum cell dose of 2.0 × 103 cells was shown to be sufficient to re-establish the leukemia in recipient NOD/scid mice. This compares favorably with previously published xenograft models that required a minimum of 2.0 × 104 CD34+CD38- cells (Cobaleda et al., 2000), 5.0 × 104 CD34+CD19- cells (Cox et al., 2004) or 5.5 × 105 CD19+ cells (Castor et al., 2005) to engraft transplanted mice and confirms the higher efficacy of intrafemoral as compared with intravenous transplantation (Mazurier et al., 2003).
Sorted cells were re-analyzed by flow cytometry and the purities of the populations were 95% (SD = 2.5%; n = 9) for CD34+CD19- cells, 97% (SD = 3.5%; n = 58) for CD34+CD19+ cells and 96% (SD = 7.0%; n = 34) for CD34-CD19+ cells. Similarly, the sort purities of the CD19+CD20-/low or CD19+CD20high blasts were above 96%. Therefore, 1 × 104 CD34+CD19+ cells with an average purity of 97% may have contained 300 CD34+CD19- or CD34-CD19+ cells and 2 × 103 CD34+CD19- cells with an average purity of 95% only 100 CD19+ cells, well below the minimal cell number necessary for leukemic engraftment. Table S2 summarizes those engraftments where the maximum contaminating population was lower than the minimally engrafting cell dose on 2.0 × 103 cells, confirming that engraftment originates from the bulk population, and not contamination.
Flow sorted populations re-establish the complete immunophenotype of the original leukemia and self-renew
To compare the maturation and self-renewal potential of the different candidate stem cell populations, we analyzed the composition of the grafts in the mice as established by these subpopulations from high and standard risk patients (Figure 2). The data show that, independent of the immunophenotype of the transplanted population, the same pattern of surface marker expression as found in the original leukemia was reestablished in the bone marrow of leukemic mice.
Figure 2. Immunophenotypic composition of the leukemic grafts in the mice.

(A) High-risk ALL. The composition of the human grafts in respect to the presence of CD34+CD19- ■, CD34+CD19+
 and CD34-CD19+
and CD34-CD19+
 cells resembles that of the original leukemias shown in C. All three subpopulations were able to reconstitute the complete phenotype of the original leukemia. However, mice transplanted with the more mature CD34-CD19+ immunophenotype engrafted with a slightly lower level of CD34+CD19- cells (0.04 vs 0.2 and 0.3%; p = 0.022) and higher levels of CD34-CD19+ lymphoid cells (36 vs 6.3 and 7.4 %; p ≤ 0.001) as compared with mice transplanted with the other two cell fractions. (B) Standard-risk ALL. The composition of the graft was similar in mice that received CD34+CD19+ and CD34-CD19+ cells. Two mice transplanted with CD34+CD19- cells did not engraft. (C) Patient samples. Distribution of the three subpopulations in the original patient samples. Error bars show one SD (not calculated for 2 standard-risk patients).
cells resembles that of the original leukemias shown in C. All three subpopulations were able to reconstitute the complete phenotype of the original leukemia. However, mice transplanted with the more mature CD34-CD19+ immunophenotype engrafted with a slightly lower level of CD34+CD19- cells (0.04 vs 0.2 and 0.3%; p = 0.022) and higher levels of CD34-CD19+ lymphoid cells (36 vs 6.3 and 7.4 %; p ≤ 0.001) as compared with mice transplanted with the other two cell fractions. (B) Standard-risk ALL. The composition of the graft was similar in mice that received CD34+CD19+ and CD34-CD19+ cells. Two mice transplanted with CD34+CD19- cells did not engraft. (C) Patient samples. Distribution of the three subpopulations in the original patient samples. Error bars show one SD (not calculated for 2 standard-risk patients).
Another important question was whether all three populations retain the capacity to re-establish the complete leukemic phenotype in vivo over a series of sequential transplantations. Figure 3 shows a representative series of three transplantations, starting with primary, unsorted mononuclear cells derived from patient #1075/04, followed by subsequent transplantations of sorted populations. This demonstrates that in both primary and subsequent transplants, regardless of immunophenotype of the transplanted population, the human leukemia in the mice very closely recapitulates the immunophenotype in the original patient. All three subpopulations, i.e. CD34+CD19-, CD34+CD19+ and CD34-CD19+ cells, were able to engraft, to proliferate, to re-establish the complete phenotype and to maintain the leukemia (self-renewal) for at least four sequential transplantations, spanning a period of ≥ 12 months.
Figure 3. All three transplanted subpopulations can self-renew and serially transfer the leukemia in the mice.
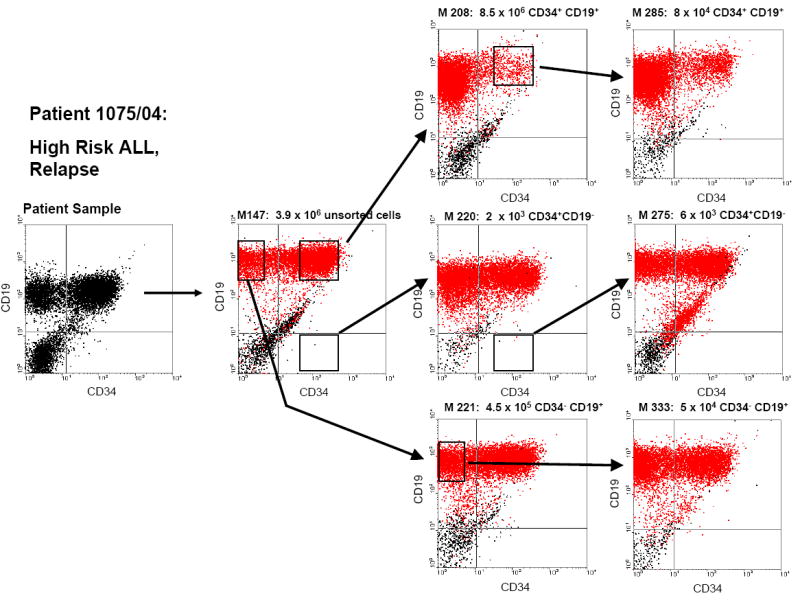
Each dot plot (except for the patient sample) represents one mouse engrafted with human leukemic cells. Sorting gates and arrows indicate the subpopulation each mouse received from its predecessor. Mouse number and the exact cell dose transplanted into that mouse are provided in the heading of each dot plot.
Sorted primary blasts retain the B precursor developmental programme and express immunoglobulin transcripts in a stage appropriate manner
Whilst B precursor populations expressing all investigated developmental surface markers are able to recapitulate the full leukemic phenotype, we wanted to know whether this is representative of an underlying biological difference. Primary patient samples were sorted by FACS to give blast populations expressing CD34+CD19+ and CD34-CD19+, with mean purity 96.8% (SD 1.98%, data not shown), for real-time RT-PCR analysis using a candidate gene approach. Figure 4 shows that, across 4 patient samples, the expression of transcription factors involved in the B precursor developmental programme mirrors that expected in normal B cell development. Principally, both populations express the early transcription factors E2A, EBF1, PAX5 and LEF1 whilst only the more mature CD34-CD19+ population shows a significant upregulation of IRF4, a transcription factor which has a role in making the immunoglobulin light chain loci accessible for recombination and expression in small pre-BII cells (Ma et al, 2006).
Figure 4. Primary patient CD34-CD19+ populations show significant upregulation of the small pre B cell specific transcription factor IRF4.
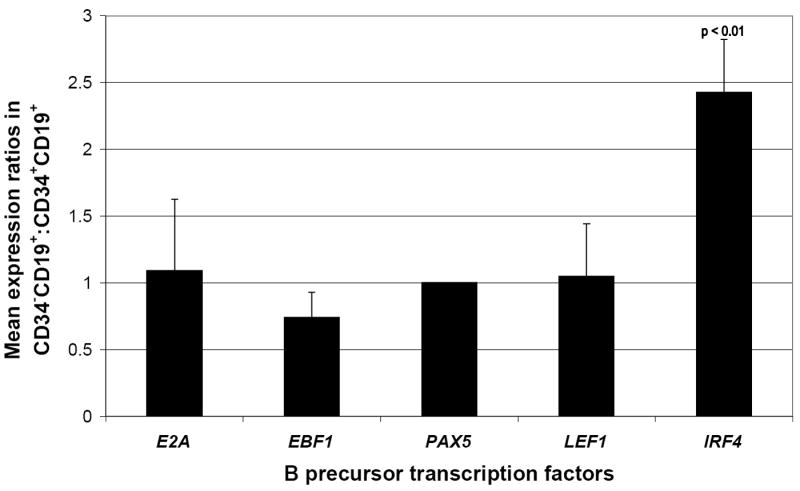
Each bar represents pooled quantitative PCR data from four (E2A n=3) patient bone marrow specimens taken at diagnosis and sorted into CD34+CD19+ and CD34-CD19+ blast populations. ΔΔCt values between CD34-CD19+ and CD34+CD19+ populations have been transformed and normalized within a patient specimen against the B cell specific transcription factor PAX5 (error bars show 1 SD). Expression of IRF4 shows significant upregulation in the more mature CD34-CD19+ population (p<0.01).
Furthermore, genome wide expression analysis has demonstrated that 5 of 13 genes which show a greater than 4 times upregulation in the CD34-CD19+ populations are immunoglobulin loci transcripts (Table 3). Additionally, CD20 and CD34 appropriately show up-regulation and down-regulation respectively in the more mature population.
Table 3. Microarray analysis of CD34+CD19+ and CD34-CD19+ primary blast populations.
Two patient bone marrow specimens taken at diagnosis and FACS sorted using CD34 and CD19 expression show an upregulation of greater than four times of the expression of immunoglobulin loci IgH, Igκ and Igλ. MS4A1 codes for the B cell differentiation marker CD20 and shows up-regulation. CD34 shows appropriate down-regulation in the CD34-CD19+ population.
| CD34-:CD34+expression ratio | |||
|---|---|---|---|
| Array ID | Transcript product | Patient L787 | Patient L812 |
| 228592_at | MS4A1 (CD20) | 11 | 92 |
| 224795_x_at | Immunoglobulin kappa locus | 14 | 33 |
| 230245_s_at | Hypothetical protein | 4 | 33 |
| 221671_x_at | Immunoglobulin kappa locus | 34 | 29 |
| 217022_s_at | Immunoglobulin heavy constant alpha 1 & 2 | 14 | 22 |
| 214677_x_at | Immunoglobulin lambda locus | 4 | 16 |
| 214836_x_at | Immunoglobulin kappa locus | 9 | 15 |
| 216834_at | Regulator of G protein signalling 1 | 10 | 7 |
| 237849_at | Transcribed locus | 5 | 6 |
| 233955_x_at | CXXC finger 5 | 6 | 5 |
| 208178_x_at | TRIO | 4 | 4 |
| 209013_x_at | TRIO | 4 | 4 |
| 1568983_a_at | cDNA clone | 4 | 4 |
|
| |||
| 209543_s_at | CD34 | 0.16 | 0.05 |
| 212002_at | Chr 1 open reading frame 144 | 0.24 | 0.18 |
| 243489_at | Transcribed locus | 0.04 | 0.18 |
Discussion
We have been able to characterize candidate human ALL stem cell populations by serial transplantation of flow sorted subpopulations in immune-deficient NOD/scid mice. Up to now, progress on understanding the hierarchy of childhood B-cell precursor ALL has been hampered by the lack of appropriate in vitro and in vivo models. In vitro, only a minority of ALL are able to survive and proliferate on stromal cell cultures (Nishigaki et al., 1997) and undergo rapid apoptosis. The few studies published to date showing successful engraftment of ex vivo manipulated subpopulations from B-cell precursor ALL gave conflicting results (Cobaleda et al., 2000; Cox et al., 2004; Castor et al., 2005; Hong et al., 2008). We, therefore, set out to develop a more sensitive and robust transplantation assay to define the cell populations that are able to maintain the ALL in vivo (leukemia-propagating stem cells).
As has been shown in previous studies (Lapidot et al., 1994; Mazurier et al., 2003; Hope et al., 2004; Castor et al., 2005) the bone marrow microenvironment appears to be sufficiently conserved between mice and men to allow the survival and proliferation of normal and leukemic human stem cell populations that cannot be maintained in vitro. However, clinically aggressive and relapsed leukemias grow better in immune-deficient mice than samples from standard-risk patients (Kamel-Reid et al., 1991; Uckun et al., 1998) and not every human leukemia is able to engraft and proliferate in mice (Bonnet et al., 1997). This may be due to the dependency of some leukemias on microenvironmental signals that are not active across species. In accordance with this experience, we saw that cells from 6 out of 7 high-risk (86%) but only 2 out of 6 standard-risk (33%) patients engrafted. Despite this, the NOD/scid mouse model represents the best available functional assay for candidate human leukemic stem cell populations, in which there appears to be only minimal pressure on the human cells to adapt to the murine microenvironment.
The intrafemoral NOD/scid serial transplantation assay described in the present study provides a highly sensitive and specific assay for transplantation of ALL stem cells. It has allowed us to achieve excellent rates of engraftment from a range of donor blast populations without preconditioning, thus avoiding co-engraftment of normal hematopoietic cells. Most importantly, the constant immunophenotype and reproduction of clinical disease over serial transplantations and many months, whether the primary transplant involved sorted or unsorted material, provides reassurance that this assay is highly representative of the biology of our target population.
The sequential expression of immunophenotypic markers of B cell differentiation has been well defined (Hystad et al., 2007; Noordzij et al., 2002). Many are present throughout much of normal B-precursor development, such as CD10, CD22 and CD38. Others can be used to define the “stages” of development of B-precursor cells to a greater degree: CD34 is expressed on hematopoietic stem cells. Down-regulation follows IgH locus rearrangement and the appearance of cytosolic IgM heavy chain; CD19 is a transcriptional target of PAX5, forming one of the earliest, B-lymphoid restricted markers. Surface expression correlates with rearrangement of VH-DJH (Zhang et al., 2006); CD20 expression is up-regulated in cells undergoing rearrangement of, or which have rearranged, the immunoglobulin light chain locus (Noordzij et al., 2002; van Zelm et al., 2005). We therefore chose to use these surface markers as a tool to identify and purify leukemic blasts mirroring subpopulations with differing maturation status.
Surprisingly, and in contrast to previous studies (Cobaleda et al., 2000; Cox et al., 2004; Castor et al., 2005), we were able to show that in high risk ALL each of the CD34+CD19-, CD34+CD19+, CD34-CD19+ populations was able to serially transplant the leukemia. In keeping with previous studies showing that standard risk ALL is restricted to the CD19+ fractions (Hotfilder et al., 2002; Castor et al., 2005; Hong et al., 2008), low levels of CD34+CD19- cells could only be purified from 2 leukemic mice and may not have constituted a real population. As expected these cells did not engraft subsequent mice. In addition to propagating leukemia, each of the transplanted fractions was able to recapitulate the complete disease immunophenotype, demonstrating the ability of blasts to move back and forth between the different populations. Again, this characteristic was maintained over serial transplantations. These results have been independently replicated by a separate series of experiments, at a second institute, using CD20 as an alternative marker of differentiation.
These findings raised the question of whether the immunophenotypically defined populations genuinely represent cells with differing biology, or whether the immunophenotype is a stochastic phenomenon. The expression patterns of sorted blasts indicate that the different populations display a pattern of B precursor transcription factors similar to that expected in normal B cell development, with upregulation of the late B precursor transcription factor IRF4 in the CD34-CD19+ population (for review see Matthias et al, 2005). Furthermore, initial expression array data show that the CD34-CD19+ population, which would be expected to contain small pre-BII cells in normal hematopoiesis, up-regulates expression of immunoglobulin loci. These findings suggest that the biology of our sorted populations may mirror, at least in part, that of their “normal” hematopoietic counterparts.
One potential concern was that serial transplantation leads to the selection of an in vivo cell line with an altered stem cell biology. However, mice transplanted with cells sorted directly from primary patient material demonstrated a consistent pattern of engraftment over multiple passages. All populations, defined using both CD34/CD19 (Table S1) and CD19/CD20 combinations, were able to re-establish the leukemia in mice. Thus, there appears to be no selection process that alters the human stem cell programme during serial transplantations in the mice.
The high sensitivity of this assay may also raise the question of the purity of the flow sorted cell populations. Transplantation of limiting dilutions with known purity has shown that, for each of the three populations, robust engraftment follows transplantation of very low cell doses at which the contaminant populations are too small to account for the leukemic engraftment. This argues strongly that engraftment originates from the intended populations, and not a minor, contaminating, stem cell population.
Although unlikely, our study does not formally rule out that there is a rare leukemic stem cell population present in all the different cell fractions. We have, however, previously shown that CD34+CD19-CD117+ cells do not appear to be part of the leukemic clone and CD19+CD117+ cells were not detectable (Baersch et al., 1999). In addition, the commonly used stem cell marker, CD133, is usually not expressed on the most mature leukemic CD34-CD19+CD20+ cells (Baersch et al., 1999). Thus, any putative rare ALL stem cell, present in all populations, could not be characterised by the established stem cell markers CD34, CD117 or CD133. The presence of a putative rare leukemic stem cell population therefore seems quite unlikely, also considering the fact that the different populations mirror normal B cell development. Thus, the observed differences between standard and high risk ALL, the consistency and reproducibility of the engraftment of all subpopulations, the sensitivity of the assay and the purity of the flow sorted cells, all indicate that these results reflect the biology of childhood ALL rather than being an artifact of our model system.
In a very elegant study, it has recently been shown that TEL/AML1 affects a CD19+ B-lymphoid progenitor cell that has partially rearranged its DJH locus (Hong et al 2008). The authors demonstrate that pre-leukemic stem cells reside only in an abnormal CD34+CD38-/lowCD19+ population, previously identified in TEL/AML-1 ALL (Castor et al, 2005). However, they did not test whether fully transformed, more mature leukemic CD34+/-CD38+CD19+ may also have acquired this stem cell phenotype. As our experiments only contain 2 engrafted mice from one patient with TEL/AML1-positive ALL, one transplanted with CD34+CD19+ and one with CD34-CD19+ cells, the question on the leukemia-propagating stem cell populations in this ALL subtype remains unresolved. It may be that complete leukemic transformation is required for B precursors to develop their full stem cell potential. Further studies are therefore required to clarify whether, unlike other leukemic subtypes transplanted in this study, TEL/AML-1-positive ALL displays stem cell activity only in a restricted population.
Our in vivo data are compatible with existing data showing a surprisingly high percentage of clonogenic cells in ALL samples (up to 20%) that are able to proliferate and self-renew in vitro on stromal cell cultures (Nishigaki et al., 1997). Similarly, in some murine leukemia models a very high frequency of 1:6 leukemic stem cells can be found (Krivtsov et al., 2006). These data are compatible with our observation that all subpopulations, i.e. cell populations with an immature stem cell-like or a more mature B-cell progenitor phenotype, contain leukemia-initiating cells. Due to the sensitivity of ALL blasts ex vivo and their tendency to undergo apoptosis, it is likely that this in vivo assay underestimates the frequency of leukemic stem cells. Therefore, despite the fact that after cell sorting, a minimum of 2 × 103 cells are needed to achieve successful leukemic engraftment, the actual frequency of leukemic stem cells may be higher, approaching that observed in the stromal cell assay.
In conclusion, we have shown that leukemic blasts expressing a range of B lineage differentiation markers are able to: engraft immunodeficient mice; reconstitute the complete leukemia phenotype (even cell populations expected to be less mature than the engrafting cells) and continue to engraft further mice with the same diverse populations over four serial transplantations and twelve months. Furthermore, we have early evidence in support of these populations being diverse at a transcriptome level and appearing to mirror their “normal” immunophenotypic counterparts. From these findings we have developed a model of B precursor ALL biology. Figure 5 shows both a schematic of normal B precursor differentiation (A) with the pattern of transcription factors, immunogenetic rearrangements, and immunophenotype, and (B), B precursor ALL behavior mirroring this “normal “ process. Whilst normal B precursor development follows a closely ordered pattern, dictated by the sequential expression of transcription factors, we show that ALL blasts are able to move back and forth within this narrow window of development, adopting biology mirroring the relevant normal populations. That all populations of lymphoid blasts should be able to initiate a self-renewal programme is perhaps not so unexpected given the ability of both large pre-BII cells and mature B cells to undergo clonal expansion. Thus, this model of malleability describes biological characteristics possessed by normal B precursor cells and therefore programmes which, presumably, remain relatively accessible following leukemic transformation.
Figure 5. Model of the malleability and self-renewal seen in B precursor ALL blasts.
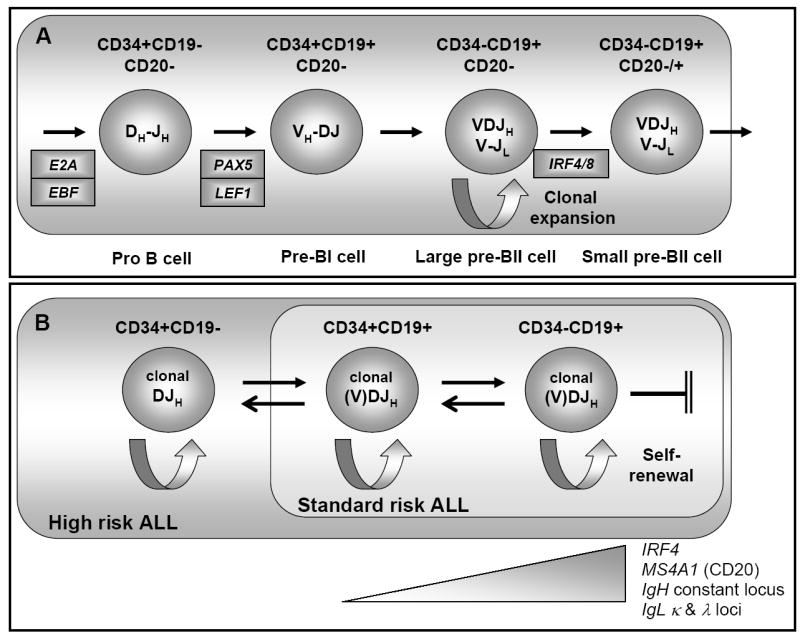
(A) Normal hematopoiesis. The development from CD34+CD19- early B progenitor, expressing transcription factors E2A and EBF1 through to CD34-CD19+CD20+/- small pre B cells expressing IRF4 and rearranging their immunoglobulin light chain loci. A period of clonal expansion in large pre B cells is shown. DJH – DH-JH segments rearranged, VH-DJH – rearrangement of the VH-DH segment, VDJH – heavy chain allele rearranged. (B) ALL blast malleability and self-renewal. The blast compartment for High and Low risk ALL is shown. Within these relatively narrow compartments, blasts are able to up-regulate and down-regulate components of the normal developmental programme, including accessing the self-renewal programme at all stages. There is however, a block to differentiation beyond the B precursor stage. Genes demonstrated as being upregulated are shown below the schema.
A key question will be to investigate the role of immature CD34+CD19- cells in drug resistance. Primitive hematopoietic stem cells are thought to protect themselves against DNA damage, e.g. by expression of certain transporter proteins (ABCB1/MDR-1/p-glycoprotein, ABCG2/BCRP) that pump a wide variety of xenobiotic toxins out of the cells. If primitive CD34+CD19- leukemic stem cells express the same stem cell-associated resistance mechanisms, this may contribute to the poor prognosis of ALL with involvement of the primitive stem cell compartment. Even if more mature ALL blasts retain the ability to self-renew, the most primitive CD34+CD19- leukemic stem cells may still provide a reservoir of drug-resistant stem cells that, unlike the more mature leukemic stem cells, survive chemotherapy and subsequently cause relapse.
This model of leukemia-propagating stem cells may prove instrumental in unraveling the exact developmental hierarchy within the different ALL subtypes, in understanding the mechanisms of in vivo chemotherapy resistance and for targeting new drugs against the cells that cause chemo resistance and relapse.
Experimental Procedures
Patient samples
Bone marrow specimens were taken from children with acute lymphoblastic leukemia either at diagnosis or at relapse (one patient #14). Patients were under the care of the Department of Pediatric Hematology and Oncology, University of Münster, Germany, The Erasmus MC Sophia Children’s Hospital, Rotterdam, The Netherlands or The Department of Paediatric Oncology, Royal Victoria Infirmary, Newcastle Upon Tyne, UK. Further patient information is given in the supplementary data.
The study was approved in Münster by the combined Ethics Committee of the Medical Faculty, University of Münster and the regional chamber of physicians (registration number: 31 VVormoor) and in Newcastle by the Newcastle & North Tyneside Ethics Committee 2 (REC reference number: 06/Q0906/79)
Antibody staining and Cell sorting
Bone marrow freshly recovered from transplanted mice and thawed mononuclear cells (1.0 × 107 to 1.6 × 108 cells) from diagnostic bone marrow samples were analyzed by flow cytometry. Cells were washed with RPMI 1640 medium containing 20% fetal calf serum (FCS) and were stained with saturating amounts of monoclonal antibodies against human cell surface antigens (Table S3) in a total volume of 0.5 –1.0mL for 20 minutes at 4°C. Flow cytometric analysis of bone marrow samples from patients and from engrafted mice was performed on a FACSCalibur flow cytometer (BD Bioscience, Heidelberg, Germany) using CellQuestPro software. For cell sorting experiments, cells were stained with anti-CD34 and anti-CD19 monoclonal antibodies (mAb) (Beckmann Coulter, Krefeld, Germany) and the cell concentration was adjusted to 107 cells per 1.5mL in RPMI 1640 containing 20% FCS. Cell sorting was performed on a FACSVantage SE cell sorter (BD Bioscience, Heidelberg, Germany). Figure S6 (A) shows the gates that were used to isolate our populations. An example of the re-analyses of the sorted populations is depicted in Figures S6 (B) – (D).
For experiments conducted in Newcastle, bone marrow samples were re-suspended in 0.2% PBSA solution (BSA; Sigma, DPBS; Cambrex) and stained with saturating amounts of monoclonal antibodies against human cell surface antigens (Table S3). In addition, samples were labeled with anti-muCD45, a pan-leukocyte marker, and anti-muTER119, an erythroid marker, antibodies, to gate out the majority of the murine background. Flow cytometric analysis was carried out on a FACSCanto II (BD Bioscience, Oxford, UK) using BDFACS Diva software. For cell sorting experiments in Newcastle samples were stained with anti-CD19 and anti-CD20 mAbs as well as mAbs to muCD45 and muTER119 and transplanted as described below (for sorting gates see Figure S5).
Animal model
NOD/scid mice (Jax® mice stock name: NOD.CB17-Prkdcscid/J mice) were provided by Prof. J. E. Dick (University of Toronto, Toronto, ON, Canada) (with permission from Leonard D. Shultz). The NOD/scid mice were bred and maintained in individually ventilated cages at the University of Münster as previously described (Baersch et al., 1997). Due to a complex immune deficiency involving B and T lymphocytes, natural killer cells, macrophages and the complement system, NOD/scid mice are almost completely unable to reject human xenotransplants (Shultz et al., 1995). In general, un-conditioned 8 - 16 week old NOD/scid mice were used for the experiments.
To further increase the sensitivity of the NOD/scid mouse model, during the course of this study we started to NK cell deplete the mice with an anti-CD122 mAb. This treatment was shown to completely eradicate any residual natural killer cell activity still present in these mice and for the first time allowed engraftment of short-term repopulating human cells (McKenzie et al., 2005). A total of 89 of 199 mice (45%) were treated intraperitoneally with 135μl of a 1.5 μg/μl anti-CD122 mAb preparation 18 - 24 hours before transplantation. The anti-CD122 mAb was purified from the hybridoma cell line, TM-β1 (gift of Dr T. Tanaka, Osaka University Medical Center, Osaka, Japan) (Tanaka et al., 1991). Purification was done using High Trap Protein G Columns (Amersham Pharmacia, Munich, Germany). The final preparation was quantified using the Bio-Rad Protein Assay (Bio-Rad, Munich, Germany) with bovine gamma globulin as standard.
For the additional experiments performed in Newcastle, NOD/scid IL2Rγnull mice (Jax® mice stock name: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) that completely lack any NK cell activity were used as recipients for human leukemic transplants (Shultz et al., 2005). All experimental manipulations with mice were performed under sterile conditions in a laminar flow hood.
Primary intrafemoral transplantations were usually performed with unsorted patient samples. For serial transplantations engrafted mice were humanely killed and both femurs and tibias were flushed with 1.5mL RPMI medium containing 20% FCS. Recovered bone marrow cells were stained with antibodies, sorted and transplanted. Overall, a total of 338 mice were transplanted with human leukemic cells (Münster cohort). 13 (4%) had to be excluded for technical reasons (mainly failed intrafemoral injections). 60 mice were transplanted with cells from the 5 non-engrafting leukemias (patients #2, 1365/01, 291/02, 384/05 and 804/05). 66 mice were transplanted with unseparated bone marrow cells from the 8 engrafting leukemias (patients #1, 9, 12, 14, 15, 862/02, 1075/04 and 1002/05). The remaining 199 mice were transplanted with ALL subpopulations purified from engrafted leukemias. These 199 mice, of which 185 received at least the minimally engrafting cell dose of 2 × 103 cells, represent the primary experimental group of this study. For a complete list of all 199 transplantations performed see Table S1.
The animal experiments in Münster were approved by the animal care committee of the local government (Bezirksregierung Münster, Aktenzeichen 50.0835.1.0 (G 25/2003)) and in Newcastle the transplantations were performed under Home Office license PPL60/3554.
Intrafemoral sample injection and bone marrow sampling
All leukemic cell samples were injected directly into the right femur of NOD/scid mice, as previously described (Mazurier et al, 2003). In brief, mice were anesthetized with an isofluorane-oxygen gas mix (2.5% isofluorane and 0.5 liters O2 per minute), and 3μg buprenorphine (Temgesic®, Essex Pharma, München, Germany) or Carprofen 50ng per 10g body weight (Rimadyl®, Pfizer, Surrey, UK) was subcutaneously injected as an analgesic. The right femur was punctured with a 25 or 27G needle and sample volumes of up to 30μL were subsequently injected with a 0.5 ml insulin syringe (27 or 30G). To sample bone marrow, primary femoral puncture with a 25 or 27G needle, was followed by bone marrow aspiration with a 25G needle into a syringe filled with 400μL of 10% heparin (Liquemin®, Hoffmann-La Roche AG, Mannheim, Germany) in PBS buffer.
Preparation of slides for fluorescence in-situ hybridization (FISH) analysis and May-Grünwald-Giemsa staining
Bone marrow cells for FISH analysis were prepared as previously described (Hotfilder et al., 2002 and 2005). In brief, 600 – 2000 cells from each population were sorted into 20 μl drops of PBS that were placed on a grease-free glass slide. Settling and adherence of the cells to the glass slide was facilitated by incubating the slides for 10 min in a moist chamber. Excess PBS was carefully removed with a paper towel. Ice-cold methanol/glacial acid (3:1, v/v) was used to fix the cells and the air-dried slides were analyzed by FISH. The histological analysis of patient samples and mouse bone marrow was done by a May-Grünwald/Giemsa staining of cytospin preparation.
FISH analysis for the TEL/AML1 gene fusions were performed on cytospin preparations of sorted cells using the LSI TEL/AML1 ES Dual Color Translocation Probe Set. In addition, for detection of MLL rearrangements the LSI MLL Dual Color Break Apart Rearrangement Probe was applied (both probes VYSIS, Abbott, Wiesbaden Germany). After a 2 minute pepsin treatment (50 μg pepsin/ml 0.01 M hydrochloric acid, pH 2.3, 37°C) slides were washed once in PBS for 3 minutes, fixed in 4% formaldehyde for 10 minutes at 4°C and after two further washes with PBS, dehydrated in a 70-100% ethanol series. Air-dried slides and probes were co-denatured for 8 minutes on a 78°C heat block and hybridized in a moist chamber at 37°C over night. Post hybridization washes and analysis of the cells were performed as described previously (Hotfilder at al., 2005).
Analysis of patient-specific clonal immunoglobulin heavy chain gene rearrangements
This analysis was done as previously described (van der Velden et al., 2007). DNA from engrafted mouse bone marrow was extracted with a DNA extraction kit (Qiagen, Hilden, Germany) (Verhagen et al.,1999). RQ-PCR was performed on the LightCycler (Roche Diagnostics, Heidelberg, Germany). For comparison and validation, initial diagnostic DNA samples of the patients were analyzed in parallel in the same experiment as positive controls. Identical crossing points of both materials confirmed specificity, origin and high tumor load of the engrafted human leukemic cells in the mice.
Transcription factor expression
Primary patient bone marrow specimens were defrosted, labeled and sorted as described above. After sorting, cells were immediately resuspended in RLT lysis buffer and homogenized using a 25G needle and syringe. Lysate was stored at -80°C prior to RNA extraction for quantitative PCR and Affymetrix expression analysis.
RNA extraction was performed using RNeasy® Plus Micro extraction columns (QIAGEN, Crawley, UK) according to the manufacturer’s instructions. cDNA synthesis was performed using RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas UK, York, UK) according to the manufacturer’s instructions. Starting reaction mixture contains 50ng total RNA and random hexamer primers.
cDNA was used immediately for quantitative RT PCR using the ABI Prism 7900HT (Applied Biosystems, Warrington, UK). Primers were designed using Primer Express® software V2.0 (Applied Biosystems) to produce transcript specific amplicons for the gene of interest. Primer sequences are given in the supplementary methods. Reactions contained 5μl Platinum® SYBR® qPCR Supermix-UDG with ROX (Invitrogen Ltd, Paisley, UK), 0.3μl primer mix at 10mM, 2.7μl RNase free water and 2μl cDNA. Experiments were run in triplicate. Data were analyzed using SDS 2.0 software (Applied Biosystems).
Affymetrix expression microarray analysis
RNA from sorted cell populations was taken forward for expression microarray analysis using the Affymetrix GeneChip® HG U133Plus 2.0 array (Gene Service Ltd, Cambridge, UK). Briefly, this process involves 2 cycles of cDNA synthesis and linear amplification. cDNA is transcribed in vitro prior to biotinylation of cRNA using the Affymetrix 2-cycle Target Labeling kit. Biotinylated cRNA is subsequently hybridized to the array chip.
Data were processed by GCRMA normalization using the Affy package supplied by Bioconductor (http://www.Bioconductor.org). Normalized data were analyzed for genes showing greater than four times difference in expression between CD34+CD19+ and CD34-CD19+ populations. The data are available online via ArrayExpress (accession number: E-MEXP-1522).
Supplementary Material
Acknowledgments
We thank Professor Andy Hall and Dr Olaf Heidenreich for their critical comments and Dr Karel Fiser for his assistance with normalizing expression array data. This work was supported by Deutsche José Carreras Leukämie-Stiftung e.V. grant R03/03 (to JV), by pump priming grants from the JGW Patterson Foundation and the North East Children’s Cancer Research Fund (to JV) and in part by NIH Cancer Core grant CA34196 (to LDS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Kelly PN, Dakic A, Nutt SL, Strasser A. Response to comment on “Tumour growth need not be driven by rare cancer stem cells”. Science. 2007;318:1722. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Baersch G, Möllers T, Hötte A, Dockhorn-Dworniczak B, Rübe C, Ritter J, Jürgens H, Vormoor J. Good engraftment of B-cell precursor ALL in NOD-SCID mice. Klin Padiatr. 1997;209:178–185. doi: 10.1055/s-2008-1043947. [DOI] [PubMed] [Google Scholar]
- Baersch G, Baumann M, Ritter J, Jürgens H, Vormoor J. Expression of AC133 and CD117 on candidate normal stem cell populations in childhood B-cell precursor acute lymphoblastic leukaemia. Br J Haematol. 1999;107:572–580. doi: 10.1046/j.1365-2141.1999.01746.x. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Castor A, Nilsson L, Astrand-Grundström I, Buitenhuis M, Ramirez C, Anderson K, Strömbeck B, Garwicz S, Békássy AN, Schmiegelow K, Lausen B, Hokland P, Lehmann S, Juliusson G, Johansson B, Jacobsen SE. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630–637. doi: 10.1038/nm1253. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer Stem Cells--Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Gutierrez-Cianca N, Perez-Losada J, Flores T, Garcia-Sanz R, Gonzalez M, Sanchez-Garcia I. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood. 2000;95:1007–1013. [PubMed] [Google Scholar]
- Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood. 2004;104:2919–2925. doi: 10.1182/blood-2004-03-0901. [DOI] [PubMed] [Google Scholar]
- Greaves MF. Stem cell origins of leukaemia and curability. Br J Cancer. 1993;67:413–423. doi: 10.1038/bjc.1993.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, Tsuzuki S, Greaves M, Enver T. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319:336–339. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- Hotfilder M, Rottgers S, Rosemann A, Jurgens H, Harbott J, Vormoor J. Immature CD34+CD19- progenitor/stem cells in TEL/AML1-positive acute lymphoblastic leukemia are genetically and functionally normal. Blood. 2002;100:640–646. doi: 10.1182/blood.v100.2.640. [DOI] [PubMed] [Google Scholar]
- Hotfilder M, Rottgers S, Rosemann A, Schrauder A, Schrappe M, Pieters R, Jurgens H, Harbott J, Vormoor J. Leukemic stem cells in childhood high-risk ALL/t(9;22) and t(4;11) are present in primitive lymphoid-restricted CD34+CD19-cells. Cancer Res. 2005;65:1442–1449. doi: 10.1158/0008-5472.CAN-04-1356. [DOI] [PubMed] [Google Scholar]
- Hystad ME, Myklebust JH, Bo TH, Sivertsen EA, Rian E, Forfang L, Munthe E, Rosenwald A, Chiorazzi M, Jonassen I, Staudt LM, Smeland EB. Characterization of early stages of human B cell development by gene expression profiling. J Immunol. 2007;179:3662–3671. doi: 10.4049/jimmunol.179.6.3662. [DOI] [PubMed] [Google Scholar]
- Kamel-Reid S, Letarte M, Doedens M, Greaves A, Murdoch B, Grunberger T, Lapidot T, Thorner P, Freedman MH, Phillips RA, Dick JE. Bone marrow from children in relapse with pre-B acute lymphoblastic leukemia proliferates and disseminates rapidly in SCID mice. Blood. 1991;78:2937–2981. [PubMed] [Google Scholar]
- Kasprzyk A, Harrison CJ, Secker-Walker LM. Investigation of clonal involvement of myeloid cells in Philadelphia-positive and high hyperdiploid acute lymphoblastic leukemia. Leukemia. 1999;13:2000–2006. doi: 10.1038/sj.leu.2401580. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumour growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Kennedy JA, Barabe F, Poeppl AG, Wang JCY, Dick JE. Comment on “Tumor growth need not be driven by rare cancer stem cells”. Science. 2007;318:1722. doi: 10.1126/science.1149590. [DOI] [PubMed] [Google Scholar]
- Krivstsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;17:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Ma S, Turetsky A, Trinh L, Lu R. IFN regulatory factor 4 and 8 promote Ig light chain κ locus activation in pre-B cell development. J Immunol. 2006;177:7898–7904. doi: 10.4049/jimmunol.177.11.7898. [DOI] [PubMed] [Google Scholar]
- Matthias P, Rolink AG. Transcriptional networks in developing and mature B cells. Nat Rev Immunol. 2005;5:497–508. doi: 10.1038/nri1633. [DOI] [PubMed] [Google Scholar]
- Mazurier F, Doedens M, Gan OI, Dick JE. Rapid myeloerythroid repopulation after intrafemoral transplantation of NOD-SCID mice reveals a new class of human stem cells. Nat Med. 2003;9:959–963. doi: 10.1038/nm886. [DOI] [PubMed] [Google Scholar]
- McKenzie JL, Gan OI, Doedens M, Dick JE. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into NOD/SCID recipients depleted of CD122+ cells. Blood. 2005;106:1259–1261. doi: 10.1182/blood-2005-03-1081. [DOI] [PubMed] [Google Scholar]
- Nishigaki H, Ito C, Manabe A, Kumagai M, Coustan-Smith E, Yanishevski Y, Behm FG, Raimondi SC, Pui CH, Campana D. Prevalence and growth characteristics of malignant stem cells in B-lineage acute lymphoblastic leukemia. Blood. 1997;89:3735–3744. [PubMed] [Google Scholar]
- Noordzij JG, de Bruin-Versteeg S, Comans-Bitter WM, Hartwig NG, Hendricks RW, de Groot R, van Dongen JJM. Composition of precursor B-cell compartment in bone marrow from patients with X-linked agammaglobulinaemia compared with healthy children. Pediatr Res. 2002;51:159–168. doi: 10.1203/00006450-200202000-00007. [DOI] [PubMed] [Google Scholar]
- Pui CH. Acute lymphoblastic leukemia in children. Curr Opin Oncol. 2000;12:3–12. doi: 10.1097/00001622-200001000-00002. [DOI] [PubMed] [Google Scholar]
- Quijano CA, Moore D, 2nd, Arthur D, Feusner J, Winter SS, Pallavicini MG. Cytogenetically aberrant cells are present in the CD34+CD33-38-19- marrow compartment in children with acute lymphoblastic leukemia. Leukemia. 1997;11:1508–1515. doi: 10.1038/sj.leu.2400754. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, Leiter EH. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–191. [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- Spiegel A, Kollet O, Peled A, Abel L, Nagler A, Bielorai B, Rechavi G, Vormoor J, Lapidot T. Unique SDF-1-induced activation of human precursor-B ALL cells as a result of altered CXCR4 expression and signaling. Blood. 2004;103:2900–2907. doi: 10.1182/blood-2003-06-1891. [DOI] [PubMed] [Google Scholar]
- Stankovic T, Weston V, McConville CM, Green E, Powell JE, Mann JR, Darbyshire PJ, Taylor AM. Clonal diversity of Ig and T-cell receptor gene rearrangements in childhood B-precursor acute lymphoblastic leukaemia. Leuk Lymphoma. 2000;36:213–224. doi: 10.3109/10428190009148843. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tsudo M, Karasuyama H, Kitamura F, Kono T, Hatakeyama M, Taniguchi T, Miyasaka M. A novel monoclonal antibody against murine IL-2 receptor beta-chain. Characterization of receptor expression in normal lymphoid cells and EL-4 cells. J Immunol. 1991;147:2222–2228. [PubMed] [Google Scholar]
- Uckun FM, Sather HN, Waurzyniak BJ, Sensel MG, Chelstrom L, Ek O, Sarquis MB, Nachman J, Bostrom B, Reaman GH, Gaynon PS. Prognostic significance of B-lineage leukemic cell growth in SCID mice: a Children’s Cancer Group Study. Leuk Lymphoma. 1998;30:503–514. doi: 10.3109/10428199809057563. [DOI] [PubMed] [Google Scholar]
- Van der Velden VHJ, Panzer-Grümayer ER, Cazzaniga G, Flohr T, Sutton R, Schrauder A, Basso G, Schrappe M, Wijkhuijs JM, Konrad M, Bartram CR, Masera G, Biondi A, van Dongen JJ. Optimization of PCR-based minimal residual disease diagnostics for childhood acute lymphoblastic leukemia in a multicenter setting. Leukemia. 2007;21:706–713. doi: 10.1038/sj.leu.2404535. [DOI] [PubMed] [Google Scholar]
- Van Hennik PB, de Koning AE, Ploemacher RE. Seeding efficiency of primitive human hematopoietic cells in nonobese diabetic/severe combined immune deficiency mice: implications for stem cell frequency assessment. Blood. 1999;94:3055–3061. [PubMed] [Google Scholar]
- Van Zelm MC, van der Burg M, de Ridder D, Barendregt BH, de Haas EFE, Reinders MJT, Lankester AC, Revesz T, Staal FJT, van Dongen JJM. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol. 2005;175:5912–5922. doi: 10.4049/jimmunol.175.9.5912. [DOI] [PubMed] [Google Scholar]
- Verhagen OJ, Wijkhuijs AJ, van der Sluijs-Gelling AJ, Szczepański T, van der Linden-Schrever BE, Pongers-Willemse MJ, van Wering ER, van Dongen JJ, van der Schoot CE. Suitable DNA isolation method for the detection of minimal residual disease by PCR techniques. Leukemia. 1999;13:1298–1299. doi: 10.1038/sj.leu.2401451. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Espinoza CR, Yu Z, Stephan R, He T, Williams GS, Burrows PD, Hagman J, Feeney AJ, Cooper MD. Transcription factor Pax5 (BSAP) transactivates the RAG-mediated VH-to-DJH rearrangement of immunoglobulin genes. Nat Immunol. 2006;7:616–624. doi: 10.1038/ni1339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.