

Abstract
Normal mammary epithelial cells are rapidly induced to G1 arrest by the widely expressed cytokine, transforming growth factor beta (TGF-β1). Studies in established breast cancer cell lines that express the estrogen receptor alpha (ERα) have demonstrated loss of this responsiveness. This inverse correlation suggests interpathway signaling important to cell growth and regulation. The adenocarcinoma breast cell line BT474, which was not growth arrested by TGF-β1, was used as a model of estrogen-inducible growth to explore interpathway crosstalk. Although BT474 cells were not growth-arrested by TGF-β1 as determined by flow cytometry analysis and 5′-bromo-3′-deoxyuridine incorporation into DNA, estrogen receptor protein levels were attenuated by 100 pM TGF-β1 after 6 h. This decrease in ERα reached 50% of untreated control levels by 24 h of treatment and was further supported by a 50% decrease in estrogen-inducible DNA synthesis. Inspection of ERα transcripts suggested that this decrease was primarily the result of altered ERα protein stability or availability. Use of the proteasome inhibitor, MG132, abolished all effects on ERα by TGF-β1. Collectively, this data supports a role for TGF-β1 in regulating the growth of otherwise insensitive breast cancer cells through modulation of ERα stability.
Keywords: breast cancer, estrogen receptor alpha, transforming growth factor beta, ubiquitin, proteasome, cell proliferation
Breast cancer is a leading cause of death among women worldwide and an estimated 203,500 new cases will be diagnosed in the US alone in 2002 [American Cancer Society, 2001]. Systemic and locally produced estrogens are intimately associated with increased risk for breast cancer development. Early menarche and late menopause increase the lifetime exposure to estrogens and as much as a 20% decrease in breast cancer risk is observed for each year this exposure is shortened [Kelsey, 1979]. By the same token, increased and sustained physical activity can reduce the total lifetime ovulatory period and has been shown to confer a significant decrease in breast cancer risk [Bernstein et al., 1994]. Numerous epidemiological studies of breast cancer risk in postmenopausal women have shown higher mean serum estrogen concentrations than disease-free control subjects [Thomas et al., 1997]. These and additional risk factors share a common theme, i.e., increased lifetime tissue exposure to estrogens. Thus, estrogen exposure is a central and reoccurring phenomenon in breast cancer etiology.
Although many risk factors have been identified, the molecular mechanisms underlying the initiation and progression of breast cancer are still elusive. Estrogens such as 17β-estradiol (E2) play a key role in the development and maintenance of the mature mammary gland. The most well characterized cellular receptor for this steroid hormone is the estrogen receptor alpha (ERα) [Moggs and Orphanides, 2001]. This receptor is expressed in a tissue specific manner and regulates growth and differentiation in response to hormonal stimulation [Dickson, 1995]. Additional co-regulators of ERα action have also been identified and studied in detail providing a foundation for the molecular mechanisms underlying the ligand-induced estrogenic response [Klinge, 2000]. Although the fundamental mode of action to elicit a response through this receptor has been characterized, little is known concerning the endogenous regulation of ERα expression.
On an average, only 7–30% of normal breast epithelial cells show expression of ERα by immunohistochemistry [Ricketts et al., 1991; Ronnov-Jessen et al., 1996]. Moreover, 60% of transformed breast epithelial cells express ERα supporting a role for hormone receptors in the selective growth of cellular subpopulations that may be important for tumor progression [Edwards et al., 1979]. Receptor availability is dictated by mechanisms regulating activity and levels in normal and in transformed breast cells. Methylation of the ERα gene promoter region may play a significant role in this process [Ferguson and Davidson, 1997]. Additionally, signals that regulate receptor protein levels also contribute to estrogen effects in the target tissue. Recent evidence supports the ubiquitin–proteasome pathway in ERα turnover [Nawaz et al., 1999]. It is not known if this pathway is utilized in generalized ERα turnover or if this mechanism is specific for malignancy and thus may contribute to tumor progression. The ubiquitin–proteasome pathway has additionally been associated with growth factor signaling in normal cellular processes [Kavsak et al., 2000; Wojcik et al., 2000].
In established breast cancer cell lines, a correlation has been observed between estrogen receptor content and sensitivity to transforming growth factor beta (TGF-β1) [Arteaga et al., 1988]. TGF-β is a cytokine growth factor secreted by many cell types that potently suppresses cell growth through autocrine and paracrine interactions [Brandt and Ebert, 1998; Roberts, 1998]. Of the three identified TGF-β isoforms, TGF-β1 has shown the most activity in epithelial cells [Silberstein and Daniel, 1987]. The primary mechanism of cell growth control by TGF-β1 is mediated by the induction of selected cyclin dependent kinase inhibitors (cdkis), thus impairing basic cell cycle machinery [Ravitz and Wenner, 1997]. In general, cells expressing ERα are unresponsive to growth-arrest signaling by TGF-β1, relative to effects in normal breast epithelial cells, while ER (−) cells are responsive [Lynch et al., 2001]. This suggests an estrogenic link to TGF-β signaling which may be involved in tumorigenesis.
Here we describe a model cellular system that implicates TGF-β1 as being instrumental in regulating growth of hormonally responsive breast cancer cells. TGF-β1 showed no effect on basal cell growth but instead appeared to suppress hormonally driven growth exclusively through ERα modulation. The thrust of this research was to uncover new signaling pathway interactions underlying cancer cell growth regulation.
MATERIALS AND METHODS
Cell Culture
Breast cell lines BT474, MDA-MB-231, T47D, and ZR75-1 were obtained from the ATCC (Rockville, MD). All cells, except MCF-10F cells, were passaged bi-weekly in B-media custom formula (MEM, Earle’s salts, 1.5 × amino acids, 2 × non-essential amino acids, L-glutamine, 1.5 × vitamins). This basal medium was supplemented with 10% FBS (Life Technologies, Brl., Rockville, MD), 2 mM L-glutamine, and 0.4% gentamycin. The immortalized MCF-10F cells [Soule et al., 1990] were cultured in DMEM: F12 supplemented with 5% horse serum (Life Technologies) treated with chelex-100 resin (BioRad, Hercules, CA) to remove calcium, 0.04 mM CaCl2, 20 ng/ml EGF (Life Technologies), 100 ng/ml cholera toxin (Life Technologies), 10 μg/ml bovine insulin (Life Technologies), and 50 ng/ml hydrocortisone. Experiments were carried out in a defined media system consisting of B-media supplemented with 1% insulin–transferrin–selenium pre-mix (Collaborative Biomedical Products, Bedford, MA) and 2 mM L-glutamine. A 10 ng/ml dose of EGF was supplemented in defined media for MCF-10F cells. No cells were used beyond passage ten.
Proliferation Assay
Cells were plated in 100 μl volumes at densities of 1 × 104 cells per well in 96-well microtiter plates using 10% FBS supplemented growth medium. After a 24 h incubation, cells were replenished with defined medium containing the necessary experimental component and incubated an additional 24 h. After 18 h, cells were dosed with 5-bromo-3′-deoxyuridine (BrdU, Boehringer Manheim, Germany) according to manufacturers instructions to allow for incorporation into DNA. Assessment of growth was determined using a Spectramax 340 microplate reader (Molecular Devices, Sunnyvale, CA) set at 370 nm.
Western Analysis
Cells were grown tosub-confluence on 100mm culture plates. The plates were rinsed and replenished with defined media in the presence or absence of experimental agent. Each plate was scraped into 1 ml of cold detachment buffer [0.25 M sucrose, 10 mM Tris (7.4), 1 mM EDTA, 0.3 mM PMSF] and pelleted at 2,000g for 10 min. The cells were then lysed by three cycles of freeze/thaw in cold lysis buffer [2 mM NaMO4, 2 mM NaVO3, 1 mM DTT, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml PMSF, 1% Triton X-100, 20 mM Tris (7.4), 2 mM EDTA, 25 mM NaF]. Cell debris was pelleted at 12,000g for 15 min and total proteins were resolved by 8% SDS–PAGE. Polyclonal antibodies to ERα (HC-20, Santa Cruz) were used for probing corresponding PVDF blots for 90 min followed by secondary antibody and ECL development. The proteosome inhibitor MG132 (Sigma-Aldrich, St. Louis, Mo) was dissolved and diluted in methanol and used at a concentration of 300 nM.
Cytotoxicity Assay
Cells were incubated in 96-well plates with or without experimental agent. Twenty-four hours following dosage, 50 μl of conditioned media was removed from each well and tested for the presence of lactate dehydrogenase (LDH) using the CytoTox 96 Assay (Promega, Madison, WI). A 100% LDH release control was achieved by three cycles of freeze–thaw on the cells to disrupt cell membrane integrity.
Flow Cytometry
Subconfluent cells were dosed with 10 nM E2, 100 pM TGF-β, or E2 + TGF-β on defined media. Cells were harvested at 24 h following dosing and fixed in 70% EtOH overnight at −20°C. Fixed cells were rinsed two times with HBSS (PBS, 1% BSA, 1 mM EDTA) and incubated with 10 μg/ml propidium iodide and 1 mg/ml RNase A at 37°C for 30 min. Samples (2 × 106 cells/ml) were refrigerated in the dark for 24 h followed by Epics Elite Scan analysis (Beckman Coulter, Palo Alto, CA). The immortalized, non-transformed breast epithelial cell line MCF-10F and the tumorigenic MDA-MB-231 cell line were used as positive controls for TGF-β1 induced G1 arrest.
RT-PCR
RT-PCR analysis was used to evaluate total isolated RNA following TGF-β1 treatment. Primers for the 650 bp ERα product were used as previously described: sense 5′-TAC TGC ATC AGA TCC AAG GG-3′; antisense 5′-ATC AAT GGT GCA CTG GTT GG-3′ [Lau et al., 1999]. Internal control gene 36B4, which is unaffected by estradiol, was used to normalize for dosing and consistent gel loading [Laborda, 1991]. Primers for the 550 bp product are: sense 5′-TTT CAG CAA GTG GGA AGG TG-3′; antisense 5′-AAA CTG CTG CCT CAT ATC CG-3′. Samples were optimized for linearity and Mg2+ concentration. RNA was isolated from treated cell cultures using the Trizol reagent (Life Technologies) and quantified by UV absorbance. A 1 μg sample of RNA was used in subsequent reverse transcription assays using superscript II reverse transcriptase (Life Technologies). A 1 μl product from each sample was then used in PCR reactions catalyzed by Platinum Taq DNA polymerase (Life Technologies). PCR was performed for ERα (26 cycles) and 36B4 (22 cycles) on an MJ Research PTC-100 Thermal Cycler under the following conditions: ERα [95°C, 1 min; 58°C, 1 min; 72°C, 1 min] and 36B4 [95°C, 45 s; 64°C, 1 min; 72°C, 1 min]. PCR products were mixed at 2:1 and were resolved by 0.9% agarose gel electrophoresis in 1 × TAE at 90 V for 3 h. The resulting gel was stained with ethidium bromide and fluorescent images were analyzed by Kodak digital science system (Eastman Kodak Company, Rochester, NY) and were quantified using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Northern Analysis
Total cellular RNA was isolated by the Trizol method (Invitrogen, Life Technologies, Carlsbad, CA). Probes to ERα and 36B4 were generated by klenow incorporation of 50 μCi of 32P-dCTP in 25 ng of DNA for 4 h at 25°C. Probes were precipitated with 95% ethanol, 3 M NaOAc, and 40 μg of glycogen at −20°C for 1 h. Pellets were collected by centrifugation and resuspended in molecular biology grade water. Probe specific activity was then determined by scintillation counting. A total of 12 × 106 cpm was used for abundant RNA species and 25 × 106 cpm was used for low level species. Following electrophoresis of RNA samples, resolved lanes were transferred to nitrocellulose membranes using the turbo blotter transfer system. Probing conditions were carried out as previously described, blocking with salmon sperm DNA. Bands were detected by Phosphor-Imager analysis.
Statistics
Statistical and graphical information was determined using GraphPad Prism software (GraphPad Software Inc., San Diego, CA) and Microsoft Excel (Microsoft Corporation, Redmond, WA). P values were calculated with the two-tailed unpaired t-test at 95% confidence.
RESULTS
E2-Induced DNA Synthesis is Negated by TGF-β1 in an ER-Dependent Manner
Three ER (+) and three ER (−) human breast cancer cell lines were evaluated for their growth responsiveness to TGF-β1. When grown in the presence of serum-free media, the ER (+) cells were unresponsive or slightly stimulated to grow by TGF-β1, while ER (−) cell growth was reduced to at least 50% of untreated samples (Table I). This data confirmed a positive correlation of ERα status to lack of TGF-β1 responsiveness. To explore possible links between TGF-β and E2 signaling, the ER (+) breast cancer cells were also initially screened for E2-inducible DNA synthesis. The cell line BT474 produced the largest induction of proliferation with E2 treatment (Fig. 1). Thus, BT474 cells were selected to dissect important interactions between TGF-β1 and E2-mediated effects on cell growth.
TABLE I.
Growth Responsiveness to TGF-β in Various Breast Cancer Cell Lines
| TGF-β1 concentration
|
||||
|---|---|---|---|---|
| ER | 0.01 nM | 0.10 nM | 1.0 nM | |
| BT20 | − | 73.7 ± 3.7* | 71.3 ± 4.8* | 48.7 ± 3.4* |
| MDA-MB231 | − | 78.9 ± 7.4* | 52.7 ± 3.5* | 48.7 ± 4.1* |
| SkBr3 | − | 70.4 ± 7.7* | 49.9 ± 8.0* | 36.2 ± 5.2* |
| T47D | + | 103.1 ± 5.3 | 93.2 ± 7.2 | 92.9 ± 3.8 |
| BT474 | + | 95.7 ± 4.7 | 97.9 ± 3.3 | 98.1 ± 4.0 |
| ZR75-1 | + | 97.6 ± 9.4 | 105.6 ± 6.5 | 107.6 ± 5.6 |
A panel of breast cancer cell lines was grown with increasing titer of TGF-β1 for 24 h in defined media. During the final 6 h, 5-bromo-2′-deoxyuridine (BrdU) was added to the media to allow for cellular uptake and DNA incorporation. At 24 h, percent of incorporated BrdU was determined at 370 nm. Untreated control absorbencies were taken as 100% growth. Values shown are percent of untreated controls and are the average of six independent treatments.
P < 0.05.
Fig. 1.
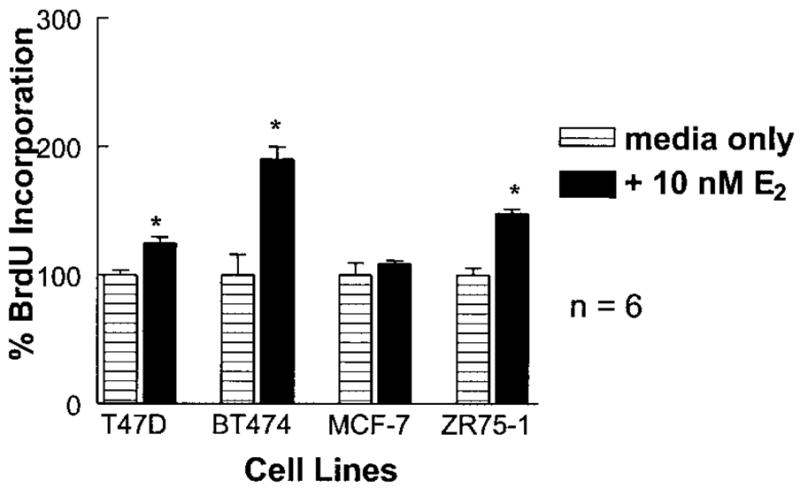
Inducible cell growth by E2 in breast cancer cell lines. Breast cancer cell lines were cultured in the presence or absence of 10 nM E2 for 24 h on defined media. At 18 h, BrdU was added to the media and at 24 h absorbencies were taken to assess DNA synthesis. All tested cell lines exhibited increased DNA synthesis in the presence of E2; however, those effects were most dramatic in cell line BT474. This data is the average of six independent treatments (*P < 0.05).
BT474 cell proliferation was determined at various TGF-β1 concentrations in the presence and absence of 10 nM E2 (Fig. 2). As expected for an ER (+) cell line, no growth inhibition was observed with increasing TGF-β1 concentration. Addition of E2 resulted in a 75% induction of DNA synthesis at 24 h, which was attenuated by a concentration gradient of TGF-β1. Addition of tamoxifen, a competitive antagonist of ERα, inhibited E2-stimulated growth, and TGF-β1 had no effect under these conditions. Collectively, these results suggested either an induction of TGF-β1 mediated growth-arrest in the presence of E2 or a reduction in E2-stimulated growth in the presence of TGF-β1. ERα appeared to underlie this effect since no change was observed in the presence of tamoxifen. These effects were observed in other ER (+) cell lines to a lesser extent (Table II).
Fig. 2.
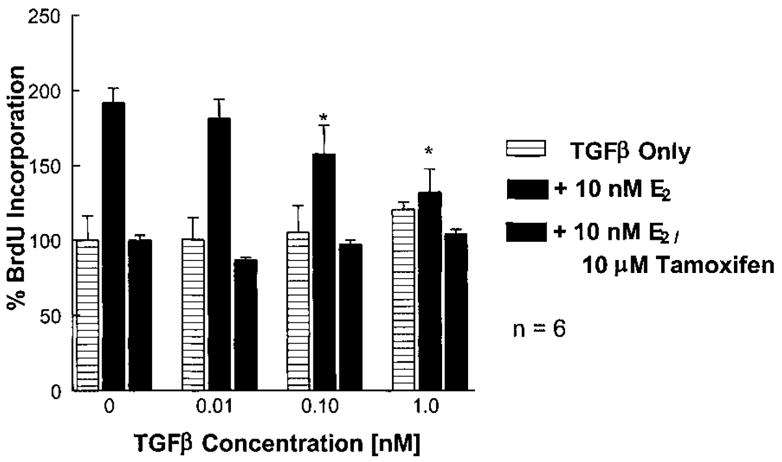
Effects of TGF-β1 on E2-induced cell proliferation. BT474 cells were cultured in the presence or absence of indicated dose for 24 h in defined media. At 18 h, BrdU was added to the conditioned media and at 24 h absorbencies were taken to assess DNA synthesis. The presence of TGF-β1 appeared to abrogate E2-induced DNA synthesis. Addition of 10 μM tamoxifen blocked E2-induced DNA synthesis and all observed effects by TGF-β1. This data is the average of six independent treatments (*P < 0.05).
TABLE II.
Additional ER (+) Cell Lines Showing TGF-β Regulation of E2-Induced Growth
| TGF-β1 concentration
|
||||||||
|---|---|---|---|---|---|---|---|---|
| 0 nM | 0.01 nM | 0.10 nM | 1.0 nM | |||||
| E2 (10 nM) | − | + | − | + | − | + | − | + |
| T47D | 100 ± 3.8 | 124.8 ± 4.9* | 113.8 ± 5.8 | 112.5 ± 3.8 | 124.7 ± 4.9 | 96.9 ± 3.0 | 135.6 ± 4.7 | 104.8 ±1.7* |
| ZR75-1 | 100 ± 5.6 | 148.3 ± 3.2* | 127.6 ± 3.8 | 138.9 ± 2.6* | 145.6 ± 4.3 | 137.5 ± 2.2 | 140.7 ± 2.2 | 114.1 ± 5.2* |
Cell lines T47D and ZR75-1 also demonstrated some signaling cross talk when initially screened in BrdU proliferation assays following a 24 h E2 treatment. Values shown are percent of untreated control BrdU incorporation into the DNA of proliferating cell cultures. Data is the average of six independent treatments at each concentration.
Asterisks denote significant differences between E2 treated and untreated samples at each TGF-β concentration. P < 0.05.
Estradiol Does Not Restore TGF-β1-Induced Growth Arrest
To better understand if TGF-β1 was active on BT474 cells only in the presence of E2 a more detailed examination of the effects on the cell cycle was warranted. TGF-β1 controls epithelial cell proliferation by inducing factors that result in G1 arrest or attenuation. Flow cytometry was used to distinguish effects of TGF-β1 on G1 phase and E2 on S phase, in the BT474 cell model. Cell lines were cultured in growth medium and treated at sub-confluence in defined media. In the immortalized and transformed ER (−) cells MCF-10F and MDA-MB-231, 100 pM TGF-β1 altered cell cycle distribution by increased percent G1 and decreased S phase (Fig. 3A, B). E2 treatment of BT474 cells resulted in nearly a 100% increase of cells in S phase as predicted, however, TGF-β1 alone had no effect on cell cycle distribution. In the presence of TGF-β1, E2-induced S phase was attenuated nearly 50% that of E2 treated samples alone (Fig. 3C). These results were consistent with BrdU incorporation studies and suggested that TGF-β1 could not control basal cell growth in BT474 cells, but instead must regulate E2-inducible growth exclusively.
Fig. 3.

TGFβ modulation of cell cycle. Standard propidium iodide uptake was used to characterize TGF-β1 induced G1 phase. A: Control non-transformed MCF-10F cells treated with or without 100 pM TGF-β for 18 h show 50% reduction in S phase and 10% induction into G1 phase. B: Example of a TGF-β1 sensitive, tumorigenic cell line, MDA-MB231, treated with or without 100 pM TGF-β for 18 h also resulting in a 10% induction into G1 phase. C: BT474 breast epithelial cell line treated with either 10 nM E2, 100 pM TGF-β, or a combination of both for 18 h. The results indicate no increase of cells in G1 phase following TGF-β1 treatment. Furthermore, a 50% reduction in E2-stimulated S phase was observed, consistent with BrdU studies.
Because TGF-β1 has been reported to induce apoptosis in some cell types, LDH was measured in the conditioned medium [Haufel et al., 1999; Rosfjord and Dickson, 1999]. No increase in released cellular LDH was detected at any concentration of TGF-β1 or TGF-β1 plus E2 (data not shown). Further, TGF-β1 did not induce genomic DNA laddering as determined by ethidium bromide staining (data not shown). Thus, TGF-β1 did not induce BT474 cell death or cell cycle arrest in the presence or absence of E2, implicating a role for TGF-β1 in estrogen receptor modulation as a novel mechanism of proliferative control.
TGF-β1 Regulates ERα Protein Levels
To investigate the role of ERα in this proliferative response, Western analysis was performed on BT474 whole cell extracts. Cells were grown to sub-confluence and dosed with varying concentrations of TGF-β1 in defined media for 24 h. These cells, which exhibited no growth response to TGF-β1 alone, appeared to respond with a 50% reduction in ERα protein levels at 100 pM TGF-β1 (Fig. 4A). As expected, cells treated with 10 nM E2 exhibited a 70% reduction in ERα. The combination of TGF-β1 and E2 did not result in additive reduction of ERα levels (Fig. 4B).
Fig. 4.

ERα protein levels in BT474 cells. Protein from BT474 cells treated with or without TGF-β1 in defined media for 24 h was isolated and probed for relative ERα expression. Total protein was isolated from BT474 cells treated for 24 h with experimental agent. Blots were probed for ERα using standard ECL detection. A: Up to a 50% reduction in ERα levels was observed at a concentration of 100 pM TGF-β1. B: An expected 70% reduction in ERα was observed in E2 only treated samples. ERα levels were not further reduced in TGF-β1 + E2 samples indicating that TGF-β1 does not act in a concerted additive fashion with E2 (*P < 0.05).
A closer inspection of ERα protein levels over a time-course of 40 h suggested an effect occurring by 6 h of treatment with TGF-β1 and maintaining suppressed ERα levels relative to untreated controls (Fig. 5). Furthermore, suppressed ERα levels were replenished following removal of the TGF-β1 and replacement with fresh defined media for an additional 24 and 48 h (data not shown). These results suggested that even though BT474 cells lack any basal growth response to TGF-β1, TGF-β1 could attenuate ERα levels thereby modulating hormonally induced growth.
Fig. 5.

Time-course effects of TGF-β1 on ERα levels. Total protein was isolated from TGF-β1 treated and untreated BT474 cell cultures at multiple time points spanning 40 h in defined media. A 50 μg quantity of total protein was resolved by 8% SDS–PAGE and the resulting PVDF membrane probed with antibody to ERα. Untreated samples showed increase in ERα levels with time. TGF-β1 treated samples showed effects occurring between 6 and 12 h that maintain suppressed ERα levels.
TGF-β1 Shows Slight Effects on ERα Transcript Levels
ERα transcript levels were likewise monitored since TGF-β1 appeared to modulate ERα protein levels. Cells were cultured identically to those used in the protein level studies and total RNA was isolated at 24 h. PCR primers to ERα and internal control gene 36B4 were then used to amplify ERα transcripts following TGF-β1 treatment. Samples treated with 100 pM TGF-β1 exhibited only a slight decrease (~10%) in the levels of the 650 bp PCR product relative to defined media controls (Fig. 6A). This decrease did not occur in cells treated with 10 pM TGF-β1 at 24 h. The phorbol ester, TPA, which has been shown to reduce ERα RNA levels in cell cultures, was used in parallel experiments at 100 nM as a positive control. These results were further validated by Northern analysis and yielded similar results (Fig. 6B).
Fig. 6.

Regulation of ERα transcript levels by TGF-β1. A: BT474 cells were treated for 24 h with 0.01 or 0.10 nM TGF-β1. Total RNA was isolated and RT-PCR was performed as described in Materials and Methods for ERα and 36B4. Combined PCR products were resolved by 0.9% agarose in 1 × TAE. Bands were visualized following ethidium bromide staining and were quantified using ImageQuant software (*P < 0.05). B: Northern analysis was carried out using a cloned fragment of human ERα labeled with α-32P-dCTP as a probe. Low endogenous levels of ERα in the BT474 cell line exceeded the level of sensitivity of this assay; however, slight increases in ERα mRNA levels over untreated samples were apparent. The ERα signal was normalized to the internal control gene 36B4, which remained constant throughout. Qualitative binding was detected by phosphorimager analysis (n = 3 for media; n = 2 for TGF-β1).
Proteasome Inhibitor MG132 Blocks ERα Modulation by TGF-β1
The inconsistency of ERα RNA and protein levels suggested that additional mechanisms might be involved in the regulation of ERα. The ubiquitin–proteasome pathway has been previously shown to play an important role in estrogen receptor turnover [Nawaz et al., 1999]. Since TGF-β1 failed to show effects on ERα transcript levels, the possibility of increased proteasomal degradation was explored. The competitive inhibitor MG132 was used to block proteasome function in BT474 cell cultures. Cells treated with TGF-β1 for 20 h showed a 50% decrease in ERα protein levels relative to untreated controls (Fig. 7A). The addition of 300 nM MG132 appeared to completely block this effect (Fig. 7B). Moreover, lanes representing cells treated with TGF-β1 had increased amounts of low molecular weight immunoreactive bands, likely indicative of proteolytic cleavage. This banding pattern was consistent with previously published reports of ERα proteolysis using anti-ER antibody, H222, and may or may not be the result of proteasome-dependent degradation [Nawaz et al., 1999].
Fig. 7.

Abrogation of TGF-β1 mediated effects on ERα. Sub-confluent cultures of BT474 cells were treated with or without 100 pM TGF-β1 in the presence or absence of 300 nM MG132 for 24 h. A: A 50% suppression in ERα levels was observed in TGF-β1 treated samples compared with media alone (n = 3). B: No suppression in ERα levels by TGF-β1 was observed relative to control samples in the presence of the proteasome inhibitor MG132 (n = 3).
DISCUSSION
Although breast cancer appears to be a multifactorial disease, estrogens have been strongly associated with the initiation and progression of this disease. Experimental evidence supports a role for estrogens in DNA synthesis and in general cellular growth-stimulatory pathways [Darbre et al., 1983; Lykkesfeldt and Briand, 1986; Mustafa and Bland, 1998; Castoria et al., 1999]. However, a full understanding of what distinctions define normal hormonally induced growth and differentiation and hormonally induced transformation will require a better understanding of interpathway regulation following hormonal stimulation. An estrogenic response requires estrogen binding to and activation of the estrogen receptor [Nilsson et al., 2001]. Therefore, control of ERα expression levels would offer a direct means of modulating E2-mediated cellular effects such as growth.
TGF-β1 is clearly instrumental to this process at the particular stage of cancer depicted in BT474 cells. TGF-β has been implicated in a wide range of effects in normal and disease processes many of which overlap or antagonize effects of estrogen action. It is important in such cellular processes as motility, growth regulation, development, and apoptosis. Loss of cell growth control by TGF-β is a common observed event in malignancy and important regulators of cell growth including p15, p21, and cdc25A are known to be transcriptionally controlled by TGF-β [Lawrence, 1996; Rooke and Crosier, 2001]. Although there have been previous reports of TGF-β1 modulating ERα levels in a breast cancer cell line [Stoica et al., 1997], to our knowledge this is the first time it has been shown in a classically TGF-β1 non-responsive cell line. We have shown a multileveled regulation, which mainly involves protein stability, but may include a transcriptional component. Screening of other hormonally responsive breast cell lines suggests that this same interpathway mechanism may exist.
While E2 has been reported to induce degradation of the ERα through the ubiquitin–proteasome pathway, this auto-regulation likely requires realized molecular end-points of E2 signaling. Additional factors that regulate the endogenous levels of ERα and thus bypass E2-mediated effects should be considered key to thegrowth of normal and cancerous cell types. Here we demonstrate antagonistic growth factor pathways mediating estrogen responsiveness through ERα modulation. This finding is consistent with other published reports of TGF-β1 effects on downstream estrogenic targets. Growth promoting hormonal targets such as cyclin D1 and c-myc, both highly associated with tumor promotion, have been shown to be negatively regulated by TGF-β via non-ERα mechanisms [Martinez et al., 2000; Seoane et al., 2001].
An important finding from this work is that lack of “growth responsiveness” to TGF-β is not synonymous with lack of “responsiveness” to TGF-β. The apparent “uncoupled” TGF-β signaling, as has been reported in other cell types [Calonge and Massague, 1999; Lu et al., 1999; Yakicier et al., 1999] warrants further examination. Regulation of selected gene expression in the absence of cell cycle control could be a result of mutation or alternative pathways activated only under select conditions. For example, specific regions of TβRII are responsible for a selected set of TGF-β responses [Chen et al., 1993] and inactivation of pRb may be important to this activity, as reported by Massague et al. [Zentella et al., 1991]. Some data on aberrant SMAD signaling coupled to a divergent response has also been reported [Liu et al., 1997]. Another likely explanation, based on reduced surface TβRII levels specific to ER (+) cell lines, could be a resultant sub-critical cdki stoichiometry sufficient to induce G1 arrest [Lynch et al., 2001].
Additional studies should address the molecular aspects of this TGF-β induced ERα degradation. Recent elegant studies by several groups have shed new light on TGF-β induction of the ubiquitin–proteasome cascade. Ubiquitin ligase components such as Smurf1/2 and the anaphase-promoting complex(APC)are activated and/or interact directly with Smad proteins leading to proteasomal degradation of target proteins [Lin et al., 2000; Stroschein et al., 2001]. These targets include Smad components, thus conferring an additional layer of auto-regulation. More importantly, Smad-induced ubiquitin targets include factors that repress or override TGF-β signaling such as the transcriptional co-repressor SnoN [Bonni et al., 2001].
The ERα has been shown here and in other reports to be a target of the Ub–proteasome pathway. A TGF-β mediated pathway to this degradation may be the result of a direct Smad2/Smurf2/ERα complex as ERα has been found directly associated with activated Smads [Yamamoto et al., 2002]. Additionally, the Ski-interacting protein (SKIP) has been shown to interact with both nuclear hormone receptors and TGF-β signaling components potentially serving an adapter role in this regulation [Baudino et al., 1998; Leong et al., 2001].
Key protein stabilities in malignancy and their destructive destination by the ubiquitin–proteasome or related pathways will no doubt be an important focus of future endeavors. The necessity of proteasomal targeting in the delicate control of cellular growth is only beginning to unfold. This research should provide a foundation to further explore new regulatory mechanisms in developmental and tumor cell biology offering new therapeutic targets. Understanding of these basic interactions may also provide a more visible picture of uncoupled TGF-β signaling exhibited by some cell types and the importance of this to hormonally responsive breast cancers.
Acknowledgments
The authors thank Dr. Melanie A. Lynch for her assistance in the cell proliferation assays.
Grant sponsor: NIH; Grant numbers: RO1 CA73698, P30 CA16058; Grant sponsor: Breast Cancer Research Fund (OSUCCC).
References
- American Cancer Society. 2001. Facts and figures.
- Arteaga CL, Tandon AK, Von Hoff DD, Osborne CK. Transforming growth factor beta: Potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cells. Cancer Res. 1988;48:3898–3904. [PubMed] [Google Scholar]
- Baudino TA, Kraichely DM, Jefcoat SC, Jr, Winchester SK, Partridge NC, MacDonald PN. Isolation and characterization of a novel coactivator protein, NCoA-62, involved in vitamin D-mediated transcription. J Biol Chem. 1998;273:16434–16441. doi: 10.1074/jbc.273.26.16434. [DOI] [PubMed] [Google Scholar]
- Bernstein L, Henderson BE, Hanisch R, Sullivan-Halley J, Ross RK. Physical exercise and reduced risk of breast cancer in young women. J Natl Cancer Inst. 1994;86:1403–1408. doi: 10.1093/jnci/86.18.1403. [DOI] [PubMed] [Google Scholar]
- Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, Wrana JL. TGF-beta induces assembly of a Smad2–Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol. 2001;3:587–595. doi: 10.1038/35078562. [DOI] [PubMed] [Google Scholar]
- Brandt R, Ebert AD. Growth inhibitors for mammary epithelial cells. Prog Mol Subcell Biol. 1998;20:197–248. doi: 10.1007/978-3-642-72149-6_10. [DOI] [PubMed] [Google Scholar]
- Calonge MJ, Massague J. Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-beta antiproliferative responses in colon cancer cells. J Biol Chem. 1999;274:33637–33643. doi: 10.1074/jbc.274.47.33637. [DOI] [PubMed] [Google Scholar]
- Castoria G, Barone MV, Di Domenico M, Bilancio A, Ametrano D, Migliaccio A, Auricchio F. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J. 1999;18:2500–2510. doi: 10.1093/emboj/18.9.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Ebner R, Derynck R. Inactivation of the type II receptor reveals two receptor pathways for the diverse TGF-beta activities. Science. 1993;260:1335–1338. doi: 10.1126/science.8388126. [DOI] [PubMed] [Google Scholar]
- Darbre P, Yates J, Curtis S, King RJ. Effect of estradiol on human breast cancer cells in culture. Cancer Res. 1983;43:349–354. [PubMed] [Google Scholar]
- Dickson RB, Lippman ME. The molecular basis of breast cancer. In: Mendelsohn J, Howley PM, Israel MA, Liotta LA, editors. The molecular basis of breast cancer. Philadelphia: W. B. Saunders Co; 1995. pp. 358–387. [Google Scholar]
- Edwards DP, Chamness GC, McGuire WL. Estrogen and progesterone receptor proteins in breast cancer. Biochim Biophys Acta. 1979;560:457–486. doi: 10.1016/0304-419x(79)90013-1. [DOI] [PubMed] [Google Scholar]
- Ferguson AT, Davidson NE. Regulation of estrogen receptor alpha function in breast cancer. Crit Rev Oncog. 1997;8:29–46. doi: 10.1615/critrevoncog.v8.i1.20. [DOI] [PubMed] [Google Scholar]
- Haufel T, Dormann S, Hanusch J, Schwieger A, Bauer G. Three distinct roles for TGF-beta during intercellular induction of apoptosis: A review. Anticancer Res. 1999;19:105–111. [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- Kelsey JL. A review of the epidemiology of human breast cancer. Epidemiol Rev. 1979;1:74–109. doi: 10.1093/oxfordjournals.epirev.a036215. [DOI] [PubMed] [Google Scholar]
- Klinge CM. Estrogen receptor interaction with co-activators and co-repressors. Steroids. 2000;65:227–251. doi: 10.1016/s0039-128x(99)00107-5. [DOI] [PubMed] [Google Scholar]
- Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic ribosomal phosphoprotein PO. Nucleic Acids Res. 1991;19:3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KM, Mok SC, Ho SM. Expression of human estrogen receptor-alpha and -beta, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci USA. 1999;96:5722–5727. doi: 10.1073/pnas.96.10.5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence DA. Transforming growth factor-beta: A general review. Eur Cytokine Netw. 1996;7:363–374. [PubMed] [Google Scholar]
- Leong GM, Subramaniam N, Figueroa J, Flanagan JL, Hayman MJ, Eisman JA, Kouzmenko AP. Ski-interacting protein interacts with Smad proteins to augment transforming growth factor-beta-dependent transcription. J Biol Chem. 2001;276:18243–18248. doi: 10.1074/jbc.M010815200. [DOI] [PubMed] [Google Scholar]
- Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. 2000;275:36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci USA. 1997;94:10669–10674. doi: 10.1073/pnas.94.20.10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SL, Kawabata M, Imamura T, Miyazono K, Yuasa Y. Two divergent signaling pathways for TGF-beta separated by a mutation of its type II receptor gene. Biochem Biophys Res Commun. 1999;259:385–390. doi: 10.1006/bbrc.1999.0788. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt AE, Briand P. Indirect mechanism of oestradiol stimulation of cell proliferation of human breast cancer cell lines. Br J Cancer. 1986;53:29–35. doi: 10.1038/bjc.1986.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA, Petrel TA, Song H, Knobloch TJ, Casto BC, Ramljak D, Anderson LM, DeGroff V, Stoner GD, Brueggemeier RW, Weghorst CM. Responsiveness to transforming growth factor-beta (TGF-beta)-mediated growth inhibition is a function of membrane-bound TGF-beta type II receptor in human breast cancer cells. Gene Expr. 2001;9:157–171. doi: 10.3727/000000001783992560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez LA, Chen Y, Pavone A, Fischer SM, Conti CJ. Deregulated expression of cyclin D1 overrides antimitogenic signals. Oncogene. 2000;19:315–322. doi: 10.1038/sj.onc.1203301. [DOI] [PubMed] [Google Scholar]
- Moggs JG, Orphanides G. Estrogen receptors: Orchestrators of pleiotropic cellular responses. EMBO Rep. 2001;2:775–781. doi: 10.1093/embo-reports/kve185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa IA, Bland KI. Physiologic effects of steroid hormones and postmenopausal hormone replacement on the female breast and breast cancer risk. Ann Surg. 1998;228:638–651. doi: 10.1097/00000658-199811000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, O’Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Ravitz MJ, Wenner CE. Cyclin-dependent kinase regulation during G1 phase and cell cycle regulation by TGF-beta. Adv Cancer Res. 1997;71:165–207. doi: 10.1016/s0065-230x(08)60099-8. [DOI] [PubMed] [Google Scholar]
- Ricketts D, Turnbull L, Ryall G, Bakhshi R, Rawson NS, Gazet JC, Nolan C, Coombes RC. Estrogen and progesterone receptors in the normal female breast. Cancer Res. 1991;51:1817–1822. [PubMed] [Google Scholar]
- Roberts AB. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111–119. doi: 10.1159/000057358. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- Rooke HM, Crosier KE. The smad proteins and TGFbeta signalling: Uncovering a pathway critical in cancer. Pathology. 2001;33:73–84. [PubMed] [Google Scholar]
- Rosfjord EC, Dickson RB. Growth factors, apoptosis, and survival of mammary epithelial cells. J Mammary Gland Biol Neoplasia. 1999;4:229–237. doi: 10.1023/a:1018789527533. [DOI] [PubMed] [Google Scholar]
- Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1, and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–408. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Daniel CW. Reversible inhibition of mammary gland growth by transforming growth factor-beta. Science. 1987;237:291–293. doi: 10.1126/science.3474783. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Stoica A, Saceda M, Fakhro A, Solomon HB, Fenster BD, Martin MB. The role of transforming growth factor-beta in the regulation of estrogen receptor expression in the MCF-7 breast cancer cell line. Endocrinology. 1997;138:1498–1505. doi: 10.1210/endo.138.4.5074. [DOI] [PubMed] [Google Scholar]
- Stroschein SL, Bonni S, Wrana JL, Luo K. Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev. 2001;15:2822–2836. doi: 10.1101/gad.912901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HV, Key TJ, Allen DS, Moore JW, Dowsett M, Fentiman IS, Wang DY. A prospective study of endogenous serum hormone concentrations and breast cancer risk in post-menopausal women on the island of Guernsey. Br J Cancer. 1997;76:401–405. doi: 10.1038/bjc.1997.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik C, Bury M, Stoklosa T, Giermasz A, Feleszko W, Mlynarczuk I, Pleban E, Basak G, Omura S, Jakobisiak M. Lovastatin and simvastatin are modulators of the proteasome. Int J Biochem Cell Biol. 2000;32:957–965. doi: 10.1016/s1357-2725(00)00044-3. [DOI] [PubMed] [Google Scholar]
- Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene. 1999;18:4879–4883. doi: 10.1038/sj.onc.1202866. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Saatcioglu F, Matsuda T. Cross-talk between bone morphogenic proteins and estrogen receptor signaling. Endocrinology. 2002;143:2635–2642. doi: 10.1210/endo.143.7.8877. [DOI] [PubMed] [Google Scholar]
- Zentella A, Weis FM, Ralph DA, Laiho M, Massague J. Early gene responses to transforming growth factor-beta in cells lacking growth-suppressive RB function. Mol Cell Biol. 1991;11:4952–4958. doi: 10.1128/mcb.11.10.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]