

Abstract
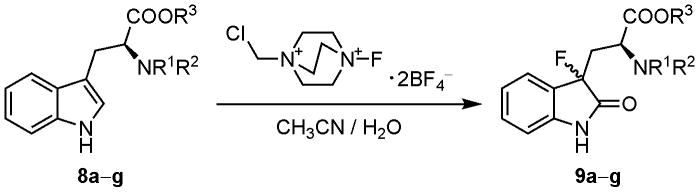
Oxidative fluorination of several protected tryptophans 8b–g with Selectfluor™ proceeded smoothly in aqueous media to give a diastereomeric mixture of the corresponding 3-fluorooxindoles 9b–g. Attempted deprotection of the 3-fluorooxindoles 9b–g under various conditions did not afford 3-(3-fluorooxindol-3-yl)-l-alanine (6). Reaction of the suitably protected tryptophan derivative 16 with Selectfluor™ produced the fluorinated product 17. Simultaneous cleavage of all protective groups of 17 under acidic conditions successfully gave the target compound 6 in excellent yield.
Keywords: Epimerization, Fluorooxindole, Hydroxyoxindole, Indole, Oxindole, Oxindolylalanine, Selectfluor™, Tryptophan
1. Introduction
The oxindole structure 1 is often found in natural products and is also formed during the metabolism of tryptophan (Fig. 1) [1,2]. Oxindoles have been recognized as probes for investigation of biological processes and leads for drug discovery [3,4]. Among many examples of oxindoles, 3-hydroxyoxindoles 2 have significance in that these frequently occur in natural products and are important structures in medicinal chemistry. Many 3-hydroxyoxindoles have useful biological and pharmacological activities [2,4]. For example, the compounds TMC-95A–D (3a–d) having a multifunctional 3-hydroxyoxindole structure inhibit the proteolytic activity of proteasome. This is a protease complex that is targeted in the design of new drugs for many diseases including cancer and autoimmune diseases [5]. 3-Oxindol-3-yl-l-alanine (4) is a tryptophan metabolite and was found to be a potent competitive inhibitor of both tryptophan synthase and tryptophanase [6]. It should be noted that the (2S,4S)- isomer of 3-(3-hydroxyoxindol-3-yl)-l-alanine (5) inhibits only tryptophan synthase and the (2S,4R)- isomer inhibits only tryptophanase [7]. Identification of this stereochemistry dependent-biological activity is possible with the 3-hydoxylated structure 5 since epimerization is blocked by the 3-substituent. Similar studies with other oxindoles such as the simple oxindole structure 4 is difficult because of epimerization at the C-4 stereogenic center [6,7].
Fig. 1.

To provide tools for investigating the relationship between biological activity and absolute configuration of epimerizable substrates, we have been studying the design, synthesis, and biological evaluation of chiral fluorinated bioorganic molecules that possess a fluorine atom at the stereogenic center [8]. Since replacement of a hydrogen of a prototype molecule with a fluorine results in minimal steric alterations, many fluorinated biomolecules and medicinal agents interact with recognition sites of enzyme and receptors in a manner similar to the fluorine-free molecules [9]. This adds to the value of these probes of stereochemistry vs. reactivity. Of course, introduction of fluorine atoms into bioactive molecules often brings about enhanced, additional and/or altered biological activities. In one example, we earlier replaced the labile proton at the stereogenic center of the epimerizable biomolecule thalidomide [10] with a fluorine atom to give a non-epimerizable analog [8a]. Substitution of a hydroxyl group of prototype molecules with fluorine can also provide the isosteric analogs because of similar electronic character and ability as hydrogen bond acceptor of fluorine to hydroxyl group [9]. In addition, apart from a hydroxyl group, a fluorine moiety is not subjected to further metabolization. For example, Prestwich et al. reported the substitution of one hydroxyl group of lysophosphatidic acids (LPA, 1- or 2-acyl-sn-glycerol 3-phosphate) with fluorine in order to prevent the acyl migration and evaluate the inherent biological activities of LPA under the acyl migration-free conditions [11].
As a part of our synthetic studies of chiral fluorinated bioorganic molecules, we earlier attempted the synthesis of 3-(3-fluorooxindol-3-yl)-l-alanine (6). The non-epimerizable compound 6 should be a suitable model to study the relationship between the stereochemistry and the biological activities of 4. Introduction of fluorinated amino acids such as 6 into peptides often changes the original conformation and biological properties [12]. Moreover, clinical application of BMS-204532 (Maxipost, 7) as a potassium channel opener [13] further encouraged us to pursue the synthesis the fluorinated tryptophan analog 6 as a potential building block for drug development.
2. Results and discussion
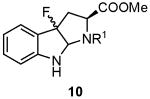
We have previously reported the synthesis of 3-fluorooxindole 9a from Nα-acetyl-l-tryptophan methyl ester (8a) by electrophilic fluorination with Selectfluor™ (Table 1, entry 1) [14]. However, we were unable to find conditions to convert 9a to the free amino acid 6. We report here the results of fluorination of oxindole substrates 8 with varying protecting groups on the fluorination reaction as well as defining substrates that allow facile deprotection to the amino acid (Table 1). In initial attempts, treatment of Nα-(tert-butoxycarbonyl)-l-tryptophan methyl ester (8b) with 3 equiv of Selectfluor™ [15] in acetonitrile/water (1/1) produced a diastereomeric mixture of the corresponding 3-fluorooxindole 9b (entry 2). However, the yield of 9b was quite low. Fluorination of the other tryptophan derivatives 8c and 8d having the same urethane protective groups gave the corresponding 3-fluorooxindoles 9c and 9d, again in rather low yields (entries 3 and 4). It should be noted that substantial amounts of the 3a-fluoropyrrolo[2.3-b]indole derivatives 10 were formed as side products during the fluorination of 8b-d. The formation of the cyclized compounds 10 was presumably due to the rather nucleophilic character of the Nb-acyloxy moiety of 8b–d compared to the Nb-acyl moiety of 8a. Indeed, Hino et al. reported that such cyclization occurs readily during electrophilic attack at the 3-position of Nb-alkoxycarbonyl protected tryptamines [16].
Table 1.
Synthesis of 3-fluorooxindoles 9a–g from protected tryptophans 8a–ga
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | indole 8 | R1 | R2 | R3 | 3-fluorooxindole 9 | yield (%) | |
| 1 | 8ab | Ac | H | Me | 9a | 70 |
|
| 2 | 8b | Boc | H | Me | 9b | 15 | |
| 3 | 8c | Cbz | H | Me | 9c | 40 | |
| 4 | 8d | Fmoc | H | Me | 9d | 42 | |
| 5 | 8e | CF3CO | H | Me | 9e | 70 | |
| 6 | 8f | Ac | H | But | 9f | 53 | |
| 7 | 8g | Boc | Boc | Me | 9g | 71 | |
Experimental conditions: Three equiv of Selectfluor™ was added to a solution of 8 in CH3CN / H2O (= 1/1) and the mixture was stirred at room temperature overnight.
Based on these results, we explored the use of the tryptophans having Nb-acyl protective groups, such as acetyl or trifluoroacetyl, which would be less nucleophilic than Nb-acyloxy groups. Indeed, when Nb-acyl protected tryptophans 8e and 8f were submitted to the same fluorination procedure, 3-fluorooxindoles 9e and 9f were obtained in 70% and 53% yields, respectively (entries 5 and 6), without detectable formation of 10. The tryptophan derivative 8g with the α-amino group being fully protected was also fluorinated to give the corresponding 3-fluorooxindole 9g in good yield (entry 7).
Although we have succeeded in the fluorination of 8b–g, stereoselectivity of the reaction was rather poor with the diastereomeric excess of 4% to 46%. Separation of the diastereomers of 3-fluorooxindoles 9d–g was carried out readily using silica gel column chromatography. In order to determine the absolute configurations, we attempted to get single crystals of 9d,e,g. However, no crystals suitable for X-ray analysis were obtained.
We then examined stepwise deprotection of 3-fluorooxindoles 9. Saponification of the diastereomeric mixture of 9b and 9c in aqueous NaOH/MeOH gave the corresponding carboxylic acids 11b and 11c in 72% and 77% yields, respectively (Table 2, entries 1 and 2). However, reaction of 9g under the same conditions led to decomposition (entry 3). This may result from lactam ring cleavage that occurs instead of the hydrolysis of the ester moiety, the reactivity of which would be lowered due to the sterically hindered neighboring Nα,Nα-di-tert-butoxycarbonylamino group. Treatment of 9f with HBr/AcOH also produced the corresponding carboxylic acid 11f in 48% yield (entry 4).
Table 2.
Deprotection of the carboxyl groups of fluorooxindoles 9b,c,f,g
 | ||||||
|---|---|---|---|---|---|---|
| entry | 3-fluorooxindole 9 | R1 | R2 | R3 | conditions | product (yield) |
| 1 | 9ba | Boc | H | Me | 0.2N NaOH/MeOH | 11b (72%) |
| 2 | 9ca | Cbz | H | Me | 0.2N NaOH/MeOH | 11c (77%) |
| 3 | 9ga | Boc | Boc | Me | 0.2N NaOH/MeOH | decomposition |
| 4 | 9fb | Ac | H | But | HBr/AcOH | 11f (48%) |
Diastereomeric mixture was used.
Less polar isomer was used.
Having established conditions to achieve ester hydrolysis, we then addressed the final stage of the synthesis, i.e., Nα-deprotection of 11b,c and 11f thus prepared. Although various conditions were applied for these compounds, we encountered difficulty in isolation of the target molecule 6 from the organic and inorganic salts mixture. Attempted simultaneous removal of all protecting groups of 9d and 9e under saponificative conditions (piperidine and/or potassium carbonate) were unsuccessful.
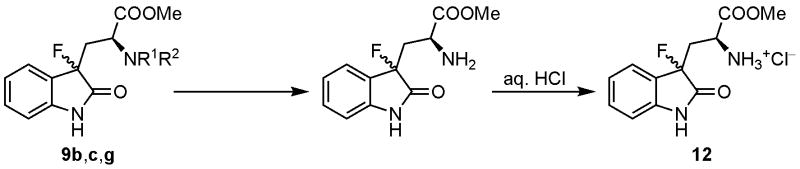
Faced with theses difficulties, we also attempted the alternative strategy that involves initial Nα-deprotection followed by ester hydrolysis. Removal of the Cbz groups of 9c occurred under hydrogenative or acidic conditions to produce the desired compound 12 although as mixtures with some inseparable products (Table 3, entries 1,2). N-Deprotection of 9d and 9e under basic conditions (piperidine and potassium carbonate) gave a complicated mixture of mostly decomposition products. Finally, treatment of 9b,g with HBr/AcOH gave the corresponding free amine, which was successfully isolated as HCl salt 12 in excellent yield (entries 3,4). However, in spite of extensive efforts, hydrolysis of the methyl ester 12 under acidic conditions either did not proceed or gave decomposition products under drastic conditions. Saponification of 12 did not yield the desired product 6, instead producing mixtures of non-fluorinated compounds.
Table 3.
Deprotection of the α-amino groups of fluorooxindoles 9b,c,g
 | |||||
|---|---|---|---|---|---|
| entry | 3-fluorooxindole 9 | R1 | R2 | conditions | yield (%) |
| 1 | 9ca | Cbz | H | HBr/AcOH | mixtureb |
| 2 | 9ca | Cbz | H | Pd-C, HCOO–NH4+, MeOH | mixtureb |
| 3 | 9ba | Boc | H | HBr/AcOH | 99% |
| 4 | 9ga | Boc | Boc | HBr/AcOH | 94% |
Diastereomeric mixture was used.
Inseparable mixture with unidentified products.
After these extensive investigations and taking into consideration all aspects of reaction conditions and isolation procedures, we eventually focused on Nα,Nα-di-(tert-butoxycarbonyl)-l-tryptophan tert-butyl ester (17) as a likely suitable precursor to the target structure. Condensation of Nα-(tert-butoxycarbonyl)-l-tryptophan (13) with tert-butanol in the presence of DCC (N,N′-dicyclohexylcarbodiimide) gave tert-butyl ester 14 in 43% yield with ee of 46% (Scheme 1). Protection of the indole nitrogen of 14 with benzyl chloroformate [17] followed by treatment with di-tert-butyl carbonate gave the fully protected tryptophan derivative 15 in moderate yield. Catalytic hydrogenation of 15 gave the fluorination precursor 16 in good yield. Compound 16 was then allowed to react with Selectfluor™ in the usual manner to produce a diastereomeric mixture of the corresponding fluorooxindole 17 in 51% yield. Finally, treatment of 17 with HBr/AcOH successfully produced the target compound 6 as HBr salt in excellent yield, although as a mixture of diastereomers.
Scheme 1.

Synthesis of 6
3. Conclusion
We have synthesized 3-(3-fluorooxindol-3-yl)-l-alanine (6) by fluorination of the suitably protected tryptophan derivative 16 with Selectfluor™ followed by simultaneous deprotection under acidic conditions. Having available this new fluorinated amino acid in unprotected form will allow several new applications. In order to evaluate the potential of 6 as a biological tool and precursor for drug development, diastereomeric and enantiomeric separations of 6 are currently underway. Use of the strategy presented here to prepare derivatives suitable for peptide incorporation will also be studied.
4. Experimental
4.1 General
Melting points were measured with a Yanaco micro melting point apparatus and are uncorrected. Spectroscopic measurements were carried out with the following instruments: optical rotations, JASCO DIP-1000 digital polarimeter; IR spectra, JEOL FT/IR-460Plus; mass spectra (MS), JEOL JMS-GCmate; high resolution mass spectra (HRMS), JEOL JMS-AX 505; 1H NMR spectra, JEOL ECX-400P (400 MHz) in CDCl3 or CD3OD with TMS (= 0.00 ppm) as an internal standard; 19F NMR spectra, JEOL ECX-400P (376 MHz) in CDCl3 or CD3OD with CFCl3 (= 0.00 ppm) as an internal standard. Column chromatography and thin layer chromatography were performed on Kanto chemical silica gel 60N (0.040–0.050 mm) or Merck 9385 silica gel 60 (0.040–0.063 mm) and on Merck 5715, respectively.
4.2 General procedure for the synthesis of the protected 3-(3-fluorooxindol-3-yl)alanine 9
Selectfluor™ (755 mg, 2.13 mmol) was added to a solution of Nα-trifluoroacetyl-l-tryptophan methyl ester 8e (223 mg, 0.710 mmol) in a mixture of acetonitrile and water (1:1, 14 mL) at room temperature. After stirring overnight, the mixture was concentrated and extracted with EtOAc. The organic layer was then dried over Na2SO4 and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluent; hexane/CHCl3/EtOAc = 5/1/1) to give the less polar isomer (80.5 mg, 0.231 mmol, 33%) and the more polar isomer (90.3 mg, 0.259 mmol, 37%) of (2S)-2-(trifluoroacetylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid methyl ester (9e). Diastereomeric excess (de) was determined to be 12% from the 1H NMR of crude mixture.: Less polar isomer: colorless solid; mp 122-123 °C; [α]D28 −12.2 (c 1.0, CHCl3); IR (KBr) v 3291, 1755, 1737, 1703, 1628 cm-1; 1H NMR (400 MHz, CDCl3) δ 2.56 (1H, ddd, J = 24.2, 15.1, 4.1 Hz), 3.05 (1H, dt, J = 15.1, 6.9 Hz), 3.81 (3H, s), 4.77 (1H, ddd, J = 8.2, 6.9, 4.1 Hz), 6.91 (1H, d, J = 7.8 Hz), 7.17 (1H, dd, J = 7.8, 7.3 Hz), 7.39 (1H, ddt, J = 7.8, 2.3, 1.4 Hz), 7.44 (1H, d, J = 7.3 Hz), 8.10 (1H, br s); 19F NMR (376 MHz, CDCl3) δ −76.48 (3F, s), −156.16 (1F, d, J = 23.8 Hz); MS (EI) m/z: 348 (M+), 289 (M+–COOMe), 269 (M+–COOMe–HF); HRMS (EI) calcd for C14H12F4N2O4 (M+): 348.0733; found 348.0714. More polar isomer: colorless solid; mp 129–130 °C; [α]D28 +15.8 (c 1.1, CHCl3); IR (KBr) 3388, 3301, 1754, 1726, 1710, 1625 cm-1; 1H NMR (400 MHz CDCl3) δ 2.64 (1H, ddd, J = 22.4, 15.1, 7.3 Hz), 2.90 (1H, dt, J = 15.1, 5.0 Hz), 3.72 (3H, s), 4.87 (1H, dt, J = 7.3, 5.0 Hz), 6.91 (1H, d, J = 7.8 Hz), 7.14 (1H, dd, J = 7.8, 7.3 Hz), 7.38 (1H, ddt, J = 7.8, 1.8, 0.9 Hz), 7.44 (1H, d, J = 7.3 Hz), 7.65 (1H, br d, J = 6.9 Hz), 7.92 (1H, br s); 19F NMR (376 MHz, CDCl3) δ −76.54 (3F, s), −153.89 (1F, dd, J = 22.4, 15.1 Hz); MS (EI) m/z: 348 (M+), 289 (M+–COOMe), 269 (M+–COOMe–HF); HRMS (EI) calcd for C14H12F4N2O4 (M+): 348.0733; found 348.0705.
4.2.1 (2S)-2-(tert-Butoxycarbonylamino)-3-(3fluoro-2-oxoindoline-3-yl)propionic acid methyl ester (9b)
Yield: 15% (as a diastereomeric mixture). De was determined to be 4% from the 1H NMR of crude mixture.: pale yellow oil; IR (neat) v 3246, 1736, 1718, 1697, 1686, 1624 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.37 (9H, s), 1.41 (9H, s), 2.57–2.79 (4H, m), 3.66 (3H, s), 3.71 (3H, s), 4.27 (1H, m), 4.53 (1H, dd, J = 12.8, 7.8 Hz), 5.14 (1H, br d, J = 7.8 Hz), 5.25 (1H, br d, J = 7.8 Hz), 6.88 (1H, d, J = 7.8 Hz), 6.90 (1H, d, J = 7.8 Hz), 7.11 (1H, t, J = 7.3 Hz), 7.13 (1H, t, J = 7.3 Hz), 7.34 (1H, t, J = 7.8 Hz), 7.36 (1H, t, J = 7.8 Hz), 7.45–7.48 (2H, m), 8.06 (1H, br s), 8.18 (1H, br s); 19F NMR (376 MHz, CDCl3) δ −152.05 (1/6F, br s), −152.30 (5/6F, br s), −152.55 (5/6F, br t, J = 14.7 Hz), −152.69 (1/6F, br s); MS (EI) m/z: 352 (M+), 296 (M+–C4H8).
4.2.2 (2S)-2-(Benzyloxycarbonylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid methyl ester (9c)
Yield = 40% (as a diastereomeric mixture). De was determined to be 16% from the 1H NMR of crude mixture.: pale yellow oil; IR (neat) v 3310, 1739, 1624 cm-1; 1H NMR (400 MHz, CDCl3) δ 2.58–2.85 (4H, m), 3.66 (3H, s), 3.72 (3H, s), 4.26 (1H, dt, J = 9.6, 4.1 Hz), 4.59 (1H, dt, J = 7.8, 5.0 Hz), 4.94 (1H, d, J = 12.4 Hz), 5.03 (1H, d, J = 12.4 Hz), 5.05 (1H, d, J = 12.4 Hz), 5.09 (1H, d, J = 12.4 Hz), 5.39 (1H, br d, J = 9.2 Hz), 5.52 (1H, br d, J = 8.2 Hz), 6.65 (1H, d, J = 7.8 Hz), 6.85 (1H, d, J = 7.8 Hz), 7.09 (1H, t, J = 7.8 Hz), 7.11 (1H, m), 7.26–7.45 (14H, m), 7.81 (1H, br s), 7.98 (1H, br s); 19F NMR (376 MHz, CDCl3) δ −152.11 (1F, t, J = 16.9 Hz), −152.39 (1F, br t, J = 10.9 Hz); MS (EI) m/z: 386 (M+), 327 (M+–COOMe).
4.2.3 (2S)-2-(9-Fluorenylmethoxycarbonylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid methyl ester (9d)
De was determined to be 8% from the 1H NMR of crude mixture.
Less polar isomer: yield = 16%; pale pink solid; mp 81–82 °C; [α]D29 −26.2 (c 1.0, CHCl3); IR (KBr) v 3298, 2954, 1739, 1624 cm-1; 1H NMR (400 MHz, CDCl3) δ 2.71–2.75 (2H, m), 3.73 (3H, s), 4.18 (1H, br t, J = 6.4 Hz), 4.28 (1H, br q, J = 7.8 Hz), 4.32 (1H, br s), 4.33 (1H, br s), 5.39 (1H, br d, J = 9.2 Hz), 6.77 (1H, d, J = 7.8 Hz), 7.11 (1H, br t, J = 7.3 Hz), 7.12 (1H, br s), 7.30–7.44 (6H, m), 7.59 (1H, d, J = 7.8 Hz), 7.62 (1H, d, J = 7.8 Hz), 7.76–7.81 (2H, m); 19F NMR (376 MHz, CDCl3) δ −152.21 (1F, br t, J = 11.7 Hz); MS (EI) m/z: 474 (M+), 456 (M+–H2O); HRMS (EI) calcd for C27H23FN2O5 (M+): 474.1591; found 474.1629.
More polar isomer: yield = 26%; pale yellow solid; mp 80–81 °C; [α]D29 −41.4 (c 1.1, CHCl3); IR (KBr) 3307, 2954, 1742, 1625 cm-1; 1H NMR (400 MHz CDCl3) δ 2.64 (1H, ddd, J = 16.9, 15.1, 7.8 Hz), 2.82 (1H, ddd, J = 17.4, 15.1, 4.6 Hz), 3.69 (3H, s), 4.19 (1H, br t, J = 6.9 Hz), 4.32 (1H, br s), 4.33 (1H, br s), 4.61 (1H, ddd, J = 7.8, 7.3, 4.6 Hz), 5.55 (1H, br d, J = 8.3 Hz), 6.85 (1H, d, J = 7.3 Hz), 7.07 (1H, t, J = 7.3 Hz), 7.28–7.46 (7H, m), 7.58 (2H, d, J = 6.9 Hz), 7.77 (2H, d, J = 7.3 Hz); 19F NMR (376 MHz, CDCl3) δ −152.30 (1F, t, J = 17.1 Hz); MS (EI) m/z: 456 (M+–H2O); HRMS (EI) calcd for C27H23FN2O5 (M+): 474.1591; found 474.1618.
4.2.4 (2S)-2-(Acetylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid tert-butyl ester (9f)
De was determined to be 46% from the 1H NMR of crude mixture.
Less polar isomer: yield = 35%; pale red oil; [α]D28 −13.4 (c 1.0, CHCl3); IR (neat) v 3284, 2981, 1739, 1658, 1625 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.44 (9H, s), 1.83 (3H, s), 2.53 (1H, ddd, J = 16.0, 14.6, 9.2 Hz), 2.68 (1H, ddd, J = 20.1, 14.6, 4.1 Hz), 4.72 (1H, ddd, J = 9.2, 7.8, 4.1 Hz), 6.11 (1H, br d, J = 7.8 Hz), 6.88 (1H, d, J = 7.8 Hz), 7.11 (1H, t, J = 7.8 Hz), 7.33 (1H, ddt, J = 7.8, 1.8, 1.4 Hz), 7.46 (1H, d, J = 7.8 Hz), 7.84 (2/3H, br s), 7.95 (1/3H, br s); 19F NMR (376 MHz, CDCl3) δ −152.43 (1F, t, J = 18.0 Hz); MS (EI) m/z: 336 (M+), 280 (M+–C4H8), 235 (M+–COOBut); HRMS (EI) calcd for C17H21FN2O4 (M+): 336.1485; found 336.1460.
More polar isomer: yield = 18%; pale red oil; [α]D29 −4.4 (c 1.0, CHCl3); IR (neat) v 3285, 2979, 1736, 1660, 1624 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.48 (9H, s), 1.95 (3H, s), 2.67 (1H, d, J = 12.8 Hz), 2.68 (1H, d, J = 12.8 Hz), 4.43 (1H, dt, J = 7.6, 6.9 Hz), 6.30 (1H, br d, J = 9.2 Hz), 6.87 (1H, d, J = 7.8 Hz), 7.13 (1H, dd, J = 7.8, 7.3 Hz), 7.35 (1H, t, J = 7.8 Hz), 7.45 (1H, d, J = 7.3 Hz), 8.23 (1H, br s); 19F NMR (376 MHz, CDCl3) δ −151.97 (1F, t, J = 13.0 Hz); MS (EI) m/z: 336 (M+), 280 (M+–C4H8), 235 (M+–COOBut); HRMS (EI) calcd for C17H21FN2O4 (M+): 336.1485; found 336.1493.
4.2.5 (2S)-2-[Bis(tert-butoxycarbonyl)amino]-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid methylester (9g)
Yield = 71% (as a diastereomeric mixture). De was determined to be 12% from the 1H NMR of crude mixture. Each diastereomer gave the following data after partial separation.
Less polar isomer: colorless solid; mp 151–152 °C; [α]D29 −62.1 (c 1.0, CHCl3); IR (KBr) v 3230, 2983, 1742, 1733, 1702, 1630 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.46 (18H, s), 2.90 (1H, ddd, J = 15.1, 11.0, 9.6 Hz), 3.02 (1H, ddd, J = 21.3, 15.1, 3.2 Hz), 3.70 (3H, s), 5.23 (1H, dd, J = 9.6, 3.2 Hz), 6.85 (1H, d, J = 7.8 Hz), 7.08 (1H, t, J = 7.8 Hz), 7.31 (1H, t, J = 7.8 Hz), 7.52 (1H, br s), 7.53 (1H, d, J = 7.8 Hz); 19F NMR (376 MHz, CDCl3) δ −153.15 (1F, br m); MS (EI) m/z: 452 (M+), 396 (M+–C4H8); HRMS (EI) calcd for C22H29FN2O7 (M+): 452.1959; found 452.1985.
More polar isomer: colorless solid; mp 140–141 °C; [α]D29 −39.7 (c 1.0, CHCl3); IR (KBr) v 3275, 2985, 1777, 1739, 1626 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.47 (18H, s), 2.90 (1H, dt, J = 15.1, 8.7 Hz), 3.08 (1H, ddd, J = 21.6, 15.1, 3.7 Hz), 3.69 (3H, s), 5.24 (1H, dd, J = 8.7, 3.7 Hz), 6.86 (1H, d, J = 7.8 Hz), 7.10 (1H, dd, J = 7.8, 7.3 Hz), 7.34 (1H, t, J = 7.8 Hz), 7.41 (1H, d, J = 7.3 Hz), 7.60 (1/2H, br s), 7.64 (1/2H, br s); 19F NMR (376 MHz, CDCl3) δ −154.39 (1F, br dd, J = 21.6, 8.7 Hz); MS (EI) m/z: 452 (M+), 396 (M+–C4H8); HRMS (EI) calcd for C22H29FN2O7 (M+): 452.1959; found 452.1967.
4.3 General procedure for saponification of the protected 3-(3-fluorooxindol-3-yl)alanine methyl ester 9b and 9c
To a solution of 3-fluorooxindole 9b (23 mg, 0.0653 mmol) in methanol (0.45 mL) was added 0.2N aqueous NaOH (0.49 mL, 0.1 mmol) at 0 °C. The mixture was stirred for 20 min at 0 °C and for 40 min at room temperature. The mixture was then concentrated and extracted with ether. The aqueous layer was acidified (pH ∼ 2) with 5% aqueous KHSO4 at 0 °C. The solution was saturated with NaCl and extracted with EtOAc. The organic layer was washed with brine and dried over Na2SO4. The solution was concentrated to give a diastereomeric mixture of (2S)-2-(tert-butoxycarbonylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid 11b as a colorless solid (16 mg, 0.0473 mmol, 72%).
IR (KBr) v 3700–2900 (br), 2980, 2932, 1728, 1624 cm-1; 1H NMR (400 MHz, CD3OD) δ 1.28 (5H, br s), 1.34 (4H, br s), 1.39 (9H, s), 2.27–2.82 (4H, m), 3.60 (1/3H, m), 3.65 (2/3H, m), 3.95 (1/3H, m), 4.01 (2/3H, dd, J = 11.0, 1.8 Hz), 6.87 (1H, d, J = 7.3 Hz), 6.88 (1H, d, J = 7.8 Hz), 7.05 (1H, dt, J = 7.8, 0.9 Hz), 7.10 (1H, t, J = 7.3 Hz), 7.24 (1H, dt, J = 7.8, 0.9 Hz), 7.34–7.38 (2H, m), 7.46 (1H, d, J = 7.3 Hz); 19F NMR (376 MHz, CD3OD) δ −150.31 (1/11F, m), −151.05 (5/11F, m), −151.47 (16/11F, m); MS (EI) m/z: 318 (M+–HF).
4.3.1 (2S)-2-(Benzyloxycarbonylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid (11c)
Diastereomeric mixture: colorless solid; IR (KBr) v 3700–3000 (br), 2924, 1725, 1624 cm-1; 1H NMR (400 MHz, CD3OD) δ 2.36 (1H, dt, J = 14.2, 10.5 Hz), 2.50–2.84 (3H, m), 4.02 (1/2H, dd, J = 10.5, 2.7 Hz), 4.09 (1/2H, dd, J = 10.5, 2.7 Hz), 4.19 (3/4H, dd, J = 10.5, 2.7 Hz), 4.26 (1/4H, dd, J = 10.5, 2.7 Hz), 4.89 (2H, s), 5.02 (2H, s), 6.83 (1H, d, J = 7.8 Hz), 6.84 (1/2H, d, J = 7.8 Hz), 6.86 (1/2H, d, J = 8.2 Hz), 6.99 (1/2H, t, J = 7.3 Hz), 7.02 (1H, t, J = 7.8 Hz), 7.07 (1/2H, t, J = 7.3 Hz), 7.17–7.46 (14H, m); 19F NMR (376 MHz, CD3OD) δ −150.29 (2/5F, m), −152.07 (1/4F, m), −152.63 (27/20F, m); MS (EI) m/z: 352 (M+–HF).
4.4 (2S)-2-(Acetylamino)-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid (11f)
To a solution of the less polar isomer of 3-fluorooxindole 9f (20 mg, 0.0595 mmol) in acetic acid (0.75 mL) was added 33% HBr/AcOH (0.75 mL) at 0 °C. The mixture was stirred for 20 min at room temperature. The mixture was poured into saturated aqueous NaHCO3 and extracted with EtOAc. The aqueous layer was acidified (pH ∼ 1) with 10% aqueous HCl at 0 °C. The solution was saturated with NaCl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by recrystallization from n-hexane/CHCl3/EtOH to give 11f as a pale brown solid (8 mg, 0.0285 mmol, 48%).
[α]D29 −27.0 (c 0.5, MeOH); IR (KBr) v 3650–2900 (br), 2923, 2853, 1733, 1625 cm-1; 1H NMR (400 MHz, CD3OD) δ 1.62 (3H, s), 2.58 (1H, dt, J = 14.2, 10.5 Hz), 2.73 (1H, ddd, J = 16.4, 14.2, 3.2 Hz), 4.52 (1H, dd, J = 10.5, 3.2 Hz), 6.89 (1H, d, J = 7.8 Hz), 7.09 (1H, t, J = 7.8 Hz), 7.34 (1H, ddt, J = 7.8, 1.8, 1.4 Hz), 7.44 (1H, d, J = 7.8 Hz); 19F NMR (376 MHz, CD3OD) δ −150.09 (1F, dd, J = 16.2, 10.8 Hz); MS (EI) m/z: 280 (M+); HRMS (EI) calcd for C13H13FN2O4 (M+): 280.0859; found 280.0860.
4.5 (2S)-2-Amino-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid methyl ester hydrochloride (12)
To a solution of 3-fluorooxindole 9g (45 mg, 0.0995 mmol) in acetic acid (0.5 mL) was added 25% HBr/AcOH (0.5 mL) at −20 °C. After stirring for 20 min at room temperature, water was added to the solution. The mixture was extracted with ether. The aqueous layer was basified (pH ∼ 11) with K2CO3 at 0 °C. The solution was saturated with NaCl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in ca. 2.5M HCl/MeOH. The solution was concentrated to give a diastereomeric mixture of 12 as a pale yellow solid (27 mg, 0.0935 mmol, 94%).
IR (KBr) v 3700–2150 (br), 3423, 1736, 1624 cm-1; 1H NMR (400 MHz, CD3OD) δ 2.54 (1H, ddd, J = 27.9, 15.6, 7.3 Hz), 2.62 (1H, ddd, J = 34.3, 16.0, 4.6 Hz), 2.92 (1H, ddd, J = 16.0, 12.4, 9.2 Hz), 3.01 (1H, ddd, J = 16.0, 12.4, 5.0 Hz), 3.79 (3H, s), 3.83 (3H, s), 4.47 (1H, dd, J = 7.3, 5.0 Hz), 4.77 (1H, dd, J = 9.2, 4.6 Hz), 6.98 (1H, d, J = 7.8 Hz), 6.99 (1H, d, J = 7.8 Hz), 7.14 (1H, t, J = 7.8 Hz), 7.16 (1H, t, J = 7.8 Hz), 7.39–7.45 (2H, m), 7.48 (1H, ddd, J = 7.8, 1.8, 1.4 Hz), 7.55 (1H, ddd, J = 7.8, 1.8, 1.4 Hz); 19F NMR (376 MHz, CD3OD) δ −154.38 (1F, dd, J = 27.9, 12.1 Hz), −155.98 (1F, dd, J = 34.2, 12.1 Hz).
4.6 (2S)-2-(tert-Butoxycarbonylamino)-3-(indol-3-yl)propionic acid tert-butyl ester (14)
To a solution of (2S)-2-(tert-butoxycarbonylamino)-3-indolylpropionic acid 13 (202 mg, 0.664 mmol) in dry CH2Cl2 (10 mL) was added tert-butanol (77 μL, 0.796 mmol), N,N′-dicyclohexylcarbodiimide (164 mg, 0.796 mmol), and 4-dimethylaminopyridine (8 mg, 0.0664 mmol) at 0 °C. The mixture was stirred for 20.5 h at room temperature. After filtration with celite, the filtrate was concentrated. The residue was dissolved by EtOAc and washed with 5% aqueous KHSO4, saturated aqueous NaHCO3, water, and brine. The organic layer was then dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent; hexane/EtOAc = 4/1) to give 14 as a colorless solid (102 mg, 0.283 mmol, 43%)
Mp 180–184 °C; 46% ee (Chiralpak IA, eluent; n-hexane/2-propanol = 9/1); [α]D29 +11.2 (c 0.24, CHCl3); IR (KBr) v 3436, 3344, 2977, 1730, 1685 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.37 (9H, s), 1.42 (9H, s), 3.23 (1H, dd, J = 15.1, 5.5 Hz), 3.27 (1H, dd, J = 15.1, 6.0 Hz), 4.54 (1H, ddd, J = 7.8, 6.0, 5.5 Hz), 5.06 (1H, br d, J = 7.8 Hz), 7.03 (1H, br d, J = 2.3 Hz), 7.11 (1H, dt, J = 7.8, 0.9 Hz), 7.19 (1H, dt, J = 8.2, 0.9 Hz), 7.35 (1H, d, J = 8.2 Hz), 7.62 (1H, d, J = 7.8 Hz), 8.07 (1H, br s); MS (EI) m/z: 360 (M+); HRMS (EI) calcd for C20H28N2O4 (M+): 360.2049; found 360.2060.
4.7 (2S)-2-[Bis(tert-butoxycarbonyl)amino]-3-(1-benzyloxycarbonylindol-3-yl)propionic acid tert-butyl ester (15)
To a solution of 14 (344 mg, 0.954 mmol) in dry CH2Cl2 (10 mL) was added benzyl chloroformate (0.125 mL, 1.43 mmol), pulverized NaOH (57.2 mg, 1.43 mmol), and benzyl tri-n-butylammonium chloride (29.8 mg, 0.0954 mmol) at 0 °C. The mixture was stirred for 15 h at room temperature. After filtration with celite, the filtrate was concentrated. The residue was dissolved by EtOAc and washed with water and brine. The organic layer was then dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent; hexane/EtOAc = 5/1) to give (2S)-2-(tert-butoxycarbonylamino)-3-(1-benzyloxycarbonylindol-3-yl)propionic acid tert-butyl ester (420 mg) as a colorless solid, which was then dissolved in dry acetonitrile (8 mL). 4-Dimethylamino pyridine (51.9 mg, 0.425 mmol) and di-tert-butyl dicarbonate (0.305 mL, 1.27 mmol) were added to the solution at 0 °C. After stirring overnight, the mixture was concentrated and dissolved in EtOAc. The solution was washed with water and dried over Na2SO4. The solvent was evaporated and the residue was purified by silica gel column chromatography (eluent; hexane/EtOAc = 8/1) to give 15 as a colorless oil (281 mg, 0.473 mmol, 50%).
[α]D29 −11.5 (c 0.39, CHCl3); IR (neat) v 2979, 2934, 1737, 1698 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.32 (18H, s), 1.48 (9H, s), 3.35 (1H, dd, J = 15.1, 10.1 Hz), 3.45 (1H, ddd, J = 15.1, 5.0, 0.9 Hz), 5.11 (1H, dd, J = 10.1, 5.0 Hz), 5.41 (1H, d, J = 11.9 Hz), 5.43 (1H, d, J = 11.9 Hz), 7.23 (1H, dt, J = 7.6, 1.4 Hz), 7.30 (1H, dt, J = 7.8, 0.9 Hz), 7.36–7.48 (6H, m), 7.54 (1H, d, J = 7.8 Hz), 8.16 (1H, br s); MS (EI) m/z: 594 (M+); HRMS (EI) calcd for C33H42N2O8 (M+): 594.2941; found 594.2933.
4.8 (2S)-2-[Bis(tert-butoxycarbonyl)amino]-3-(indol-3-yl)propionic acid tert-butyl ester (16)
To a solution of 15 (658 mg, 1.11 mmol) in methanol (20 mL) was added 5% palladium on carbon (132 mg) at room temperature. The mixture was stirred for 33 h under hydrogen at room temperature. The palladium on carbon was filtered and the filtrate was concentrated. The residue was purified by silica gel column chromatography (eluent; hexane/EtOAc = 6/1) to give 16 as a colorless solid (405 mg, 0.879 mmol, 80%).
Mp 110–112 °C; [α]D27 −25.0 (c 0.33, CHCl3); IR (KBr) v 3347, 3327, 2981, 1782, 1735 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.32 (18H, s), 1.49 (9H, s), 3.40 (1H, dd, J = 15.1, 10.1 Hz), 3.54 (1H, ddd, J = 15.1, 5.0, 0.9 Hz), 5.09 (1H, dd, J = 10.1, 5.0 Hz), 7.01 (1H, br d, J = 2.3 Hz), 7.09 (1H, ddd, J = 8.2, 7.3, 0.9 Hz), 7.16 (1H, ddd, J = 8.2, 6.9, 0.9 Hz), 7.32 (1H, d, J = 8.2 Hz), 7.60 (1H, d, J = 7.3 Hz), 8.02 (1H, br s); MS (EI) m/z: 460 (M+); HRMS (EI) calcd for C25H36N2O6 (M+): 460.2573; found 460.2596.
4.9 (2S)-2-[Bis(tert-butoxycarbonyl)amino]-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid tert-butyl ester (17)
Fluorination of 16 (2.08 g, 4.52 mmol) in the same manner as that of 8b–g produced a diastereomeric mixture of 17 (1.13 g, 2.29mmol, 51%) as a colorless oil after purification by silica gel column chromatography (eluent; hexane/EtOAc = 6/1∼4/1). De was determined to be 14% from the 1H NMR of crude mixture.
IR (neat) v 3295, 3008, 2981, 2935, 1788, 1739, 1697, 1625 cm-1; 1H NMR (400 MHz, CDCl3) δ 1.40 (18H, s), 1.45 (18H, s), 1.46 (18H, s), 2.84–3.07 (4H, m), 4.89 (1H, ddd, J = 9.6, 3.7, 0.9 Hz), 5.03 (1H, dd, J = 9.6, 3.2 Hz), 6.86 (1H, d, J = 7.8 Hz), 6.87 (1H, d, J = 7.3 Hz), 7.06 (1H, t, J = 7.3 Hz), 7.10 (1H, t, J = 7.8 Hz), 7.30 (1H, ddt, J = 7.8, 1.8, 1.4 Hz), 7.34 (1H, tt, J = 7.3, 1.4 Hz), 7.42 (1H, d, J = 7.8 Hz), 7.45 (1H, d, J = 7.3 Hz), 7.98 (1H, br s), 8.17 (1H, br s); 19F NMR (376 MHz, CDCl3) δ –153.08 (1F, dd, J = 20.2, 10.8 Hz), −153.30 (1F, dd, J = 19.3, 8.1 Hz); MS (EI) m/z: 494 (M+), 438 (M+–C4H8).
4.10 (2S)-2-Amino-3-(3-fluoro-2-oxoindoline-3-yl)propionic acid hydrobromide (6)
To a solution of 17 (25 mg, 0.0506 mmol) in acetic acid (0.5 mL) was added 25% HBr/AcOH (0.5 mL) at 0 °C. The mixture was stirred for 20 min at room temperature and then concentrated in vacuo. The residue was dissolved in water and the residual acetic acid was azeotropically removed under reduced pressure to give a diastereomeric mixture of 6 as a pale brown solid (15 mg, 0.470 mmol, 93%).
IR (KBr) v 3700–2400 (br), 3430, 1732, 1718, 1625 cm-1; 1H NMR (400 MHz, CD3OD) δ 2.46 (1H, ddd, J = 27.9, 16.0, 8.7 Hz), 2.61 (1H, ddd, J = 37.1, 16.5, 3.7 Hz), 2.91 (1H, ddd, J = 16.3, 11.4, 9.6 Hz), 3.03 (1H, ddd, J = 16.0, 12.8, 3.7 Hz), 4.50 (1H, dd, J = 8.7, 3.7 Hz), 4.76 (1H, dd, J = 9.6, 3.7 Hz), 6.97 (1H, d, J = 7.8 Hz), 6.98 (1H, d, J = 7.8 Hz), 7.14 (1H, t, J = 7.8 Hz), 7.16 (1H, t, J = 7.8 Hz), 7.41 (1H, tt, J = 7.8, 1.4 Hz), 7.43 (1H, tt, J = 7.8, 1.4 Hz), 7.47 (1H, d, J = 7.8 Hz), 7.54 (1H, d, J = 7.8 Hz); 19F NMR (376 MHz, CD3OD) δ −156.57 (1F, dd, J = 27.9, 13.0 Hz), −156.70 (1F, dd, J = 37.1, 11.4 Hz).
Acknowledgments
KLK acknowledges support from the intramural research funds of NIDDK, NIH.
Footnotes
Dedicated to the memory of Professor Toshio Satoh of Tokushima Bunri University who made many lasting contributions to the field of medicinal chemistry.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Ma J, Hecht SM. Chem Commun. 2004:1190–1191. doi: 10.1039/b402925a. [DOI] [PubMed] [Google Scholar]; (b) Kam T, Choo Y. Phytochemistry. 2004;65:603–608. doi: 10.1016/j.phytochem.2003.12.014. [DOI] [PubMed] [Google Scholar]; (c) Pandey R, Singh SC, Gupta MM. ibid. 2006;67:2164–2169. doi: 10.1016/j.phytochem.2006.06.017. [DOI] [PubMed] [Google Scholar]; (d) Kogure N, Ishii N, Kitajima M, Wongseripipatana S, Takayama H. Org Lett. 2006;8:3085–3088. doi: 10.1021/ol061062i. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kagata T, Saito S, Shigemori H, Ohsaki A, Ishiyama H, Kubota T, Kobayashi J. J Nat Prod. 2006;69:1517–1521. doi: 10.1021/np0602968. [DOI] [PubMed] [Google Scholar]; (b) Suzuki H, Morita H, Shiro M, Kobayashi J. Tetrahedron. 2004;60:2489–2495. [Google Scholar]
- 3.(a) Dermatakis A, Luk K, DePinto W. Bioorg Med Chem Lett. 2003;13:1873–1881. doi: 10.1016/s0968-0896(03)00036-1. [DOI] [PubMed] [Google Scholar]; (b) Natarajan A, Fan Y, Chen H, Guo Y, Iyasere J, Harbinski F, Christ WJ, Aktas H, Halperin JA. J Med Chem. 2004;47:1882–1885. doi: 10.1021/jm0499716. [DOI] [PubMed] [Google Scholar]; (c) Pandit B, Sun Y, Chen P, Sackett DL, Hu Z, Rich W, Li C, Lewis A, Schaefera K, Lia P. Bioorg Med Chem. 2006;14:6492–6501. doi: 10.1016/j.bmc.2006.06.017. [DOI] [PubMed] [Google Scholar]; (d) Li H, Zhang X, Tan J, Chen L, Liu H, Luo X, Shen X, Lin L, Chen K, Ding J, Jiang H. Acta Pharmacol Sin. 2007;28:140–152. doi: 10.1111/j.1745-7254.2007.00473.x. [DOI] [PubMed] [Google Scholar]
- 4.(a) Tokunaga T, Hume WE, Umezome T, Okazaki K, Ueki Y, Kumagai K, Hourai S, Nagamine J, Seki H, Taiji M, Noguchi H, Nagata R. J Med Chem. 2001;44:4641–4649. doi: 10.1021/jm0103763. [DOI] [PubMed] [Google Scholar]; (b) Tokunaga T, Hume WE, Nagamine J, Kawamura T, Taiji M, Nagata R. Bioorg Med Chem Lett. 2005;15:1789–1792. doi: 10.1016/j.bmcl.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 5.(a) Koguchi Y, Kohno J, Nishio M, Takahashi K, Okuda T, Ohnuki T, Komatsubara S. J Antibiot. 2000;53:105–109. doi: 10.7164/antibiotics.53.105. [DOI] [PubMed] [Google Scholar]; (b) Kohno J, Koguchi Y, Nishio M, Nakao K, Kuroda M, Shimizu R, Ohnuki T, Komatsubara S. J Org Chem. 2000;65:990–995. doi: 10.1021/jo991375+. [DOI] [PubMed] [Google Scholar]; (c) Borissenko L, Groll M. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 6.(a) Phillips RS, Miles EW, Cohen LA. Biochemistry. 1984;23:6228–6234. doi: 10.1021/bi00320a052. [DOI] [PubMed] [Google Scholar]; (b) Idem J Biol Chem. 1985;260:14665–14670. [PubMed] [Google Scholar]; (c) DM Kiick, Phillips RS. Biochemistry. 1988;27:7339–7344. doi: 10.1021/bi00419a024. [DOI] [PubMed] [Google Scholar]; (d) Roy M, Miles EW, Phillips RS, Dunn MF. ibid. 1988;27:8661–8669. doi: 10.1021/bi00423a023. [DOI] [PubMed] [Google Scholar]; (e) Phillips RS, Bender SL, Brzovic P, Dunn MF. ibid. 1990;29:8608–8614. doi: 10.1021/bi00489a016. [DOI] [PubMed] [Google Scholar]
- 7.Labroo RB, Cohen LA. J Org Chem. 1990;55:4901–4904. [Google Scholar]
- 8.(a) Takeuchi Y, Shiragami T, Kimura K, Suzuki E, Shibata N. Org Lett. 1999;1:1571–1573. [Google Scholar]; (b) Chem Abstr. 2002;136:247496v. [Google Scholar]; (c) Takeuchi Y, Shibata N, Suzuki E, Iimura Y, Kosasa T, Yamanishi T, Sugimoto H. WO 2002020482 PCT Int Appl. ; (c) Fujisawa H, Fujiwara T, Takeuchi Y, Omata K. Chem Pharm Bull. 2005;53:524–528. doi: 10.1248/cpb.53.524. [DOI] [PubMed] [Google Scholar]; (d) Takeuchi Y, Fujisawa H, Fujiwara T, Matsuura M, Komatsu H, Ueno S, Matsuzaki T. Chem Pharm Bull. 2005;53:1062–1064. doi: 10.1248/cpb.53.1062. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ojima I, McCarthy JR, Welch JT. Biomedical Frontiers of Fluorine Chemistry. ACS Symposium Series 639; Washington, DC: American Chemical Society; 1996. [Google Scholar]; (b) Filler R, Kobayashi Y. Biomedical Aspects of Fluorine Chemistry. Kodansha/Elsevier Biomedical; Tokyo: 1982. [Google Scholar]
- 10.(a) Blaschke G, Kraft HP, Fickentscher K, Koehler F. Arzneim-Forsch/Drug Res. 1979;29:1640–1642. [PubMed] [Google Scholar]; (b) Nishimura K, Hashimoto Y, Iwasaki S. Biochem Biophys Res Commun. 1994;199:455–460. doi: 10.1006/bbrc.1994.1250. [DOI] [PubMed] [Google Scholar]; (c) Winter W, Frankus E. Lancet. 1992;339:365. doi: 10.1016/0140-6736(92)91684-z. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Qian L, Prestwich GD. J Org Chem. 2003;68:5320–5330. doi: 10.1021/jo020729l. [DOI] [PubMed] [Google Scholar]
- 12.Jäckel C, Koksch B. Eur J Org Chem. 2005:4483–4503. [Google Scholar]
- 13.Hewawasam P, Gribkoff VK, Pendri Y, Dworetzky SI, Meanwell NA, Martinez E, Boissard CG, Post-Munson DJ, Trojnacki JT, Yeleswaram K, Pajor LM, Knipe J, Gao Q, Perrone R, Starrett JE., Jr Bioorg Med Chem Lett. 2002;12:1023–1026. doi: 10.1016/s0960-894x(02)00101-4. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi Y, Tarui T, Shibata N. Org Lett. 2000;2:639–642. doi: 10.1021/ol991400y. [DOI] [PubMed] [Google Scholar]
- 15.(a) Banks RE. J Fluorine Chem. 1998;87:1–17. [Google Scholar]; (b) Lal GS, Pez GP, Syvret RG. Chem Rev. 1996;96:1737–1755. doi: 10.1021/cr941145p. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi M, Hino T. Tetrahedron. 1981;37:1487–1494. [Google Scholar]
- 17.Kiso Y, Inai M, Kitagawa K, Akita T. Chem Lett. 1983:739–742. doi: 10.1248/cpb.31.1818. [DOI] [PubMed] [Google Scholar]