

SUMMARY
Neuromodulators such as acetylcholine, serotonin, and noradrenaline are powerful regulators of neocortical activity. Although it is well established that cortical inhibition is the target of these modulations, little is known about their effects on GABA release from specific interneuron types. This knowledge is necessary to gain a mechanistic understanding of the actions of neuromodulators because different interneuron classes control specific aspects of excitatory cell function. Here, we report that GABA release from fast-spiking (FS) cells, the most prevalent interneuron subtype in neocortex, is robustly inhibited following activation of muscarinic, serotonin, adenosine, and GABAB receptors—an effect that regulates FS cell control of excitatory neuron firing. The potent muscarinic inhibition of GABA release from FS cells suppresses thalamocortical feedforward inhibition. This is supplemented by the muscarinic-mediated depolarization of thalamo-recipient excitatory neurons and the nicotinic enhancement of thalamic input onto these neurons to promote thalamocortical excitation.
INTRODUCTION
The neocortex is the target of extensive ascending projections releasing neurotransmitters such as acetylcholine (ACh), serotonin (5-HT, or 5-hydroxytryptamine), dopamine, and noradrenaline and is exposed to activity-dependent levels of substances such as adenosine and GABA. These and other neuromodulators have a profound influence on the function of the cerebral cortex due to their effects on the excitability and synaptic properties of neocortical neurons. Moreover, these modulatory systems are frequent therapeutic targets for the treatment of such conditions as anxiety disorders, depression, schizophrenia, and Alzheimer’s disease (Gordon and Hen, 2004; Gray and Roth, 2007; Kasa et al., 1997; McCormick, 1993). Therefore, understanding the cellular mechanisms of action of these neuromodulators is exceedingly important. In cortical structures, this is a challenging task due to the diversity of cellular elements.
Inhibitory neocortical interneurons (INs) releasing the neurotransmitter γ-aminobutyric acid (GABA) are a major target of these modulators (Bacci et al., 2005; Beaulieu and Somogyi, 1991; Smiley and Goldman-Rakic, 1996). Although GABAergic INs are a minority of the neuronal population of the neocortex (10%-20%), they have profuse local axonal arborizations such that a single GABAergic IN can control hundreds, if not thousands, of excitatory cells. Interneurons have key roles in regulating the organization, function, and dynamics of cortical circuits (Buzsaki et al., 2004; Freund and Katona, 2007; McBain and Fisahn, 2001) and are believed to be involved in the pathophysiology of neuropsychiatric disorders such as epilepsy, autism, depression, and schizophrenia (Cossart et al., 2005; Gray and Roth, 2007; Levitt et al., 2004). However, cortical INs constitute a highly diverse group of neurons, with subtypes controlling specific aspects of excitatory cell function (Buzsaki et al., 2004; Freund and Katona, 2007; Kawaguchi and Kubota, 1997; Markram et al., 2004). Knowledge of the effects of neuromodulators on specific interneuron subtypes is very incomplete but is necessary to understand how the effects of these neuromodulators on inhibitory processes contribute to their effects on cortical networks and behavior.
Of particular interest are the effects of neuromodulators on fast-spiking (FS) basket cells, the predominant IN subtype in the mammalian neocortex. FS cell axons preferentially target the soma and proximal dendrites of principal neurons, forming multiple powerful synapses with a high probability of release (Freund and Katona, 2007; Kawaguchi and Kubota, 1997; Markram et al., 2004). Consequently, and given the prevalence of FS cells among GABAergic interneurons, GABA release from FS cells likely constitutes the dominant inhibitory system in neocortex. Although inhibition from dendritic-targeting interneurons can control the efficacy and plasticity of excitatory inputs onto PCs, perisomatic inhibition is ideally suited to control the output and synchronization of excitatory neurons, (Bartos et al., 2007; Freund and Katona, 2007).
Several studies have found that, in contrast to other types of INs, the excitability of FS cells is affected little, if at all, by neuromodulators, leading to the view that these cells tend to operate as a near constant “clockwork” for cortical network oscillations (reviewed in Freund and Katona, 2007). However, in addition to their presence in somatodendritic membranes, receptors for many neuromodulators are also present in presynaptic terminals where they can powerfully regulate neurotransmitter release. In prefrontal cortex, GABA release from synaptic terminals of FS cells is regulated by dopamine (Gao et al., 2003); however, the effects on FS cell terminals of other neuromodulators that have widely distributed receptors in neocortex have not been investigated.
In this study, we investigated the modulation of GABA release from FS INs in somatosensory cortex. We first screened for agents that modulate the inhibitory postsynaptic currents (IPSCs) evoked in L5 pyramidal cells (PCs) by extracellular stimulation using an assay biased toward detecting modulation of GABA release from FS cells. Four out of ten agents tested were found to effectively inhibit eIPSC amplitude via presynaptic mechanisms: muscarine, serotonin, adenosine, and baclofen. We then used paired recordings from synaptically connected FS-to-PC pairs to confirm that the modulations occur at FS cell synapses. The results of activation of muscarinic receptors were particularly strong, and experiments in thalamocortical (TC) slices showed powerful suppression of feedforward inhibition and, as a result, regulation of the window of integration and summation of thalamic inputs and hence the dynamics of the thalamocortical circuit. The effect on feedforward inhibition is complemented by a muscarinic-mediated depolarization of thalamo-recipient excitatory neurons and a nicotinic-mediated enhancement of TC EPSPs on excitatory (but not inhibitory) neurons to facilitate TC excitation of the cortex.
RESULTS
Screening for Modulators of Perisomatic Inhibition of Neocortical Pyramidal Cells
To screen for modulators of GABA release from FS cells, we recorded the IPSCs evoked by extracellular stimulation (eIPSCs) in L5 PCs of somatosensory cortex using a paired-pulse stimulation protocol (Figures 1A and 1B1). IPSCs were recorded in the presence of inhibitors of glutamatergic neurotransmission. eIPSCs reversed near ECl and were blocked by GABAA receptor antagonists. At interpulse intervals ranging from 20 to 500 ms, paired-pulse depression was always observed, with an average paired-pulse ratio (PPR, calculated as mean IPSC2/mean IPSC1) of 0.84 ± 0.04 (n = 165) for the 20 Hz stimulation used in most experiments. Pharmacological agents were applied directly onto the soma of the recorded cell using fast local puffs through a second patch pipette located ∼5-10 μm away from the PC soma (Figure 1A). This restricted drug action to an ∼50 μm radius from the puffing pipette tip (see Figure S1 available online), therefore sampling effects on perisomatic inputs.
Figure 1. Presynaptic Modulation of Evoked IPSCs by Local Activation of mAChRs.
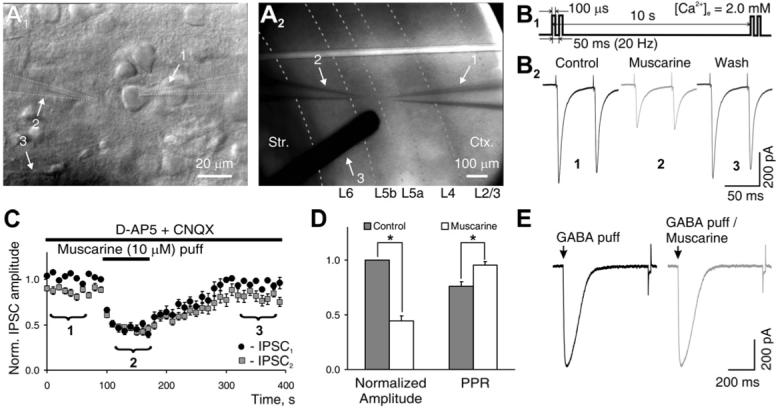
(A1) Recording from an L5 PC: 1, patch pipette; 2, puffing pipette; 3, shadow from bipolar concentric stimulation electrode with 125 μm tip diameter. (A2) Photomicrograph of a 300 μm thick coronal slice of the rat somatosensory cortex with recording electrode positioned in L5a and electrical stimulation electrode placed laterally in L5. Dotted lines indicate borders between layers. (1-3 as in [A1]).
(B1) Paired-pulse stimulating protocol used for studying modulatory effects of locally applied compounds. (B2) eIPSCs recorded from L5 PCs and their modulation by locally puffed muscarine, averaged over the time windows denoted as 1, 2, and 3 in (C). Experiments were performed in the presence of AMPA/NMDA receptor blockers to isolate inhibitory synaptic transmission.
(C) Time course of changes in normalized eIPSC amplitude to first (black symbols) and second (gray symbols) stimuli produced by muscarine puff (data averaged from five experiments).
(D) Population data from all experiments (n = 20), representing the changes in normalized mean amplitude of first IPSC and paired-pulse ratio (PPR) in control (gray column) and during muscarine puff (white column).
(E) Currents evoked by a brief (1 ms) local application of GABA (100 μM) to an L5 PC in control (left panel) and in the presence of a bath-applied muscarine (right panel). Traces are averages of ten trials. Summary graphs show mean ± SEM, *p < 0.01, paired t test. Ctx, cortex; Str, striatum.
We used this experimental paradigm to screen several neuromodulators known to activate signaling pathways following association with G protein-coupled receptors (GPCRs). We found that the majority of the agents (6 out of 10) had no detectable effect on the eIPSCs (Table 1). However, four of the compounds, muscarine, adenosine, serotonin, and baclofen, produced consistent effects on the eIPSCs, suggesting robust modulation of inhibitory synaptic transmission.
Table 1.
The Agonists of Different GPCRs Tested with the Assay Described in the Text
| Agonist | Muscarine | Serotonin | Baclofen | Adenosine | t-ACPD | Norepinephrine | HTMT | WIN 55,212-2 | SOM-14 | nsCCK-8 |
|---|---|---|---|---|---|---|---|---|---|---|
| Receptor | mAChRs | 5-HT (1-7) | GABAB | Adenosine (A1-A3) | Group I and II mGluRs | α,β adreno-receptors | H1 and H2 | CB1 and CB2 | Somatostatin | Cholecystokinin |
| Concentrations tested | 1-20 μM | 1-20 μM | 1-10 μM | 10-50 μM | 1-20 μM | 10-50 μM | 1-20 μM | 1-5 μM | 1-10 μM | 1-10 μM |
| Effect | Inhibition of perisomatic GABA release | Inhibition of perisomatic GABA release | Inhibition of perisomatic GABA release | Inhibition of perisomatic GABA release | No change of perisomatic GABA release | No change of perisomatic GABA release | No change of perisomatic GABA release | No change of perisomatic GABA release | No change of perisomatic GABA release | No change of perisomatic GABA release |
| Number of cells | 24 | 20 | 15 | 10 | 5 (p > 0.85*) | 10 (p > 0.95*) | 5 (p > 0.9*) | 7 (p > 0.9*) | 5 (p > 0.8*) | 5 (p > 0.85*) |
All drugs were locally perfused onto L5 PCs in somatosensory cortex.
Paired t test.
Locally applied muscarine (1-20 μM), an agonist of muscarinic acetylcholine receptors (mAChRs), produced a robust and reversible reduction of eIPSC amplitude in 24 out of 24 cells tested (Figures 1B and 1C). On average, 10 μM muscarine inhibited the amplitude of eIPSCs by 62% ± 8% (Figure 1D; range 45%-86%, p < 0.001, paired t test, n = 20). This effect of muscarine is likely of presynaptic origin because muscarine did not produce changes in the rates of rise and decay of the eIPSCs or the input resistance (Rin) of the postsynaptic cell. Moreover, inhibition of the IPSC by muscarine was accompanied by an increase in the PPR (Figure 1D). In addition, muscarine had no effect on the currents elicited in PCs by brief local application of GABA (p > 0.8, n=5, Figure 1E).
Serotonin and adenosine produced smaller but consistent effects on the eIPSCs. Local application of serotonin (1-10 μM) produced a reversible inhibition of eIPSCs in 16 out of 20 cells tested (Figure 2A). On average 10 μM serotonin inhibited the amplitude of eIPSCs by 27% ± 6% (Figure 2D, range 19%-35%, p < 0.001 paired t test, n = 13). Increasing the serotonin concentration 5-fold to 50 μM did not produce additional inhibition. Adenosine (10-50 μM) produced a reversible inhibition of the eIPSCs in 10 out of 10 cells tested (Figure 2B), with an average reduction of eIPSC amplitude of 39% ± 6% for 50 μM (Figures 2B and 2D, range 29%-55%, p < 0.0001 paired t test, n = 7). The effects of adenosine and serotonin on the amplitude of the eIPSCs were also of presynaptic origin because there were no changes in the kinetics of the eIPSCs, the Rin of the postsynaptic cell, or the currents elicited by local application of GABA (p > 0.9, n = 5 in each case, Figures 2A and 2B), and both resulted in an increase in the PPR (Figure 2E). Activation of GABAB receptors by local application of baclofen (1-10 μM) potently and reversibly inhibited the amplitude of eIPSCs in 10 out of 10 cells tested (Figure 2C), with 10 μM producing an average reduction of the eIPSCs of 75% ± 5% (Figures 2C and 2D, range 55%-91%, p < 0.0001 paired t test, n = 7). As in the case of the previous modulators, baclofen did not change the rates of rise and decay of the eIPSCs or the Rin of the post-synaptic cell and produced an increase in the PPR (Figure 2E). Furthermore, baclofen application did not affect the amplitude of the currents elicited by local application of GABA onto the PC (p > 0.9, n = 5, Figure 2C), indicating a presynaptic mechanism of action. The effect of baclofen was prevented by preapplication of the specific GABAB receptor antagonist CGP55845 (Figure S2B).
Figure 2. Presynaptic Modulation of eIPSCs by Local Activation of 5-HT, Adenosine, and GABAB Receptors.

(A) Serotonin modulation of eIPSCs. (Left panel) eIPSCs recorded from L5 PCs and their modulation by locally puffed serotonin, averaged over the time windows denoted as 1, 2, and 3 in the middle panel. (Middle panel) Time course of changes in normalized first and second eIPSC amplitude in response to serotonin puff (data averaged from five experiments). (Right panel) Effect of serotonin on currents induced by GABA puff.
(B and C) Same as (A), but for the effect of adenosine and baclofen, respectively.
(D) Population data describing the effect of serotonin (n = 13), adenosine (n = 7), and baclofen (n = 7) on the amplitude of eIPSCs.
(E) Changes in paired-pulse ratios produced by the above-mentioned compounds. Summary graphs show mean ± SEM; *p < 0.01; **p < 0.001, paired t test.
It is known that muscarine can depolarize a subtype of somatostatin-containing INs known as LTS cells (but not FS cells, see below), which could lead to an increase in cortical GABA concentration (Beierlein et al., 2000). Elevated GABA could inhibit GABA release from the INs responsible for the eIPSC by activating presynaptic GABAB receptors. However, the GABAB receptor blocker CGP55845 did not affect the action of muscarine on the eIPSC (Figure S2A).
It should be noted that the aforementioned recordings from PCs were made with a Cs+-based intracellular solution that effectively blocks many K+ channels, such as G protein-activated inward rectifier K+ channels (GIRKs), that otherwise might be targets of the modulators investigated here. The effects of muscarine and baclofen in cells recorded with physiological intracellular solutions are described later.
FS Basket Cells Are the Major Contributors to the eIPSC
We reasoned that the assay we used in the previous experiments is biased toward detecting effects of modulators on the eIPSC resulting from GABA release from FS INs based on the following arguments.
First, although the axons of many interneuron types are stimulated with an extracellular electrode, most of the eIPSC in L5 PCs is likely to arise from FS cells because they are the largest (∼ 50%) population of INs in L5 and, given the perisomatic localization of their terminals, should produce larger somatically recorded IPSCs (Kawaguchi and Kubota, 1997; Markram et al., 2004). To explore this quantitatively, we compared the properties of unitary synaptic connections onto L5 PCs from identified FS basket cells and the second largest (30%-40%) class of L5 INs, the somatostatin-positive Martinotti cells (MCs) (Figure 3; Kawaguchi and Kubota, 1997; Markram et al., 2004). The strength of the unitary synaptic connections measured at the PC soma was about 10-fold smaller for MC-to-PC pairs compared to FS-to-PC pairs (13 ± 5.4 pA versus 129 ± 24 pA, p = 0.0004, Mann-Whitney U test). Moreover, the synaptic delay and the latency to peak of the unitary IPSCs (uIPSCs) were much longer and less synchronous in MC-to-PC versus FS-to-PC pairs (Figure 3D). These observations suggest a minor contribution of MCs to the peak amplitude of the eIPSC measured in L5 PC somata. Furthermore, the peak of somatically recorded uIPSC from MCs occurs after the peak of the eIPSC (4.46 ± 0.3 versus 2.6 ± 0.5 ms, n = 10).
Figure 3. FS Basket Cells Mediate Most of the eIPSC in L5 PCs.

(A-D) Properties of unitary synaptic connection of FS and Martinotti cells onto L5 PCs. (A) Representative high-frequency nonaccommodating firing pattern of a L5 FS cell. (Lower panel) Depressing uIPSCs in a PC produced by stimulation of an FS cell with five brief current injections (0.5 nA, 2 ms) at 50 Hz (ratio between fifth and first IPSC = 0.25 ± 0.05, Rin(FS) = 89 ± 15 MΩ, τmembr(FS) = 9.2 ± 1.5 ms, n = 8). (B) Representative firing pattern of an L5 MC, showing low spike threshold and rebound spikes. (Lower panel) Facilitating uIPSCs in an L5 PC produced by stimulation of an MC at 50 Hz (IPSC5/IPSC1 = 0.91 ± 0.07, Rin(MC) = 241 ± 55 MΩ, τmembr(MC) = 19.2 ± 3.5 ms, n = 5). (C) Morphological reconstruction of a connected MC-to-PC pair, blue dots indicate putative synaptic contacts between MC axon (red) and PC dendrite (blue). Dendrites of MC shown in green, the axon of PC in gray. (D) (Left) Quartile plot of uIPSCs amplitudes at 30°C. The central dot is the mean, the central line is the median value, the box edges mark the interquartiles, and the bars mark the limits of the sample distribution. (Center, right) Distribution histograms of synaptic delay and the latency to the peak of uIPSCs measured relative to the peak of presynaptic action potential. The data were pooled from five connected pairs (ten trials each). *p < 0.001 Mann-Whitney U test.
(E and F) Ca2+ channels mediating extracellularly evoked GABA release. (E) (Right) Representative example showing modest inhibition of eIPSCs in L5 PC by N-type Ca2+ channel blocker and lack effect of this treatment on the baclofen-induced reduction of eIPSCs. (Left) Blocking P/Q-type Ca2+ channels dramatically inhibits eIPSCs and suppresses the effect of baclofen. (F) P/Q-type Ca2+ channel blocker insensitive component of the eIPSC shows facilitating profile (top) as well as fast kinetics of rise and decay similar to total eIPSC (bottom). Black traces, control; blue, after ω-Aga-Tx-IVa.
Second, it has been shown in the hippocampus that FS cells utilize P/Q-type Ca2+ channels for synaptic transmission, while other interneurons making synapses near the soma of pyramidal cells utilize N-type Ca2+ channels (Hefft and Jonas, 2005; Wilson et al., 2001). We investigated whether FS cells in barrel cortex also utilize exclusively P/Q-type Ca2+ channels for synaptic transmission. As illustrated in Figure S3, unitary FS-PC connections in L5 barrel cortex were insensitive to bath application of the Cav2.2 (N-type) Ca2+ channel blocker ω-conotoxin-GVIa (1 μM) but were completely and irreversibly blocked by the Cav2.1 (P/Q-type) Ca2+ channel antagonist ω-agatoxin-IVa (500 nM). We then tested the effect of blocking N- or P/Q-type Ca2+ channels on the eIPSCs. Blocking N-type Ca2+ channels reduced the amplitude of eIPSCs by 15% ± 4% (Figure 3E, n = 5, p < 0.01, paired t test). In contrast, blocking P/Q-type Ca2+ channels dramatically reduced the amplitude of the eIPSCs by 89% ± 5% (Figure 3E, n = 3, p < 0.001, Wilcoxon test). Interestingly, the component of the eIPSC remaining after blocking P/Q-type Ca2+ channels displayed facilitating short-term dynamics, instead of the depression seen for the total eIPSC (Figure 3F). Hence, the contribution of the N-type Ca2+ channel-dependent eIPSC to the first total eIPSC of the train is about 10%; however, it rises during repetitive stimulation reaching ∼30% at the fifth stimulus at 50 Hz. This component also showed fast kinetics of rise and decay (Figure 3F), suggesting it does not arise from Martinotti cells, but from a population of non-FS interneurons that form proximal synapses on the PC. This component could arise from CCK basket cells, which display facilitating synapses (Galarreta et al., 2004) or other non-FS cells making proximal connections on the pyramidal cell. Next, we explored the effects of the neuromodulators on the eIPSCs in the presence of the Ca2+ channel blockers. We found that the N-type Ca2+ channel blocker did not affect the magnitude of the baclofen-induced inhibition of eIPSCs (Figure 3E, 71% ± 6% versus 75% ± 5%, n = 3, p > 0.8, Wilcoxon test) or the magnitude of the muscarine effect (51% ± 7% versus 55% ± 8%, n = 2, data not shown), further suggesting that their actions occur on FS cell terminals.
Third, we found that local somatic application of low doses (1 mM) of tetraethylammonium (TEA), which blocks Kv3 potassium channels in FS cell terminals, increased the amplitude of eIPSCs by a factor of 1.88 ± 0.11 (range 1.58-2.01, n = 8) without affecting the shape of the IPSC (Figure S4). The magnitude of the observed effect of TEA is similar to that reported for the effect of 1 mM bath-applied TEA on the unitary IPSC in FS-to-PC connections (1.62 ± 0.25; Goldberg et al., 2005), further supporting the contention that the majority of the eIPSCs on L5 PCs arises from FS cells. Finally, as shown below, all four neuromodulators that affected the eIPSC had quantitatively similar effects on unitary FS-to-PC connections.
Modulation of Unitary FS-to-PC Synaptic Transmission
To confirm that GABA release from FS cell synapses is regulated by the four neuromodulators affecting the eIPSC, we performed dual whole-cell recordings from connected FS-to-PC pairs in L5. We found that bath-applied serotonin, muscarine, adenosine, and baclofen inhibited the amplitude of the uIPSC to a similar extent as the eIPSC (Figure 4, n = 5 in each case, p < 0.01, paired t test), without affecting uIPSC kinetics. We also monitored the PPR and the coefficient of variation (CV) of uIPSC amplitudes. While changes in CV do not unequivocally reflect a presynaptic mechanism, these two parameters are widely used to evaluate changes in neurotransmitter release probability (Zucker and Regehr, 2002). We found that all four reagents increased the PPR and the CV of uIPSCs (Figure 4, p < 0.01, paired t test), supporting a presynaptic mechanism of action.
Figure 4. Modulation of FS-to-PC Connections.

(A) (Left) Inhibition of uIPSCs triggered by brief current injections delivered to a L5 FS cell (0.5 nA, 2 ms at 20 Hz) and recorded in a nearby PC by bath application of muscarine (10 μM). Responses shown are the average of ten sweeps. (Right) Population data showing normalized mean amplitude of first IPSC, paired-pulse ratio, and coefficient of variation in control (filled column) and during muscarine application (white column).
(B-D) same as in (A), but for serotonin (10 μM), adenosine (50 μM), and baclofen (10 μM), respectively. Summary graphs show mean ± SEM; *p < 0.01 paired t test.
The experiments described so far were carried out in infragranular layers. We asked whether the two most potent modulators of GABA release from FS cells in these layers—muscarine and baclofen—would exhibit the same effect in supragranular layers. As shown in Figure S5, muscarine and baclofen inhibited unitary synaptic transmission from FS-to-L3 PCs similarly to their action in infragranular layers, suggesting that the modulation of perisomatic GABA release occurs in FS cells throughout somatosensory cortex.
Two Pathways for the Modulation of GABA Release
Observation of the time course of drug action on the eIPSC suggested that the effect of serotonin was slower than the effects of muscarine, adenosine, or baclofen (Figures 1 and 2). However, the stimulation frequency (0.1 Hz) used in those experiments did not provide sufficient resolution to accurately measure the rates of the modulatory effects. Therefore, we increased the time resolution of our assay by stimulating at 0.5 Hz while decreasing the extracellular Ca2+ concentration to 1.3 mM (Figure 5). Reducing extracellular Ca2+ was necessary to lower the release probability and minimize short-term synaptic depression at these rates of stimulation. We found that the effect of local application of serotonin developed after a 6.5 ± 0.9 s delay, and the time course of the modulation, fitted with the logistic Hill function, yielded a time constant of 17.5 ± 4.2 s (n = 5, r2 = 0.85). On the other hand, the effects of muscarine, adenosine, and baclofen showed no delay, and their time constants were 7 ± 2.0 s (n = 5, r2 = 0.89), 8 ± 3.1 s (n = 5, r2 = 0.94), and 6 ± 3.9 s (n = 5, r2 = 0.96), respectively. Such a “slow” and “fast” time course of eIPSC inhibition suggests that two different mechanisms of modulation are involved.
Figure 5. Two Types of Modulation of GABA Release.

(A) A protocol used for studying the time course of modulation. Note reduced extracellular [Ca2+].
(B) “Slow” and delayed modulation of eIPSCs amplitude by locally applied serotonin and “fast” eIPSC modulation by local application of muscarine, adenosine, and baclofen. eIPSCs were normalized and averaged from five experiments. The datasets were fitted with the logistic Hill function, with “zero” time at the beginning of drug application. The time constants of the fit are shown in red.
(C and D) “Slow” but not “fast” modulation requires activation of protein kinases. (C) Representative example (top left) demonstrating the absence of serotonin effect on eIPSCs amplitude after preincubation of slice with staurosporine for 2 hr. (Other panels) Examples of staurosporine inability to preclude the effects of muscarine, adenosine, and baclofen. Drug concentrations are the same as in (B). (D) Population data comparing drug effects with (gray columns) or without (white columns) staurosporine preincubation. Shown are means ± SEM; *p < 0.01, unpaired t test.
To explore this conclusion further, we asked whether protein phosphorylation is required for the effects of each modulator. We incubated the slices for 2 hr with staurosporine (1 μM), a protein kinase (PK) inhibitor able to block many PKs (PKA = PKC > PKG > CaMK) (Hidaka and Kobayashi, 1993). We found that the effect of serotonin on the eIPSC was the only modulation affected by staurosporine (2% ± 4.0% inhibition of the eIPSC in the presence of staurosporine versus 27% ± 6% inhibition in control slices, Figures 5C and 5D, n = 10, p < 0.01, unpaired t test). The inhibitory effects of muscarine, adenosine, and baclofen were not affected by the treatment with staurosporine (Figures 5C and 5D, p > 0.5, unpaired t test). This suggests that the “slow” modulation observed with serotonin involves a PK-dependent second messenger pathway, while the “fast” modulations produced by muscarine, baclofen, and adenosine are independent of PK activity and likely involve the membrane-delimited inhibition of presynaptic Ca2+ channels (Hille, 2001; however, see Discussion).
These two types of signaling mechanisms are mediated by distinct G proteins activated by specific receptor subtypes. The membrane-delimited modulation typically requires association of the target molecules, such as the α1 subunit of neuronal Ca2+ channels, with the βγ subunits of pertussis toxin-sensitive Gi/o protein (Dolphin, 2003; Mirotznik et al., 2000). While it is known that adenosine A1 receptors and GABAB receptors are coupled to Gi/o proteins (Wettschureck and Offermanns, 2005), the large molecular diversity of serotonin and muscarine receptors requires further analysis to determine the receptor type that mediates the modulation of GABA release.
Pharmacological studies presented in supplementary material (Figures S6 and S7) suggest that the effect of serotonin on the eIPSC is mediated by 5-HT2 receptors. These receptors are coupled to Gq/11 protein that activates the PLC pathway and subsequently PKC (Wettschureck and Offermanns, 2005), supporting the hypothesis that PK activity mediates the action of serotonin on IPSCs. On the other hand, the effect of muscarine appears to be mediated by M2 and M4 receptors, which are coupled to Gi/o proteins, supporting the conclusion that the muscarinic effect on eIPSCs involves the membrane-delimited pathway.
Modulation of GABA Release from FS Cells and the Control of Excitatory Cell Activity
We showed that four neuromodulators robustly inhibit GABA release from FS cells—muscarine and baclofen being particularly effective. The significance of this reduction in perisomatic inhibition was tested by investigating how muscarine affects the ability of FS cells to control the activity of neocortical PCs (Figure 6). These experiments used intracellular Cl- concentrations to match the physiological Cl- reversal potential (∼-70 mV; Cossart et al., 2005). Layer 3 PCs were depolarized to produce sustained action potential (AP) discharge at a rate of ∼5 Hz, by a 30 pA current injection. A train of APs (30 spikes at 40 Hz) evoked in a single connected FS cell was able to dramatically reduce the firing probability of the PC (by 75% ± 7%, p < 0.01, n = 4, Wilcoxon test). Bath application of 10 μM muscarine significantly reduced the IPSPs recorded in the postsynaptic PC (Figure 6A, inset) and as a result prevented the GABA-mediated silencing of PC firing (Figures 6A and 6C). We also investigated how modulators change the effect of perisomatic inhibition on the discharge of PCs evoked by afferent stimulation. Strong stimulation of white matter (Figure 6B, inset) was required to drive deep L3 PCs to firing threshold. Spike probability was severely reduced (by 93% ± 5%, p < 0.01, n = 4, Wilcoxon test, Figures 6B and 6C) by a few APs evoked in a single connected FS IN. Bath application of muscarine significantly relieved the inhibitory control of PC discharge imposed by the FS IN (31% ± 7% versus 93% ± 5% firing suppression, p < 0.01 Wilcoxon test, Figures 6B and 6C).
Figure 6. Modulation of the Inhibitory Control of PC Activity.

(A) Representative recording from a connected pair of an L3 PC and an FS IN. (Top) An overlay of 15 sweeps recorded from the PC following injection of 30 pA depolarizing current (4 s). A train of 30 APs at 40 Hz in the connected FS IN (bottom) silenced PC firing. Bath application of muscarine relieved the silencing of the PC (middle panel). (Inset) Muscarine-induced reduction in uIPSPs (scale bar = 1.5 mV/150 ms, Vm(PC) = -55 mV, ECl- = -70 mV).
(B) Same as in (A), but APs in the PC (Vm = -60 mV) were evoked by white matter (w.m.) stimulation (inset). Five APs in FS IN preceding w.m. stimulation (bottom) reliably suppressed AP generation in PC (top). Bath application of muscarine prevented FS cell-mediated suppression of PC firing (middle).
(C) Probability of PC firing calculated as the ratio of number of APs in PC during FS IN firing relative to this number in the absence of FS IN stimulation (black bar). The same ratio in the presence of muscarine (blue bar). *p < 0.01, n = 4, Wilcoxon test. Shown are means ± SEM.
Modulation of Thalamocortical Integration
The robust inhibition by muscarine of GABA release from neocortical FS cells suggests that this modulation might contribute to mediating the powerful actions of ACh on cortical function. To investigate the physiological significance of this modulation, we studied a neocortical circuit in which synaptic transmission from FS cells has a well-defined role. It is known that individual thalamocortical (TC) afferents contact both excitatory neurons and FS interneurons in L4 and deep L3. Activation of both excitatory and inhibitory cells establishes a simple disynaptic circuit that provides powerful, local feedforward inhibition (Figures 7A and 8F). The latency between the onset of the TC excitation of excitatory cells and the onset of feedforward inhibition results in a temporal integration window (IW) during which the excitatory neurons can integrate TC inputs. This window is critical for the processing of sensory information in the neocortex (Cruikshank et al., 2007; Gabernet et al., 2005; Miller et al., 2001; Swadlow, 2003; Wilent and Contreras, 2004, 2005).
Figure 7. Neuromodulators Regulate Thalamocortical Feedforward Inhibition and Integration.

(A) A cartoon of TC slice preparation depicting the schematics of the feedforward inhibitory circuit and the recording arrangement.
(B) Overlay of 15 EPSC/IPSC sequences (Vh = -45 mV) before, during, and after local perfusion of muscarine (10 μM). Bold line is the average of the currents (excluding failures) under each condition. The “window of integration” is denoted as IW (see text).
(C) Population data representing muscarine effect on IW (n = 6). Shown are means ± SEM.
(D) Application of baclofen (10 μM) eliminates the outward feedforward inhibitory component of EPSC/IPSC sequence and strongly inhibits the excitatory component (traces are averages of ten sweeps).
(E) Ten superimposed EPSP/IPSP sequences (L3/4 EC, Vm = -45 mV) with sim-EPSPs triggered at different intervals (at 1, 3, 5, and then every 4 ms) after thalamic stimulation, with (right) and without (left) local application of muscarine. Bold line is the average of 15 EPSP/IPSP sequences without sim-EPSCs injection (shown superimposed in the top-right inset). (Bottom) Time course of sim-EPSC injection.
(F) Summation of thalamic response and sim-EPSPs calculated as the ratio of the value of the response minus the value of averaged EPSP/IPSP sequence at the time of sim-EPSP peak (a) to the amplitude of sim-EPSP (b, top-left inset in [E]). Shown are means ± SEM; *p < 0.01, paired t test.
Figure 8. Modulation of Specific Components of the Feedforward TC Circuit.
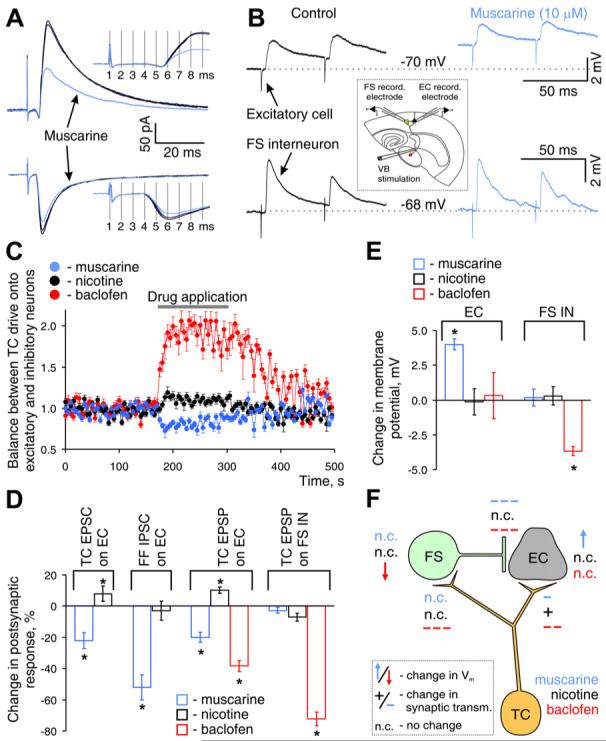
(A) Electrical segregation of feedforward inhibitory and excitatory components of the excitatory-inhibitory sequence produced by TC stimulation and their modulation by locally applied muscarine. PSCs were recorded in L3 ECs held at EAMPA (∼0 mV, top) and ECl- (∼-70 mV, bottom). Insets show the absence of muscarine-induced changes in the latency of each PSC.
(B) TC EPSPs simultaneously recorded in a GFP-positive FS IN and a nearby EC in L3/4 (traces represent the averages of ten sweeps) and their modulation by bath application of muscarine ([inset] diagram of experimental design).
(C) Modulator-induced shift in the balance of TC excitatory drive onto ECs and FS INs, computed as the ratio of the amplitude of TC EPSP in EC over TC EPSP in FS IN, both normalized to values before drug application. Plots are the averages of four experiments.
(D) Population data of modulator effect on post-synaptic response, plotted relative to corresponding value before drug application. TC, thalamocortical; FF fedforward.
(E) Population data on change in resting membrane potential of ECs and FS INs produced by bath application of the indicated modulator.
(F) Schematic diagram summarizing effects of neuromodulators on each component of feed-forward TC circuit. “+” or “-” denotes potentiation or inhibition of synaptic transmission, with the strength of the effect indicated by the number of symbols; arrows denote change in Vm (up depolarization). Summary graphs show mean ± SEM. p < 0.01, paired t test.
We recorded sequences of excitatory/inhibitory postsynaptic events from deep L3/L4 excitatory cells (ECs) evoked by stimulation of the ventrobasal (VB) nucleus of the thalamus in the thalamocortical slice preparation and studied the effect of muscarine on the apparent IW measured as the duration of the net inward current in EPSC/IPSC sequences (Figures 7B and 7C). Local muscarine application significantly decreased the disynaptic inhibitory response and consequently increased the apparent IW by 55% ± 6% (Figures 7B and 7C; n = 6, p = 0.017, Wilcoxon test).
The effect of muscarine was not due to failure in the firing of FS cells in response to TC stimulation, because we could clearly distinguish such failures by the absence of the outward (inhibitory) component in EPSC/IPSC sequences (see, for example, the two lower traces in the middle panel in Figure 7B, and note the absence of the notch produced by the onset of disynaptic IPSC). Failures occurred occasionally in the presence or absence of muscarine, but on average, we did not observe a muscarine-induced increase in failure rate of FS IN firing in response to TC stimulation (p > 0.8, n = 6; failure rates were 15% ± 6% in absence and 18% ± 7% in presence of muscarine). This stands in contrast to the effects of activation of GABAB receptors on TC responses. Application of baclofen (10 μM), which produces potent inhibition of GABA release from FS cells, also prolonged the apparent IW (n = 3, Figure 7D). However, in this case there was a complete elimination of the inhibitory component of the TC response, as can be appreciated by the lack of an outward current in the presence of baclofen (Figure 7D). Therefore, the action of this drug on the IW seems to result primarily from an inhibitory effect on the TC-to-FS IN connection that prevents FS cell from firing (see below).
To appreciate the effects of the muscarinic modulation of TC responses on the integrative properties of excitatory thalamo-recipient cells, we investigated how simulated EPSPs (sim-EPSPs) summate with the TC-evoked disynaptic EPSP/IPSP sequence (Figure 7E). Individual EPSPs on thalamo-recipient ECs are small, and hence EPSP summation is necessary to excite these cells and initiate cortical excitation (Cruikshank et al., 2007). sim-EPSPs were generated in ECs by injecting prerecorded TC EPSCs in current-clamp mode. We found that summation of sim-EPSPs with the TC response depended on the timing of TC EPSC injection. There is a narrow IW (range 1-2 ms, n = 4) during which sim-EPSP can effectively summate with the TC response, followed by a refractory period with a pronounced sublinear summation (13 ± 0.8 ms required for 90% recovery, n = 4, Figures 7E and 7F), likely explained by the potent shunt produced by perisomatic GABA release (Gabernet et al., 2005). Local application of muscarine significantly increased the summation efficacy during the initial IW (113% ± 8% versus 84% ± 6%, n = 4, p = 0.027, Wilcoxon test) and decreased the duration of the refractory period (Figures 7E and 7F; 9 ± 0.6 ms required for 90% recovery, n = 4), likely a result of the muscarinic reduction of the GABA-mediated shunt (Figure 7E, right inset).
We next investigated the effects of neuromodulators on the other elements of the feedforward thalamocortical circuit (see summary diagram in Figure 8F) in order to appreciate how the observed effects on feedforward inhibition may combine with other effects to modulate thalamic excitation of the cortex. We first studied the effect of local muscarine application on the isolated excitatory and inhibitory component of the EPSC/IPSC sequence by recording PSCs at the reversal potential for the respective neurotransmitter current. The isolated disynaptic inhibitory component was much more sensitive to muscarine than the monosynaptic excitatory component (54% ± 8% versus 18% ± 4% reduction, n = 4, Figures 8A and 8D). Muscarine did not change the latency or time to peak of either the monosynaptic excitatory or the disynaptic inhibitory component (Figure 8A, insets), suggesting modulation of the same synaptic connections rather than recruitment of distinct connections following drug application.
Second, we investigated the effect of muscarine on the TC drive to FS and excitatory cells in layers 3/4, by simultaneously recording TC EPSPs in both cell types (Figures 8B-8D). Stimulation of TC afferents produced significantly larger EPSPs in FS INs than in ECs (7.4 ± 2.1 mV versus 2.3 ± 0.6 mV, n = 12, p < 0.01 paired t test), as previously reported (Cruikshank et al., 2007). Bath application of muscarine shifted the balance of TC drive by slightly inhibiting TC EPSPs on ECs, while not affecting TC EPSPs on FS INs (Figures 8B-8D). In contrast, activation of GABAB receptors inhibited the TC EPSP on FS INs twice as potently as the TC EPSP on ECs (Figures 8C and 8D, right panel, and Figure S8). We also studied the effect of muscarine on the membrane properties of FS INs and ECs. Muscarine depolarized ECs and slightly decreased Rin; while not affecting the membrane properties of FS INs (Figure 8E and Figure S9).
Because ACh activates both nicotinic and muscarinic receptors, we also investigated the effects of nicotine on the various components of the TC feedforward circuit. Contrary to the effects of muscarine, 5 μM nicotine increased the amplitude of TC EPSPs on ECs without affecting the TC drive to FS INs (Figure S10E and Figures 8C and 8D). Furthermore, a local nicotine puff increased the amplitude of the excitatory component of the excitatory-inhibitory PSC sequence in response to TC stimulation (Figure 8D and Figure S10B), but did not affect the disynaptic GABA release from FS cells or the apparent IW (Figure S10C). Nicotine application did not affect the membrane potential or Rin of the FS INs or the ECs (Figure 8E and Figure S9).
Thus, in summary, our results show that cholinergic action facilitates thalamocortical excitation of primary somatosensory cortex primarily by reducing feedforward inhibition. This effect is complemented by a nicotinic-mediated increase in the TC drive onto thalamo-recipient ECs as well as the muscarinic-mediated increase in the excitability of these cells (Figure 8F). In contrast, activation of GABAB receptors inhibits all synapses in the circuit, but most potently TC excitation of FS INs and perisomatic GABA release, therefore effectively excluding FS cells from generating feedforward inhibition.
DISCUSSION
In this study, we developed an assay to screen for modulators of GABA release from FS basket cells. We found that activation of four out of ten G protein-coupled receptors (GPCRs) tested— muscarinic, serotonin, adenosine, and GABAB receptors—produced, via two different mechanisms, robust inhibition of perisomatic GABA release. We confirmed that these modulations occur on terminals of FS basket cells using paired recordings from synaptically connected FS-to-pyramidal cell pairs.
These modulators have powerful effects on the function of the neocortex, and it is likely that the observed effects on GABA release from FS cells, the source of the dominant inhibitory system in neocortex controlling the output of principal cells, contribute significantly to their actions on cortical function. The effects of muscarine, which were particularly strong, are of special interest, given the importance of cholinergic modulation of cortical activity. We showed that the potent inhibition of GABA release from FS cells produced by activation of muscarinic cholinergic receptors regulated the dynamics of thalamocortical activation.
FS Basket Cells Mediate Most of the eIPSC in L5 PCs
To screen for neuromodulators of GABA release from FS cells, we tested the effects of locally applied reagents on the somatically recorded eIPSC on L5 PCs in somatosensory cortex. We show that this assay is biased toward detecting modulation of GABA release from FS cells based on evidence that the large majority (∼90%) of the eIPSC under our recording conditions is mediated by GABA release from this class of interneurons.
Dendritically targeting Martinotti cells contribute little to the somatically recorded eIPSC given the amplitude and timing of their unitary currents; however, these neurons may play an important role in the local dendritic integration or be a source of a potent recurrent inhibition following high-frequency repetitive PC activity (Kapfer et al., 2007; Silberberg and Markram, 2007).
Another class of basket cells—CCK-positive basket cells—also have a minor contribution to the amplitude of the eIPSC, given their low abundance in L5 (∼5% of all interneurons) and significantly fewer number of perisomatic contacts on large PCs in L5. Moreover, CCK basket cells receive less glutamatergic input and are more difficult to drive than FS cells (Freund and Katona, 2007). Consistent with this conclusion, there was no detectable effect on the eIPSC upon activation of presynaptic cannabinoid (CB1) receptors (Table 1), which are present in a large proportion of CCK-basket cell terminals and mediate inhibition of neurotransmitter release (Bodor et al., 2005).
Together, our data strongly indicate that under our experimental conditions GABA release from FS basket cells constitutes the majority of the eIPSC in L5 PCs, suggesting that FS cells are responsible for the dominant part of the inhibition converging onto PC somata evoked by electrical stimulation of nearby cells and axons. Monitoring eIPSC in these neurons can be used reliably and efficiently to screen modulators of GABA release from FS INs and to study kinetic and pharmacological properties of these modulations without having to resort to more complicated paired recordings.
Mechanisms of Modulation of GABA Release from FS Cell Terminals
Interestingly, we found that the modulations characterized in this study appear to be mediated by two different mechanisms, although all four modulators produced inhibition of GABA release. We showed that the effect of serotonin had a relatively slow onset and involved the activation of protein kinases (PKs) by Gq/11 proteins coupled to 5-HT2 receptors, which are known to be present in cortex and specifically in GABAergic PV-containing basket interneurons (Jakab and Goldman-Rakic, 2000). Serotonin binding to 5-HT2 receptors leads to the activation of phospholipase C (PLC-β), an enzyme that generates two intracellular second messengers: inositol (1,4,5) trisphosphate and diacylglycerol. These signals in turn produce an elevation in intracellular Ca2+ and activation of protein kinase C (PKC), respectively (Raymond et al., 2001). However, the ultimate target of this pathway in FS cell terminals is not known and may include, among others, activation of Ca2+-dependent K+ channels or PKC-dependent inhibition of P/Q-type Ca2+ channels (Raymond et al., 2001).
On the other hand, the modulation of GABA release produced by muscarine, adenosine, and baclofen displayed fast onset and independence of PK activation, implicating the involvement of membrane-delimited mechanisms. These mechanisms utilize association of βγ subunits of G proteins (Gβγ) with effector targets (Hille, 2001). Known effectors of this type of modulation include G protein-activated inwardly rectifying K+ channels (GIRKs) as well as N- or P/Q-type voltage-gated Ca2+ channels. It is unlikely that modulation of GIRKs is involved in the regulation of GABA release from FS cells, because these channels typically have a postsynaptic localization in the neocortex and hippocampus (Luscher et al., 1997). On the other hand, direct inhibition of the CaV2 family of Ca2+ channels by the Gβγ subunit is a well-described phenomenon (for review see Dolphin, 2003) and has been implicated in the presynaptic suppression of neurotransmitter release in several instances, including the endocannabinoid-mediated inhibition of GABA release from CCK-containing interneurons (Wilson et al., 2001). Fast, Gβγ -mediated inhibition of CaV2 channels stems from the activation of Gi/o and not Gq/11 protein (Mirotznik et al., 2000; Wettschureck and Offermanns, 2005), the type of G protein activated by adenosine A1, GABAB, and the M2/4 receptors identified in our study. However, we cannot rule out the possibility that Gβγ affects directly the vesicle fusion machinery, as has been recently observed in some preparations (Blackmer et al., 2005).
Cholinergic Modulation of Feedforward Inhibition
ACh is one of the most important neuromodulators in the brain, critical for normal cognitive function. Most neocortical ACh originates from axons projecting from the nucleus basalis of Meynert (NB). Cholinergic modulation of cortical structures is important for multiple functions, including attention, learning, memory, and the cortical processing of sensory information (for review see Lucas-Meunier et al., 2003; McCormick, 1993). Impairment of cortical cholinergic function is considered crucial to the pathology of Alzheimer’s disease (Kasa et al., 1997).
ACh can regulate neuronal excitability via actions on somatodendritic muscarinic and nicotinic receptors as well as neurotransmitter release through actions on receptors present in presynaptic terminals. We observed that muscarine but not nicotine depolarized ECs in L3-4 of barrel cortex, but neither muscarine nor nicotine affected the intrinsic properties of FS INs. These results are in agreement with a recent report showing that cholinergic agonists do not affect the intrinsic excitability of FS cells in various cortical areas and different layers (Gulledge et al., 2007).
Cholinergic regulation of transmitter release in neocortex has received less attention. Several studies point to ACh effects on inhibitory neurotransmission (for review see Krnjevic, 2004), yet, to our knowledge, no studies have addressed the cholinergic regulation of transmitter release from perisomatically targeting IN subtypes in neocortex. Here, we show that muscarine is a potent inhibitor of GABA release from FS cells in somatosensory cortex in both infragranular and supragranular layers. We found that the feedforward disynaptic inhibitory response to thalamocortical stimulation is much more sensitive to mAChR activation than the monosynaptic excitatory response, resulting in an overall increase in thalamic excitation of the neocortex.
It is believed that ACh enhances attention by increasing the influence of sensory stimuli relative to internal cortical processing by enhancing the weight of feedforward afferent inputs while suppressing excitatory feedback connections (Hasselmo and Giocomo, 2006; Hsieh et al., 2000; Oldford and Castro-Alamancos, 2003). The net increase in TC synaptic excitation of thalamo-recipient neurons due to the muscarinic suppression of feedforward inhibition described here should be a significant contributor to these effects of ACh. The observed muscarinic-mediated depolarization of excitatory thalamo-recipient neurons will facilitate the ability of the elevated TC synaptic excitation to drive these cells to firing threshold.
Moreover, reduction of perisomatic inhibition produced by activation of M2/4 AChRs in FS cell terminals provides a mechanism for broadening stimuli tuning of cortical cells by increasing the time window during which excitatory cells can integrate sensory inputs, making cortical neurons sensitive to stimuli to which they normally do not respond. This idea is supported by the observation that iontophoresis of ACh to the somatosensory cortex of awake and drug-free animals produced an enlargement of whisker receptive fields (Delacour et al., 1990). Similarly, ACh broadened frequency tuning in auditory cortex (McKenna et al., 1989). Both effects are mediated by the activation of mAChRs.
Here, we show that nicotine potentiated TC synaptic transmission onto ECs (see also Gil et al., 1997) but did not affect TC drive onto FS INs. This intriguing observation is consistent with immunohistochemical results in macaque monkey visual cortex showing that nicotinic receptors are present on TC synapses on excitatory but not inhibitory cells (Disney et al., 2007).
FS-mediated perisomatic inhibition of ECs was unaffected by nicotine (Figure 8). Thus, activation of nicotinic AChRs in TC afferents to ECs and somatic muscarinic receptors on the ECs will complement the reduced muscarinic-mediated perisomatic inhibition to enhance cortical responsiveness to sensory stimuli. Furthermore, modulation of feedforward inhibition provides control over the temporal summation of TC inputs by changing the duration of the integration window and refractory period. The specific subcellular localization of AChRs—nicotinic in presynaptic TC afferents contacting EC dendrites, M2 in FS basket cell output synapses (Chaudhuri et al., 2005; Hajos et al., 1998), and M1 in ECs (Volpicelli and Levey, 2004)—may be an important factor in the specificity of the responses to ACh.
Modulation of GABA Release by Baclofen, Adenosine, and Serotonin
We showed that baclofen, adenosine, and serotonin also inhibited perisomatic GABA release from FS cells. GABAB and adenosine receptors are expected to mediate modulations by agents that are predominantly intrinsic to the neocortex and regulated by cortical activity. GABAB receptor activation is involved in numerous neuronal processes, including regulation of the induction of long-term potentiation (Davies et al., 1991) and modulation of rhythmic activity in the hippocampus (Scanziani, 2000) as well as seizure generation (Schuler et al., 2001). GABAB receptors are widely distributed in presynaptic terminals throughout the brain (Bowery et al., 2002), and baclofen, which is known to activate GABAB receptors, has been shown to inhibit neurotransmitter release of many cell types, including GABAergic interneurons. However, GABAB receptors were believed to be absent in FS cells (Freund and Katona, 2007). Yet, we find that baclofen produced the most potent inhibition of GABA release from FS cells, and we show that this effect is mediated by GABAB receptors. We also observed that baclofen had a potent effect on the TC connection with FS cells (in agreement with Porter and Nieves, 2004), effectively excluding these cells from the feedforward inhibitory circuit. The physiological significance of the effect of baclofen on these and other synapses remains to be investigated and, specifically, whether physiological concentrations of GABA activate GABAB receptors to the same extent as baclofen.
Effects of adenosine on inhibitory synaptic transmission in neocortex have not been previously reported. Physiologically, adenosine concentrations in the neocortex increase during periods of activity as a result of either increases in its intracellular levels or the enhanced corelease of ATP during synaptic transmission and its subsequent conversion to adenosine by extracellular ecto-5′-nucleotidase (Dunwiddie and Masino, 2001). While not as potent as the effect of muscarine or baclofen, the effect of adenosine on perisomatic GABA release was consistently observed and is potentially interesting as a homeostatic feedback mechanism to suppress inhibition following periods of increased activity. On the other hand, antagonizing adenosine receptors with caffeine (Basheer et al., 2004) would regulate PC output by reducing the inhibitory actions of adenosine.
Serotonin is an important modulatory neurotransmitter in the brain playing a role in both normal physiology and, importantly, in pathologies such as migraine, depression, fear and anxiety, obsessive compulsive disorders, schizophrenia, and addiction (Gray and Roth, 2007). It is released in neocortex by axons originating from neurons in the mesencephalic dorsal and median raphe nuclei that preferentially target interneurons (Smiley and Goldman-Rakic, 1996). There is a large diversity of serotonin receptors, including seven families of metabotropic receptors and a single family of ionotropic receptors (5HT3), with 5-HT1,2,3,6 being most abundantly present in the cortex (Raymond et al., 2001). However, 5-HT3 receptors are not present in FS interneurons (Morales and Bloom, 1997). Serotonin has been shown to have diverse effects on the excitability of inhibitory neurons in neocortex (Foehring et al., 2002; Xiang and Prince, 2003; Zhou and Hablitz, 1999), but to our knowledge, it has not been previously reported to inhibit GABA release from FS cells. It remains to be investigated how this effect is integrated with the other cellular effects of serotonin. The 5-HT2 receptor-mediated serotonergic suppression of inhibition of PCs described here could have important implications to understanding the role of serotonin in a variety of physiological and pathological states.
EXPERIMENTAL PROCEDURES
Recording of Extracellularly Evoked IPSCs
Acute brain slices (300 μm) were prepared from 16- to 21-day-old mice following institutional animal care guidelines essentially as described previously (Goldberg et al., 2005). Synaptic currents were recorded in whole-cell voltage clamp of L5 PCs of mouse primary somatosensory (“barrel”) cortex visually identified under IR-DIC by their large somata and pia-oriented apical dendrite. During recording, slices were superfused with a solution containing (in mM) 125 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2, MgCl2, and 10 glucose (equilibrated with 95% O2/5% CO2). In some experiments, Ca2+ was reduced to 1.3 mM, and Mg2+ was increased to 2.7 mM. A concentric bipolar-stimulating electrode (125 μm in diameter; FHC) was placed laterally (∼100 μm) to the recording pipette within L5 (Figure 1A). IPSCs were evoked by 0.1 ms pulses, typically 20-60 μA, to keep IPSC magnitude low. Recordings were performed at 27°C-30°C with patch pipettes (2.5-4.5 MΩ) filled with intracellular solution containing (in mM) 120 CsMeSO4, 10 CsCl, 9.0 NaCl, 4.0 Mg2+-ATP, 0.3 Na+-GTP, 10 HEPES, 0.5 EGTA, and 5 lidocaine N-ethyl bromide (QX-314), with pH adjusted to 7.40 with CsOH. All eIPSCs were recorded in the presence of 10 μM CNQX and 50 μM D-APV to block excitatory synaptic transmission. The predicted ECl under these conditions was ∼-50 mV, and eIPSCs were recorded as inward currents during hyperpolarizations of the membrane potential to -80 mV (Figure 1B). Unless stated otherwise, all drugs were locally applied through a second patch pipette located 5-10 μm from the cell body of PC using Picospritzer III (Parker Corp) (Figure S1).
To minimize voltage-clamp errors, we used low-resistance pipettes and series resistance compensation, and we recorded at potentials close to the chloride equilibrium potential (ECl) to reduce eIPSC amplitudes. A cell was rejected if series resistance (Rs) after break-in was >20 MΩ (typically, 10-15 MΩ), if Rs could not be compensated to <10 MΩ (typically, 5-8 MΩ), or if Rs changed by >20% during the experiment. Series resistance was rigorously monitored after each evoked response. Reported values for membrane potential were not corrected for the liquid junction potential (∼-12 mV). Currents were recorded with an Axopatch 200B amplifier (Molecular Devices), sampled at 20 kHz, low-pass filtered at 5 kHz, and digitized at 16 bit resolution (Digidata 1322A; Molecular Devices). Data analysis and the source of pharmacological agents are described in Supplemental Data.
Paired Recordings and Morphological Reconstructions
Simultaneous whole-cell recordings were performed from INs and PC pairs in L5 and deep L3 of primary somatosensory cortex using patch electrodes (4-6 MΩ) filled with a solution containing the following (in mM): 65 KCl, 65 K-gluconate, 10 HEPES, 0.5 EGTA, 4.0 Mg-ATP, 0.3 Na-GTP, 5 QX-314, and 1% biocytin; pH 7.4, with KOH. Use of this solution predicts an ECl· of ∼-20 mV. A GAD67-EGFP G42 mouse expressing EGFP in PV-expressing interneurons (Goldberg et al., 2005) was used to aid in the identification of FS cells (Figure S3A). A GIN mouse line expressing EGFP in a subset of somatostatin-containing interneurons (Oliva et al., 2000) was used for the identification of MCs. Presynaptic cells were recorded in the bridge mode of an Axoclamp 2B amplifier (Molecular Devices), while postsynaptic cells were recorded in voltage-clamp mode with an Axopatch 200B amplifier (Molecular Devices). For the studies in Figure 6, we used an intracellular solution with a predicted ECl·∼-70 mV (similar to that used in TC Recordings below, but without QX-314).
After recording, slices were fixed with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (NaPB) overnight at 4°C for subsequent histological analysis (see Supplemental Data).
Thalamocortical Recordings
Somatosensory thalamocortical slices (400 μm thick, 35° tilt from coronal plane) were obtained from mice aged postnatal day 14-16 as previously described (Agmon and Connors, 1991). To activate thalamic afferents, extracellular stimuli were delivered to the VB through a concentric bipolar-stimulating electrode (Figure 7A). Stimulation intensities were chosen to be just above the threshold for EPSP detection (range 30-60 μA). Disynaptic EPSP/IPSP or EPSC/IPSC sequences were recorded in L3/4 ECs (Vm = -45 mV); monosynaptic TC EPSPs in FS INs and ECs were recorded at Vm = -70 mV. Patch electrodes (4-6 MΩ) were filled with a solution containing the following (in mM): 121 K-gluconate, 9 KCl, 10 HEPES, 0.5 EGTA, 4.0 Mg-ATP, and 0.3 Na-GTP, 5 QX-314, pH 7.4. The predicted ECl under these conditions was ∼-70 mV. Feedforward eIPSCs reversed near ECl and eEPSCs reversed at +5 mV (data not shown).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health Grants NS30989 and NS045217 and NSF IBN-0314645 to B.R. We thank Dr. Z. Josh Huang for the gift of G42 transgenic mice; Brian Clark for help with neuronal reconstructions; Edward Zagha for help with thalamocortical slice preparation; and Dr. Ethan Goldberg, Edward Zagha, and Brian Clark for their comments on the manuscript and useful discussions.
REFERENCES
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA. Modulation of neocortical interneurons: extrinsic influences and exercises in self-control. Trends Neurosci. 2005;28:602–610. doi: 10.1016/j.tins.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Prog. Neurobiol. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Beaulieu C, Somogyi P. Enrichment of cholinergic synaptic terminals on GABAergic neurons and coexistence of immunoreactive GABA and choline acetyltransferase in the same synaptic terminals in the striate cortex of the cat. J. Comp. Neurol. 1991;304:666–680. doi: 10.1002/cne.903040412. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW. A network of electrically coupled interneurons drives synchronized inhibition in neocortex. Nat. Neurosci. 2000;3:904–910. doi: 10.1038/78809. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein betagamma directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat. Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyiri G, Mackie K, Ledent C, Hajos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J. Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M, Bonner TI, Enna SJ. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid(B) receptors: structure and function. Pharmacol. Rev. 2002;54:247–264. doi: 10.1124/pr.54.2.247. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Geisler C, Henze DA, Wang XJ. Interneuron diversity series: circuit complexity and axon wiring economy of cortical interneurons. Trends Neurosci. 2004;27:186–193. doi: 10.1016/j.tins.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Chaudhuri JD, Hiltunen M, Nykanen M, Yla-Herttuala S, Soininen H, Miettinen R. Localization of M2 muscarinic receptor protein in parvalbumin and calretinin containing cells of the adult rat entorhinal cortex using two complementary methods. Neuroscience. 2005;131:557–566. doi: 10.1016/j.neuroscience.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABA autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- Delacour J, Houcine O, Costa JC. Evidence for a cholinergic mechanism of “learned” changes in the responses of barrel field neurons of the awake and undrugged rat. Neuroscience. 1990;34:1–8. doi: 10.1016/0306-4522(90)90299-j. [DOI] [PubMed] [Google Scholar]
- Disney AA, Aoki C, Hawken M. Gain modulation by nicotine in macaque V1. Neuron. 2007;56:701–713. doi: 10.1016/j.neuron.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol. Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Foehring RC, van Brederode JF, Kinney GA, Spain WJ. Serotonergic modulation of supragranular neurons in rat sensorimotor cortex. J. Neurosci. 2002;22:8238–8250. doi: 10.1523/JNEUROSCI.22-18-08238.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Erdelyi F, Szabo G, Hestrin S. Electrical coupling among irregular-spiking GABAergic interneurons expressing cannabinoid receptors. J. Neurosci. 2004;24:9770–9778. doi: 10.1523/JNEUROSCI.3027-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J. Neurosci. 2003;23:1622–1630. doi: 10.1523/JNEUROSCI.23-05-01622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J. Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Hen R. The serotonergic system and anxiety. Neuromolecular Med. 2004;5:27–40. doi: 10.1385/NMM:5:1:027. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophr. Bull. 2007;33:1100–1119. doi: 10.1093/schbul/sbm074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J. Neurophysiol. 2007;97:2215–2229. doi: 10.1152/jn.00493.2006. [DOI] [PubMed] [Google Scholar]
- Hajos N, Papp EC, Acsady L, Levey AI, Freund TF. Distinct interneuron types express m2 muscarinic receptor immunoreactivity on their dendrites or axon terminals in the hippocampus. Neuroscience. 1998;82:355–376. doi: 10.1016/s0306-4522(97)00300-x. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Giocomo LM. Cholinergic modulation of cortical function. J. Mol. Neurosci. 2006;30:133–135. doi: 10.1385/JMN:30:1:133. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat. Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Kobayashi R. Use of protein (serine/threonine) kinase activators and inhibitors to study protein phosphorylation in intact cells. In: Hardie DG, editor. Phosphorylation - A Practical Approach. Oxford University Press; Oxford: 1993. pp. 89–107. [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Third Edition Sinauer Associates, Inc.; Sunderland, MA: 2001. [Google Scholar]
- Hsieh CY, Cruikshank SJ, Metherate R. Differential modulation of auditory thalamocortical and intracortical synaptic transmission by cholinergic agonist. Brain Res. 2000;880:51–64. doi: 10.1016/s0006-8993(00)02766-9. [DOI] [PubMed] [Google Scholar]
- Jakab RL, Goldman-Rakic PS. Segregation of serotonin 5-HT2A and 5-HT3 receptors in inhibitory circuits of the primate cerebral cortex. J. Comp. Neurol. 2000;417:337–348. doi: 10.1002/(sici)1096-9861(20000214)417:3<337::aid-cne7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kapfer C, Glickfeld LL, Atallah BV, Scanziani M. Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat. Neurosci. 2007;10:743–753. doi: 10.1038/nn1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb. Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Krnjevic K. Synaptic mechanisms modulated by acetylcholine in cerebral cortex. Prog. Brain Res. 2004;145:81–93. [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network. Pflugers Arch. 2003;446:17–29. doi: 10.1007/s00424-002-0999-2. [DOI] [PubMed] [Google Scholar]
- Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat. Rev. Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Actions of acetylcholine in the cerebral cortex and thalamus and implications for function. Prog. Brain Res. 1993;98:303–308. doi: 10.1016/s0079-6123(08)62412-7. [DOI] [PubMed] [Google Scholar]
- McKenna TM, Ashe JH, Weinberger NM. Cholinergic modulation of frequency receptive fields in auditory cortex: I. Frequency-specific effects of muscarinic agonists. Synapse. 1989;4:30–43. doi: 10.1002/syn.890040105. [DOI] [PubMed] [Google Scholar]
- Miller KD, Pinto DJ, Simons DJ. Processing in layer 4 of the neocortical circuit: new insights from visual and somatosensory cortex. Curr. Opin. Neurobiol. 2001;11:488–497. doi: 10.1016/s0959-4388(00)00239-7. [DOI] [PubMed] [Google Scholar]
- Mirotznik RR, Zheng X, Stanley EF. G-Protein types involved in calcium channel inhibition at a presynaptic nerve terminal. J. Neurosci. 2000;20:7614–7621. doi: 10.1523/JNEUROSCI.20-20-07614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Bloom FE. The 5-HT3 receptor is present in different subpopulations of GABAergic neurons in the rat telencephalon. J. Neurosci. 1997;17:3157–3167. doi: 10.1523/JNEUROSCI.17-09-03157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldford E, Castro-Alamancos MA. Input-specific effects of acetylcholine on sensory and intracortical evoked responses in the “barrel cortex” in vivo. Neuroscience. 2003;117:769–778. doi: 10.1016/s0306-4522(02)00663-2. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr., Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J. Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, Nieves D. Presynaptic GABAB receptors modulate thalamic excitation of inhibitory and excitatory neurons in the mouse barrel cortex. J. Neurophysiol. 2004;92:2762–2770. doi: 10.1152/jn.00196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JR, Mukhin YV, Gelasco A, Turner J, Collinsworth G, Gettys TW, Grewal JS, Garnovskaya MN. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol. Ther. 2001;92:179–212. doi: 10.1016/s0163-7258(01)00169-3. [DOI] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Schuler V, Luscher C, Blanchet C, Klix N, Sansig G, Klebs K, Schmutz M, Heid J, Gentry C, Urban L, et al. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)) Neuron. 2001;31:47–58. doi: 10.1016/s0896-6273(01)00345-2. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Goldman-Rakic PS. Serotonergic axons in monkey prefrontal cerebral cortex synapse predominantly on interneurons as demonstrated by serial section electron microscopy. J. Comp. Neurol. 1996;367:431–443. doi: 10.1002/(SICI)1096-9861(19960408)367:3<431::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Swadlow HA. Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb. Cortex. 2003;13:25–32. doi: 10.1093/cercor/13.1.25. [DOI] [PubMed] [Google Scholar]
- Volpicelli LA, Levey AI. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004;145:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- Wilent WB, Contreras D. Synaptic responses to whisker deflections in rat barrel cortex as a function of cortical layer and stimulus intensity. J. Neurosci. 2004;24:3985–3998. doi: 10.1523/JNEUROSCI.5782-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilent WB, Contreras D. Dynamics of excitation and inhibition underlying stimulus selectivity in rat somatosensory cortex. Nat. Neurosci. 2005;8:1364–1370. doi: 10.1038/nn1545. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Prince DA. Heterogeneous actions of serotonin on interneurons in rat visual cortex. J. Neurophysiol. 2003;89:1278–1287. doi: 10.1152/jn.00533.2002. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ. Activation of serotonin receptors modulates synaptic transmission in rat cerebral cortex. J. Neurophysiol. 1999;82:2989–2999. doi: 10.1152/jn.1999.82.6.2989. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.