

Abstract
The protein product of the ocular albinism type 1 gene, named OA1, is a pigment cell-specific G protein-coupled receptor exclusively localized to intracellular organelles, namely lysosomes and melanosomes. Loss of OA1 function leads to the formation of macromelanosomes, suggesting that this receptor is implicated in organelle biogenesis, however the mechanism involved in the pathogenesis of the disease remains obscure. We report here the identification of an unexpected abnormality in melanosome distribution both in retinal pigment epithelium (RPE) and skin melanocytes of Oa1-knock-out (KO) mice, consisting in a displacement of the organelles from the central cytoplasm towards the cell periphery. Despite their depletion from the microtubule (MT)-enriched perinuclear region, Oa1-KO melanosomes were able to aggregate at the centrosome upon disruption of the actin cytoskeleton or expression of a dominant-negative construct of myosin Va. Consistently, quantification of organelle transport in living cells revealed that Oa1-KO melanosomes displayed a severe reduction in MT-based motility; however, this defect was rescued to normal following inhibition of actin-dependent capture at the cell periphery. Together, these data point to a defective regulation of organelle transport in the absence of OA1 and imply that the cytoskeleton might represent a downstream effector of this receptor. Furthermore, our results enlighten a novel function for OA1 in pigment cells and suggest that ocular albinism type 1 might result from a different pathogenetic mechanism than previously thought, based on an organelle-autonomous signalling pathway implicated in the regulation of both membrane traffic and transport.
INTRODUCTION
In mammals, pigmentation of the skin, hair and eyes results from the presence and distribution of melanins. These black-brown and yellow-red pigments are synthesized by specialized pigment cells, including skin melanocytes and retinal pigment epithelium (RPE), within dedicated intracellular organelles, named melanosomes (1). Melanosomes are commonly classified as lysosome-related organelles (2,3); however, they differ from lysosomes and are distinct from the canonical endocytic pathway, displaying unique structural and functional features (4). Melanosomes are thought to originate from endosomal precursors (5) and subsequently undergo a series of maturation stages, each characterized by typical ultrastructural morphology and melanin content: stage II and III correspond to non-pigmented and partially pigmented immature melanosomes, respectively, and stage IV to fully melanized mature organelles (6,7).
Pigmented melanosomes move from the perinuclear region towards the cell periphery, from where they are eventually transferred to surrounding keratinocytes in the skin. The transport process is dependent both on microtubules (MTs), by means of kinesin and dynein motors, and on actin filaments (AFs), by means of a tripartite complex comprising the monomeric GTPase Rab27a, its effector melanophilin, and the actin-based motor myosin Va in melanocytes (for review see 8), or Rab27a, MyRIP and myosin VIIa in RPE (9,10). Studies in mammalian melanocytes and in the related fish and frog melanophores have revealed that these two cytoskeletal systems regulate the distribution of melanosomes by generating opposite and competitive forces, as in a ‘tug of war’, in which MTs promote perinuclear accumulation and AFs support peripheral dispersal of the organelles (11). Accordingly, MT depolymerization or dynein down-regulation result in centrifugal AF-dependent displacement of melanosomes (12,13), while disruption of actin-based motility by drugs or inactivation of the Rab27a/melanophilin/myosin Va complex causes centripetal MT-dependent redistribution of the organelles (8,14). Similarly, in the absence of a functional Rab27a/MyRIP/myosin VIIa complex, melanosomes redistribute from the actin-rich apical region of RPE cells to the MT-filled cell body, indicating that melanosome movement and distribution in the retina and skin melanocytes are regulated by a conserved mechanism (15,16).
Inherited defects of melanogenesis in humans comprise a heterogeneous group of diseases, collectively defined as albinism, and characterized by variable hypopigmentation of the skin and severe developmental defects of the eyes (17). Ocular albinism type 1 (OA1; MIM 300500) represents the most common form of ocular albinism and is transmitted as an X-linked trait, with affected males exhibiting severe reduction of visual acuity, nystagmus, strabismus, photophobia, iris translucency, hypopigmentation of the retina, foveal hypoplasia and misrouting of the optic tracts, resulting in loss of stereoscopic vision (17). Cutaneus changes are usually mild or absent in ocular albinism. Nevertheless, the histopathological examination of the skin may be useful in the diagnosis of the disease, due to the typical occurrence of giant melanosomes (macromelanosomes) both in RPE and skin melanocytes of affected patients, suggesting that ocular albinism is in fact an oculocutaneus disorder of melanosome biogenesis (18).
The protein product of the human OA1 and mouse Oa1 genes, named OA1 (also known as GPR143), is an integral membrane glycoprotein that is exclusively expressed by melanocytes and RPE (19,20) and shares structural and functional similarities with G protein-coupled receptors (GPCRs). In fact, OA1 shows seven transmembrane domains and homologies with members of the GPCR superfamily, including residues highly conserved in most GPCRs, some of which are the site of albinism-causing mutations (21–23). Furthermore, OA1 binds heterotrimeric Gi, Go and Gq proteins, as revealed by coimmunoprecipitation and in vitro binding assays, and co-localizes with Gi proteins in normal human melanocytes (21). Finally, OA1 was found to interact with a yeast/mammalian G protein chimera in a yeast-based signalling assay (24), and to efficiently activate heterotrimeric G proteins and bind the GPCR-adaptors arrestins in a mammalian expression system, thus behaving as a bona fide GPCR (25).
However, unlike canonical GPCRs, OA1 is not localized to the cell surface, but is exclusively detectable on the membrane of intracellular organelles, namely late-endosomes/lysosomes and melanosomes (19–21). In addition, we recently identified two separate sorting signals that are both necessary and sufficient for the intracellular localization of OA1 in melanocytic and non-melanocytic cells (26). These findings support the idea that OA1 is a resident GPCR of lysosomes and melanosomes, and that, according with its established topological orientation with the C-terminus towards the cytoplasm (21,23), a putative ligand should exist within the organelle lumen. The downstream pathway triggered by OA1 remains mysterious and can only be hypothesized based on the presence of macromelanosomes resulting from loss-of-function of the receptor in human patients, as well as in a mouse model of the disorder (27), suggesting a defect in organelle biogenesis.
Although the eyes appear more severely affected than the skin in ocular albinism, the RPE represents a difficult system to study melanosome biogenesis, due to the short prenatal time window of melanogenesis in vivo, and to the poor melanogenic ability of cultured cells from post-natal eyes (28). In contrast, skin melanocytes are highly and continuously active in melanosome production both in vivo and in culture, and melanocyte cell lines have been extensively used to uncover pathways leading to organelle morphogenesis in pigment cells. Therefore, in order to unravel the role of OA1 in melanosome biogenesis, we coupled the examination of Oa1-KO RPE at early developmental stages in vivo with the generation and analysis of Oa1-KO melanocyte cultures. Our results indicate that OA1 regulates not only the biogenesis, but also the motility of melanosomes.
RESULTS
Oa1-KO RPE displays abnormal distribution of melanosomes towards the apical surface
Our previous studies of RPE from Oa1-KO mice have shown that the cytological abnormalities typically observed in ocular albinism, namely reduced number and increased size of mature melanosomes, are first observed at or after birth (27,29). However, the anomalies of the visual system leading to optic misrouting manifest at earlier developmental stages (30). We reasoned that embryonic Oa1-KO RPE might display subtle melanosomal abnormalities that remained undetected, since extensive quantitative analyses at prenatal stages have never been carried out. Therefore, we performed an ultrastructural evaluation of mouse retinas collected from wild-type and Oa1-KO mice at embryonic (E) days 15.5, 18.5 and at birth (post-natal day 0, P0). At these stages, which precede the formation of macromelanosomes, mature melanosomes are sufficiently abundant in both wild-type and Oa1-KO RPE, allowing a statistically relevant comparison of organelle size, density and distribution. The results of this analysis are shown in Figure 1.
Figure 1.
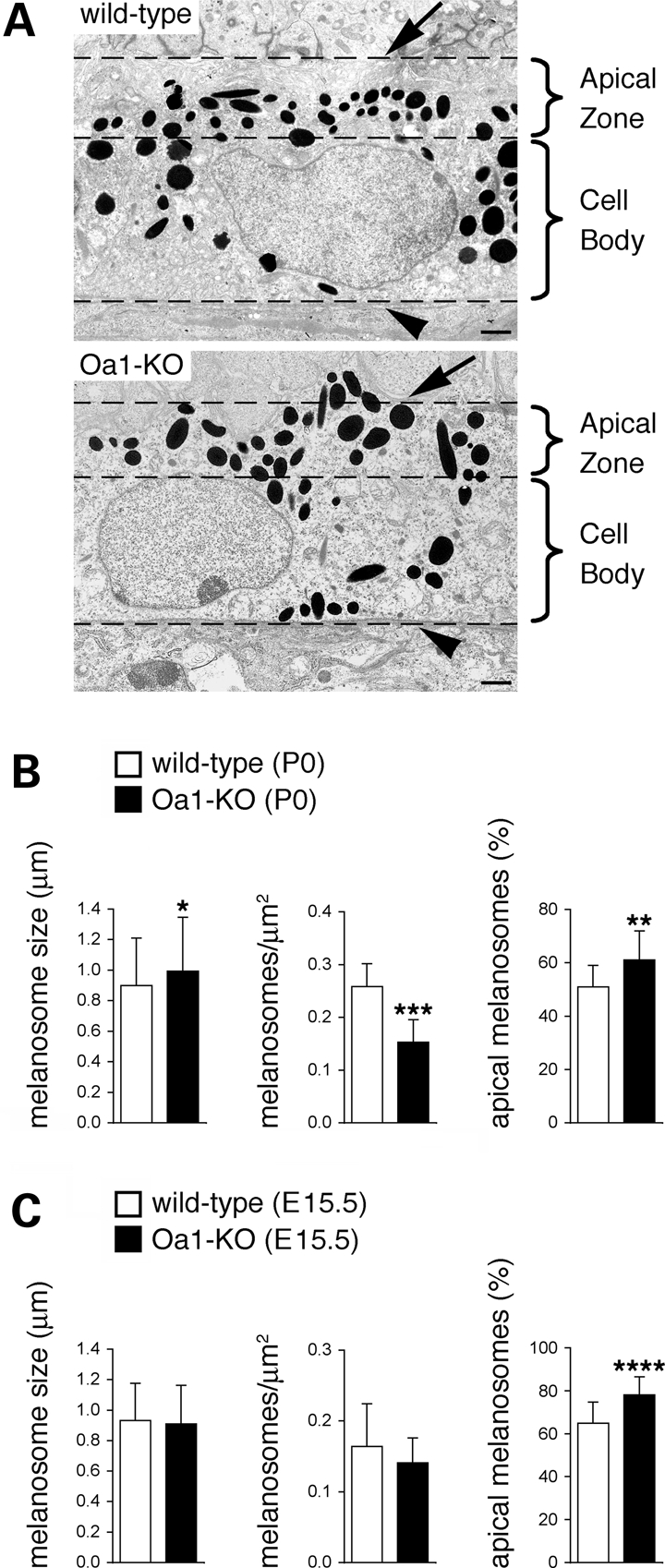
Melanosomes are more concentrated towards the apical surface in Oa1-KO RPE. (A) Micrographs of RPE cells from wild-type and Oa1-KO mice at P0. At this stage, RPE apical processes (and photoreceptor outer segments) are not developed yet and therefore could not be evaluated. Dashed lines indicate the boundaries of apical and basolateral regions of RPE cells as defined to quantify melanosome distribution (see below and Materials and Methods). In Oa1-KO RPE, a greater fraction of melanosomes appears located in the apical region (this is particularly evident in areas devoid of nuclei). Arrows, apical membrane; arrowheads, basal membrane. Bars, 1 µm. (B and C) Quantification of melanosome size, density and distribution in wild-type and Oa1-KO RPE at P0 (B), and E15.5 (C). Melanosome size histograms represent the mean ± SD of the major organelle diameter measured for 120–140 (B) or 215–230 (C) melanosomes belonging to different sections and with an apparent size ≥0.5 µm. Melanosome density histograms represent the mean ± SD of the number of organelles per μm2 obtained from 7–8 (B) or 23–25 (C) independent RPE sections. Melanosome distribution histograms represent the mean ± SD of the percent of organelles located in the apical area (apical third of the selected area, see Materials and Methods) over the total number of organelles in 20–25 independent RPE sections. Total number of melanosomes counted to determine melanosome distribution: wild-type =1132 (B) and 753 (C); Oa1-KO = 891 (B) and 575 (C). *P < 0.02, **P < 0.002, ***P < 0.001, ****P < 0.00001 (unpaired Student's t-test assuming equal variances). At both stages, including E15.5, when the organelles are not enlarged nor significantly reduced in number yet, melanosomes are more abundant towards the apical region of RPE cells in Oa1-KO compared with wild-type mice.
At birth (Fig. 1A and B), mature melanosomes showed comparable size in wild-type and Oa1-KO RPE, although the latter displayed a slight increase in melanosome size (by 10%), possibly indicating that the pathological process subsequently leading to the formation of the giant organelles is already initiated (Fig. 1B). In contrast, the number of organelles per square micron was dramatically reduced in Oa1-KO RPE (∼40% less than wild-type; Fig. 1B), consistent with previous observations (29). In addition, although a greater fraction of melanosomes was located in the apical region compared with the basolateral side in all samples, this displacement appeared more striking in Oa1-KO RPE (Fig. 1A). To quantify this difference, we calculated the percent of organelles located in the apical area in several independent sections (see Materials and Methods). We found that, while in wild-type RPE over half of all melanosomes were located in the apical region, in Oa1-KO RPE this value was increased by 20% (Fig. 1B). Results on organelle size, density and distribution were essentially identical at E18.5, with the percent of apical melanosomes increasing from 65% in wild-type to 82% in Oa1-KO RPE (P < 0.0001; number of RPE sections = 20; data not shown). In contrast, the size and number of mature melanosomes were not significantly different between wild-type and Oa1-KO RPE at E15.5. Interestingly, even at this stage the percent of apical melanosomes was higher in Oa1-KO than wild-type RPE (Fig. 1C), suggesting that this abnormal organelle distribution might represent an early event in the pathogenesis of the disease.
Oa1-KO melanocytes show abnormal distribution of melanosomes towards the cell periphery
To verify whether skin melanocytes display abnormal melanosome distribution, as RPE cells, we generated immortalized melanocyte cell lines from homozygous Oa1+/+ (melan-A, indicating wild-type agouti), heterozygous Oa1+/− (melan-Oa1+/−) and homozygous Oa1−/− (melan-Oa1−/−) female mice, and initially analyzed them by bright field optical microscopy. In fact, this approach allows the identification of melanosomes as the only visible organelles, thanks to the dark melanin pigment. Despite their agouti A/A genotype, in culture all these cells displayed pigment colour similar to genetically black (a/a) melanocytes as expected, since the agouti gene is not expressed by melanocytes and the culture conditions stimulate eumelanogenesis. Relative to Oa1+/+ and Oa1+/− cells, which appeared indistinguishable (see Materials and Methods and Supplementary Material, Fig. S1, for cell line characterization), Oa1−/− melanocytes showed the presence of macromelanosomes (Fig. 2A) and a reduced density of pigmented organelles, consistent with a lower melanin content (∼75–50% less than that of wild-type, depending on passage; not shown). Larger melanosomes, detectable in 30–50% of Oa1−/− cells, showed variable size and were typically surrounded by apparently normal melanosomes, although cells containing mostly giant organelles were also observed.
Figure 2.

Melanosomes are displaced towards the cell periphery in Oa1-KO melanocytes. (A) Representative bright field optical pictures of the indicated melanocyte cell lines, in which melanosomes, thanks to the melanin pigment, represent the only visible dark objects. In addition to the presence of larger melanosomes (see inset for 3× magnification), Oa1−/− melanocytes show exclusion of the pigmented organelles from the perinuclear region and their concentration at the cell periphery. (B) Melanosomes are colocalized with the cell nuclei, visualized by Hoechst staining. The merge shows poor overlap between melanosomes and the cell nuclei in Oa1−/− cells. (C) Melanosomes are colocalized with the Golgi apparatus, visualized by anti-giantin antibodies. Arrowheads in bright field pictures and merge point to the melanosomal enrichment in the Golgi area in Oa1+/−, but not Oa1−/−, melanocytes. Bars, 15 µm.
The reduced melanosome density in Oa1−/− melanocytes was particularly evident in the central cytoplasm, while the pigmented organelles appeared relatively more concentrated at the periphery (Fig. 2A–C). The low-density region corresponded to the perinuclear area, as shown by nuclear co-staining (Fig. 2B), and was centred on the Golgi apparatus (Fig. 2C). In contrast, in wild-type melanocytes, melanosomes were evenly distributed throughout the cytoplasm, either delimiting tightly or shielding the nuclear envelope, and were often more concentrated around the Golgi apparatus, while the equivalent region appeared ‘empty’ in Oa1−/− cells (Fig. 2C, arrowheads). This distribution phenotype was observed in the majority of Oa1−/− melanocytes and was virtually absent in Oa1+/+ and Oa1+/− cells, suggesting that it could represent a key consequence of OA1 deficiency. Finally, electron microscopy analysis of Oa1−/− melanocytes revealed the presence of morphologically normal melanosomes at all maturation stages (Fig. 3, arrows and arrowheads), together with the occurrence of giant completely melanized organelles with an average diameter about 3-folds larger than normal (Fig. 3, asterisks). Overall, these findings are consistent with the present and previous studies on Oa1-KO mouse retinas (27,29) and confirm the suitability of the cultured mouse melanocytes as in vitro model of Oa1-KO pigment cells.
Figure 3.

Both normal and giant melanosomes are found in Oa1-deficient melanocytes. Ultrastructural analysis of the indicated cell lines, in which mature fully pigmented melanosomes appear as black organelles due to melanin electron density and display either elliptical or spherical shapes depending on the section. Oa1−/−LOA1SN and Oa1−/−LOA1Δ18SN, cell lines derived from transduction of Oa1−/− melanocytes with retroviral vectors carrying the wild-type or mutant OA1, respectively. Similarly to wild-type, Oa1−/− and Oa1−/−LOA1Δ18SN melanocytes show immature (arrows) and mature (arrowheads) melanosomes of normal size. However, differently from wild-type, these cells also show the presence of macromelanosomes (asterisks). Bars, 1 µm.
The peripheral displacement of melanosomes depends on OA1 function
In order to confirm that the melanosomal phenotype observed in Oa1−/− cells is due to OA1 loss-of-function, we transfected them with expression vectors for wild-type or mutant human OA1. In transient transfections, 24 h of OA1 expression were not sufficient to rescue the abnormal melanosomal size, which might require organelle turnover (Fig. 4A, pR/OA1wt; arrows). In contrast, this short expression time was sufficient to correct the abnormal distribution of melanosomes, leading to a concentration of pigmented organelles in the perinuclear/Golgi area where OA1 is also normally enriched (Fig. 4A, pR/OA1wt; arrowheads). Recovery of the perinuclear distribution involved only a subset of melanosomes, most often tiny and lightly pigmented and possibly newly formed, while a conspicuous fraction of melanosomes, including the giant ones, were not redistributed towards the cell centre and remained at the periphery (Fig. 4A, pR/OA1wt). As expected, no melanosome redistribution was observed by transient expression of mutant OA1 proteins, either carrying a missense mutation identified in patients with ocular albinism (22) (Fig. 4A, pR/OA1T232K; arrowhead), or deleted of a critical region for GPCR activity (OA1Δ18, not shown; see below).
Figure 4.

Expression of the OA1 cDNA rescues the Oa1-KO phenotype. Oa1−/− melanocytes were transfected with a plasmid vector (A) or infected with a retroviral vector (B) expressing either wild-type (pR/OA1wt and LOA1SN) or mutant (pR/OA1T232K or LOA1Δ18SN; both these mutants display a subcellular distribution indistinguishable from wild-type) human OA1. Expression of the recombinant proteins was analyzed 24 h after transfection (A), or following G418 selection through several passages (B), by indirect immunofluorescence with antibodies specific to human OA1. Transient expression of wild-type OA1 is not sufficient to eliminate the giant melanosomes (A, pR/OA1wt; arrows), which disappear only upon stable transduction (B, LOA1SN). Redistribution of melanosomes towards the perinuclear/Golgi area, where OA1 is also normally enriched, is induced by wild-type OA1 both in transiently and stably transduced Oa1−/− cells (A, pR/OA1wt, and B, LOA1SN; arrowheads). No correction of melanosomal size or distribution was observed with mutant OA1 at any time (A, pR/OA1T232K, and B, LOA1Δ18SN; arrowheads). Bars, 15 µm.
To obtain a complete phenotypic rescue, Oa1−/− melanocytes were stably transduced with retroviral vectors carrying either the wild-type human OA1 (LOA1SN), or a mutant that carries a deletion of 18 amino acids in the third cytosolic loop (LOA1Δ18SN), and that had been previously shown to lack G-protein activation abilities, despite an apparently normal subcellular distribution (25). The resulting Oa1−/−LOA1SN and Oa1−/−LOA1Δ18SN melanocyte lines showed stable OA1 expression in 80–90% of cells by immunofluorescence analysis (not shown). Optical and electron microscopy examination revealed that in Oa1−/−LOA1SN cells the size of melanosomes was homogeneous and comparable to Oa1+/− melanocytes (Figs 3 and 4B). In addition, the organelles recovered to an even cytoplasmic distribution with increased concentration in the perinuclear/Golgi region (Fig. 4B, arrowheads). In contrast, no rescue of macromelanosomal phenotype and abnormal melanosomal distribution was observed in Oa1−/−LOA1Δ18SN melanocytes (Figs 3 and 4B). Finally, the requirement for OA1 in normal melanosome distribution was further confirmed by RNA interference (RNAi) studies, which reproduced the Oa1-KO phenotype in Oa1+/− melanocytes (data not shown).
Oa1-KO melanocytes behave similarly to wild-type upon disruption of AF-based transport
All together, the above findings indicate that both in RPE cells and skin melanocytes the absence of OA1 function determines an alteration of melanosome distribution that appears opposite to that observed upon disruption of AF-mediated transport and suggests a dysfunction in melanosome motility along MTs and/or AFs. In both parental Oa1+/− and Oa1−/− melanocytes, as well as transduced Oa1−/−LOA1SN and Oa1−/−LOA1Δ18SN cells, the MT cytoskeleton displays a similar radial arrangement, with the greatest concentration of tubulin filaments in the perinuclear region (Fig. 5A and Supplementary Material, Fig. S2). This organization appears in striking contrast with the distribution of melanosomes, which are enriched at the centrosome in wild-type cells, while are excluded from this area in Oa1-deficient melanocytes (Fig. 5A, arrowheads). Conversely, in all cell types AF staining is more prominent at the cell periphery (Fig. 5A and Supplementary Material, Fig. S2), as previously reported (31), suggesting that abnormal melanosome distribution is not a consequence of cytoskeletal disorganization.
Figure 5.

Melanosomes redistribute according to MT density towards the nucleus upon disruption of the actin network in wild-type and Oa1-KO melanocytes. (A) Melanosome distribution and cytoskeletal organization. Pictures show the typical distribution of melanosomes, visualized in bright field, compared with tubulin or actin filaments, visualized by indirect immunofluorescence with anti-tubulin antibodies or phalloidin staining, respectively, in Oa1+/− and Oa1−/− cells. Black/white circles indicate the perinuclear area, as defined in organelle tracking analyses (see Materials and Methods), which is typically enriched in MTs (and melanosomes in wild-type, but not mutant, cells), while AF are more abundant at the cell periphery. Arrowheads point to the position of the centrosome. In Oa1−/− melanocytes, despite a similar cytoskeletal organization, melanosomes appear excluded from the MT-enriched perinuclear region. (B) Melanosome redistribution upon disruption of the actin cytoskeleton. Shown are representative bright field pictures of Oa1+/− and Oa1−/− cells prior to treatment (untreated); after 1 h of cytochalasin D treatment (cyto D 1 h); and, following removal of the drug and extensive washing, allowed to recover for 1 h (recovery 1 h). In both cell types, melanosome similarly redistribute upon AF disruption and recover to the original distribution after withdrawal of the drug. (C) Melanosome redistribution after 1 h of cytochalasin D treatment is compared with the residual AF labelling by phalloidin under the plasma membrane, confirming the absence of generalized retraction of cell margins. Bars, 15 µm.
To test whether the Oa1-KO phenotype is due to inability of melanosomes to move on tubulin filaments, we eliminated the contribution of actin-based transport by using AF-disrupting drugs. Treatment with cytochalasin D (which caps AFs inducing reorganization of the actin cytoskeleton in short actin patches; see Supplementary Material, Fig. S2) resulted in a similar perinuclear concentration of melanosomes both in wild-type and Oa1-deficient cells, either parental or transduced, with rapid recovery (within 1 h) of the initial distribution upon drug withdrawal (Fig. 5B and data not shown). Similar results were obtained with latrunculin A, although higher cell toxicity was observed (not shown). As alternative approach, we inhibited actin-based transport by transfecting wild-type and Oa1-deficient melanocytes with a plasmid encoding a dominant-negative construct for myosin Va, consisting in the melanocyte-specific tail domain of the motor (pEGFP-MC-LT) (31). Constructs for EGFP alone or the brain-specific tail domain of myosin Va (pEGFP-BR-LT) (32) were used as negative controls. Following 24–72 h of expression, pEGFP-MC-LT was able to induce a striking perinuclear distribution of melanosomes in both wild-type and Oa1-deficient cells (Fig. 6A and Supplementary Material, Fig. S3A), reminiscent of the dilute phenotype (null at the Myo5a gene locus). In contrast, no or minor effects were generated by pEGFP or pEGFP-BR-LT, consistent with previous observations (32). Quantification of the dilute-like phenotype in pEGFP-MC-LT expressing cells revealed a comparable behaviour of wild-type and Oa1-deficient cell lines, either parental or transduced (Fig. 6B), supporting the notion that lack of OA1 function does not impair MT-based motility per se.
Figure 6.

Melanosomes aggregate to the central cytoplasm upon disruption of myosin Va function and efficiently recruit its tail domain in wild-type and Oa1-KO melanocytes. (A) Representative optical pictures showing the redistribution of melanosomes upon expression of a dominant-negative construct for myosin Va. Oa1+/− and Oa1−/− cells were transfected with pEGFP-MC-LT, driving the expression of the EGFP-tagged melanocyte-specific tail domain of myosin Va. The pEGFP-BR-LT plasmid, driving the expression of the brain-specific tail domain of myosin Va, was used as negative control. After 24–72 h, the distribution of melanosomes in bright field was assessed in EGFP-positive cells. The white line marks the edges of transfected cells. Bars, 15 µm. (B) Quantification of melanosome aggregation in parental and transduced pEGFP-MC-LT transfected cells. Results represent the mean ± SD of 2-3 independent experiments (total number of cells counted for each line: 80–250). The dilute-like phenotype was observed in a similarly high percent of transfected wild-type and Oa1-deficient melanocytes. (C) Representative optical pictures showing the colocalization of melanosomes with the melanocyte-specific tail domain of myosin Va. Oa1+/− and Oa1−/− cells were transfected with pEGFP-MC-LT and analyzed before the appearance of a complete dilute-like phenotype. Melanosomes are typically surrounded by a ring of fluorescence corresponding to the EGFP-MC-LT construct in both wild-type and Oa1-KO cells. In the insets (4× magnification), arrows point to examples of colocalization. Bars, 15 µm. (D) Quantification of melanosome colocalization with the EGFP-MC-LT fusion protein in parental and transduced transfected cells. Results represent the mean ± SD of the data obtained from 10 different cells pooled from 2–3 independent experiments (total number of melanosomes counted for each cell line: 250–300). No significant differences were found between wild-type and Oa1-deficient melanocytes.
The behaviour of wild-type and Oa1-deficient melanocytes also suggested that myosin Va was similarly recruited to melanosomes in both cell types. To directly confirm this expectation, we performed co-localization studies in cells transfected with pEGFP-MC-LT before the appearance of a completely aggregated phenotype. As shown in Figure 6C and Supplementary Material, Figure S3B, we observed that the EGFP-MC-LT fusion protein distributed in a ring-like pattern in most transfected cells and was found to label a high fraction of melanosomes in both Oa1+/− and Oa1−/− melanocytes (Fig. 6C), as well as Oa1−/−LOA1SN and Oa1−/−LOA1Δ18SN melanocytes (Supplementary Material, Fig. S3B). Quantification of co-localization in different transfected cells revealed an identical fraction of mature melanosomes positive for EGFP-MC-LT in wild-type and Oa1-deficient cell lines, either parental or transduced (Fig. 6D). In addition, consistent with the observation that all melanosomes, independently on their size (Fig. 6A and Supplementary Material, Fig. S3A; Fig. 7), were redistributed towards the nucleus upon pEGFP-MC-LT transfection in Oa1-deficient melanocytes, both normal and giant melanosomes were found to extensively co-localize with the fusion protein (Fig. 6C and Supplementary Material, Fig. S3B). These data suggest the presence of an intact binding site for myosin Va on Oa1-KO melanosomes and do not support the possibility that lack of OA1 function grossly interferes with the correct recruitment of the Rab27a/melanophilin/myosin Va tripartite complex on the organelles.
Figure 7.

Melanosomes display defective motility in Oa1-KO melanocytes. Representative examples of melanosome paths covered during the 90 s time frame in Oa1+/− and Oa1−/− cells, either in untreated conditions (NT), or after nocodazole treatment, or following expression of the dominant-negative myosin Va construct (EGFP-MC-LT). In untreated cells, melanosomes were analysed either within (NT in), or outside (NT out) of the perinuclear area, which is delimited by the black 15 µm-radius circle in the pictures. In the insets (2.5× magnification) representative trajectories are shown in red. The longest paths occur in untreated Oa1+/− cells (particularly in the perinuclear area) and in pEGFP-MC-LT-transfected Oa1+/− and Oa1−/− cells, while melanosomes appear mostly stationary in untreated Oa1−/− cells and in both cell types upon nocodazole treatment. Bars, 5 µm.
Oa1-KO melanocytes exhibit reduced melanosome motility in the presence of intact AF-based transport
The depletion of melanosomes from the MT-enriched perinuclear region and their relative concentration in the AF-enriched cell periphery in Oa1-KO melanocytes suggest a defect of MT or a prevalence of AF-based transport. However, Oa1-deficient melanosomes are able to efficiently move on MTs towards the nucleus upon AF removal and show a similar capacity of recruiting the myosin Va tail domain compared with organelles in wild-type cells. To determine whether melanosomes do actually display abnormal motility in the absence of OA1, we performed time-lapse video microscopy in living cells. In order to reduce the incidence of cell line-dependent variables, we compared both parental Oa1+/+ and Oa1+/− versus Oa1−/− lines, and transduced Oa1−/−LOA1SN versus Oa1−/−LOA1Δ18SN lines. Bright field movies were obtained by capturing two pictures per second for 90 s from melanocytes, either untreated or following treatment with AF- or MT-disrupting drugs, or following transfection with pEGFP-MC-LT.
The direct observation of movies acquired in untreated conditions revealed that in general melanosomes display variable and intermittent motility, with slow movements and pauses interrupted by bursts of faster movements, as described previously (9,31). Moreover, both in Oa1+/− (or Oa1+/+) and Oa1−/− melanocytes, melanosomes within a few microns from the nucleus were more motile than those towards the cell periphery (Fig. 7 and Supplementary Material, Movies 1–4). This distinction presumably depends on the different distribution of MTs, concentrated in the central cytoplasm and responsible for fast long-range bidirectional movements, relative to AFs, predominant at the cell periphery and responsible for local short-range movements (7,33). However, in wild-type melanocytes the organelles showed a definitively higher motility compared with Oa1-KO cells, particularly in the perinuclear region, but also towards the periphery (Fig. 7 and Supplementary Material, Movies 1–4). Nocodazole treatment lead to a general paralysis of melanosome movements in all cell lines (Fig. 7), suggesting that most appreciable movements are in fact MT-dependent. Conversely, pEGFP-MC-LT transfection or cytochalasin D treatment resulted into a similarly intense organelle motility throughout the cytoplasm in both wild-type and Oa1-KO cells (Fig. 7, Supplementary Material, Movies 5 and 6, and not shown). Similar results were obtained in transduced cell lines (not shown).
To quantify these observations, we performed organelle tracking studies. Given the gradient of motility from the central region to the periphery of cells in untreated conditions, we made a distinction between the organelles localized close to the nucleus and those at some distance (Fig. 7; see Materials and Methods for further details). In addition, since Oa1−/− and Oa1−/−LOA1Δ18SN melanocytes often display a considerable heterogeneity in melanosome dimensions, and this physical variable could influence organelle motility, we compared melanosomes of equivalent size in wild-type versus Oa1-deficient lines (Supplementary Material, Fig. S4 and Table S1; see Materials and Methods for further details). Figure 8 shows the quantification of results obtained in various conditions. To define the characteristics of MT and AF-dependent movements in our system, we first pooled all melanosome steps obtained from Oa1+/− melanocytes, either untreated or following various treatments, and binned them in three increasing speed intervals: 0.0–0.2, 0.2–0.4 and ≥0.4 μm/s (Fig. 8A, white, light grey and dark grey bars, respectively).
Figure 8.

Quantification of melanosome motility in wild-type and Oa1-KO melanocytes. (A) Speed distribution profile obtained by pooling and binning in increasing speed intervals, as indicated, all melanosome steps from Oa1+/− melanocytes, or from Oa1−/− melanocytes. Cells were analyzed either untreated and selected inside or outside of the perinuclear area (NT in and out, respectively); or after treatment with nocodazole to deplete the MTs; or following expression of the dominant-negative myosin Va construct (EGFP-MC-LT), or treatment with cytochalasin D (Cyto D) to disrupt AF-based transport. Number of melanosomes tracked: NT in = 103, NT out = 100, nocodazole = 90, EGFP-MC-LT and Cyto D = 50 (obtained from several different cells: 10–12 for NT and nocodazole; 5–7 for EGFP-MC-LT and Cyto D). Data are expressed as percent of total steps. In both melanocyte lines, speeds ≥0.4 μm/s (dark grey) are totally abolished by nocodazole, indicating that they represent a pure MT-dependent component. Oa1−/− melanocytes show a decreased motility in untreated conditions, yet recover completely upon disruption of AF-based transport. (B) Average total path of melanosomes from the same tracking analysis described above for Oa1+/− versus Oa1−/− melanocytes, and from similar analyses performed in Oa1+/+ versus Oa1−/− melanocytes, and in transduced Oa1−/−LOA1SN versus Oa1−/−LOA1Δ18SN lines. For each wild-type and Oa1-deficient lines compared in the graphs, tracked melanosomes were of comparable size (see Supplementary Material, Table S1). Results represent the mean ± SEM of the data from 40–100 melanosomes (considering one melanosome path as one data point). Compared with their wild-type counterpart, Oa1-deficient melanocytes show a significantly reduced average displacement in untreated conditions, while they completely recover upon pEGFP-MC-LT transfection, or cytochalasin D treatment. Transduced lines showed poor motility at the cell periphery (possibly due to the drug treatment necessary for maintenance of the transgenes), so differences were only appreciable in the perinuclear area. *P < 0.004, **P < 0.00001 (unpaired Student's t-test assuming equal variances).
In untreated Oa1+/− melanocytes, organelle tracking within the perinuclear area (Fig. 8A, NT in) resulted in 55% of steps corresponding to pauses or slow movements under 0.2 μm/s, 38% of steps corresponding to movements between 0.2 and 0.4 μm/s, and the remaining 7% of steps corresponding to faster movements. Consistent with a progressive depletion of MTs towards the cell periphery, organelle tracking outside the perinuclear area (Fig. 8A, NT out) resulted in a reduction of melanosome motility, which was further decreased upon nocodazole treatment (Fig. 8A, Nocodazole). In the latter condition, we observed an almost complete disappearance of steps corresponding to fast speeds ≥0.4 μm/s, suggesting that they actually reflect true MT-dependent motility. In contrast, in pEGFP-MC-LT-transfected or cytochalasin D-treated Oa1+/− cells, melanosome motility was recovered to values similar to that displayed by perinuclear organelles in untreated melanocytes (Fig. 8A, EGFP-MC-LT and Cyto D). When Oa1−/− melanocytes were subjected to similar analysis, an overall lower motility of melanosomes was observed in untreated conditions. In fact, the values obtained within (NT in) and outside (NT out) the perinuclear area in Oa1−/− cells were closely resembling those found outside the perinuclear area (NT out) and upon nocodazole treatment in Oa1+/− cells, respectively. However, when AF-based transport was disrupted by pEGFP-MC-LT transfection or cytochalasin D treatment, melanosomes in Oa1−/− melanocytes completely recovered their motility to levels comparable to organelles in wild-type cells. Similar results were obtained in Oa1−/−LOA1SN versus Oa1−/−LOA1Δ18SN cells and in untreated Oa1+/+ versus Oa1−/− melanocytes (not shown; analysis of Oa1+/+ melanocytes upon transfection or drug treatment was not possible due to cell suffering).
To determine the significance of these findings, we calculated the total paths covered by single melanosomes. Melanosome values for each wild-type line were averaged and compared with those obtained from the respective Oa1-deficient counterpart (Fig. 8B). In untreated conditions, Oa1−/− and Oa1−/−LOA1Δ18SN melanocytes showed a profound and significant reduction of melanosome motility compared with their wild-type counterparts. These differences could mainly be attributed to MT-based transport, since (i) the higher motility of wild-type melanosomes was easier to appreciate in the MT-enriched perinuclear region (Figs 7 and 8B, compare melanosome paths in untreated conditions in Oa1+/− versus Oa1−/− cells and in transduced lines); (ii) fastest movements ≥0.4 μm/s, reflecting MT-dependent motility, were more represented in wild-type than Oa1-deficient melanocytes (Fig. 8A) and (iii) the contribution of AFs to melanosome paths appeared negligible, since the average displacement calculated upon nocodazole treatment was 1.5–3.0 µm/90 s in all cell lines (not shown).
When the same analysis was conducted considering only melanosomes showing fast movements (≥0.4 μm/s), which represent a greater fraction in wild-type (54–91%, depending on cell line and distance from nucleus) than Oa1-deficient melanocytes (16–61%), differences were partially levelled (data not shown). These findings suggest that the main defect in Oa1-deficient melanocytes consists in a lower frequency (rather than velocity) of MT-based movements. However, in the absence of actin-based transport, Oa1-deficient melanosomes not only redistributed at the cell centre as in wild-type cells, but also showed equivalent motility on MTs. In fact, upon pEGFP-MC-LT transfection or cytochalasin D treatment differences were basically abolished, with melanosomes in Oa1-deficient cells showing a recovery of motility equivalent to that observed in the perinuclear area of wild-type cells (Fig. 8B). Finally, we evaluated whether Oa1-deficient melanosomes move towards the cell centre or the periphery at different rates compared with wild-type organelles. The ratio between centrifugal and centripetal displacement was determined for single melanosomes and values for each wild-type line were averaged and compared with those obtained from the respective Oa1-deficient counterpart. No significant differences were found between wild-type and Oa1-deficient cells, either at steady-state or following AF disruption (Supplementary Material, Fig. S5), suggesting that OA1 might not affect the balance between kinesin and dynein motors. From these data we conclude that in the absence of OA1 function melanosomes move less efficiently and especially less frequently on the tubulin cytoskeleton, however, this deficiency is not intrinsic of the MT-based transport system and manifests exclusively in the presence of intact actin-based transport.
DISCUSSION
The histological hallmark of ocular albinism is the presence of macromelanosomes in pigment cells of the skin and eyes, suggesting a defect in melanosome biogenesis. However, the process leading to macromelanosome formation and the actual role of the giant organelles in the pathogenesis of the disease remain unclear. In fact, the macromelanosomal phenotype displays variable expressivity in vitro and in vivo (34,35) and, at least in the mouse, manifests during development subsequent to optic misrouting, implying that it might represent only an epiphenomenon of the disease (27). In the search for the primary role of OA1 in ocular albinism, we identified an abnormally skewed distribution of melanosomes towards the apical pole of the RPE in Oa1-KO mice at embryonic stages preceding the formation of macromelanosomes and the reduction of organelle number. An equivalent abnormality was replicated in melanocyte cultures obtained from the skin of Oa1-KO mice. In fact, Oa1-KO melanocytes showed not only abnormal size, but also abnormal distribution melanosomes, with depletion of the organelles from the perinuclear area and accumulation towards the cell periphery. Despite their exclusion from the central cytoplasm at steady-state, Oa1-KO melanosomes were able to aggregate at the centrosome upon disruption of the actin cytoskeleton or expression of a dominant-negative construct for myosin Va. Finally, melanosome tracking analyses revealed a significant reduction of MT-based motility exclusively in the presence of intact AF-mediated transport, consistent with the idea that OA1 might regulate the dynamic equilibrium between the two cytoskeletal systems. These results were independent on the physical size of melanosomes, thus revealing a novel function for OA1 in organelle transport that might be of relevance for the disease pathogenesis.
OA1 regulates melanosome transport on MTs
Both in melanophores and mammalian melanocytes, the distribution of melanosomes results from the dynamic balance between AF-based and MT-based transport. In fish or frog melanophores, the switch between these two transport systems is believed to depend on signalling events, including cAMP and protein kinases, that direct the coordinated dispersion or aggregation of melanosomes in response to extracellular stimuli and result in colour adaptation (for review see 36). In contrast, in mammalian melanocytes, melanosome transport is associated with melanogenesis and is thought to proceed mostly in a perinuclear to centrifugal route, adapted to ensure the peripheral accumulation of mature melanosomes prior to keratinocyte transfer (7). In this context, locally operating signalling mechanisms would allow the organelles to move independently of one another, perhaps depending on their maturation stage. The observation that not all melanosomes are redistributed in Oa1−/− cells upon transient expression of OA1 (Fig. 4A) suggests that OA1 does not function through diffuse cytosolic signalling, but rather in an organelle autonomous fashion.
In the competition between the actin and tubulin cytoskeletal systems, OA1 seems to facilitate the functional interaction of pigmented melanosomes with MTs, which could determine by itself the accumulation of organelles in the perinuclear area. In fact, when actin-mediated capture at the cell periphery is down-regulated, melanosomes typically redistribute according to MT density and therefore in the central cytoplasm where the bulk of tubulin filaments reside (31). Based on our data and because of the ‘tug of war’ between the two transport systems involved, we cannot distinguish if stimulation of MT-based transport is a direct effect of OA1 activity, or the indirect result of inhibition of actin-based transport. OA1 might act by tethering melanosomes to tubulin filaments, thereby stabilizing the productive association between cargo and cytoskeletal route, as hypothesized for the dynactin complex (37). Alternatively, OA1 could counteract the capture of melanosomes by the AF system and as a consequence enhance the frequency by which the organelles move along MTs (11,31), while in the absence of OA1 melanosome entrapment by the actin network at the cell periphery would prevail.
The exact molecular mechanism by which OA1 accomplishes its effects on transport remains unclear. Our analyses indicate a normal ability of Oa1-KO melanosomes not only to move on MTs, but apparently also to recruit the Rab27a/melanophilin/myosin Va tripartite complex responsible for actin-mediated capture, suggesting that the receptor might play a regulatory—rather than structural—role in the concerted effort of MT and AF-based motors required for proper organelle distribution. Switching between MT and AF-based systems is regulated by cytoplasmic cAMP levels in fish and frog melanophores (38,39). Furthermore, in the same animal models, melanosome-associated MAP kinase has been shown to regulate organelle transport on MTs (40). Since OA1 appears to couple with Gαi in melanocytes (21), it is possible that it regulates the local concentration of the second messenger, thereby using an equivalent signalling mechanism at the single organelle level. Alternatively, OA1 might contribute to the recruitment of signalling complexes on the melanosomal membrane, including specific adaptors or kinases involved in transport. Consistent with the in vivo motility studies, we found that OA1 interacts efficiently with tubulin in co-immunoprecipitation assays (M.V.S. and P.B., unpublished observations), suggesting that MTs represent a downstream effector of the receptor. However, the same immunoprecipitates did not contain any molecular motors or regulators known to direct the movement of melanosomes along MTs, such as dynectin, dynein and conventional kinesin (M.V.S. and P.B., unpublished data). Therefore, the role of this interaction in melanosome motility remains to be established, and further studies will be necessary to define the signal transduction pathway triggered by OA1 and its downstream effectors at the melanosomal membranes.
OA1 plays a role in both melanosome biogenesis and transport
Organelle biogenesis and transport are tightly interconnected processes (for review see 41,42) and the same protein regulators, as the Rab GTPases, can control both pathways through the assembly of multiprotein complexes on specific subcellular compartments (43,44). Our findings with the intracellular GPCR OA1 now add another class of GTPases to the list of regulators of organelle biogenesis and transport, namely heterotrimeric G proteins, which are best known as plasma membrane transducers, but have also been implicated in various membrane trafficking events in the endomembrane system (45). Does OA1 regulate melanosome biogenesis and transport independently, or one as consequence of the other? The abnormal melanosome distribution in Oa1-KO RPE at stages preceding the appearance of giant organelles and the defective motility of normal size melanosomes in Oa1-KO melanocytes, as well as the apparently normal ability of Oa1-deficient melanosomes to recruit the myosin Va tail domain—suggesting proper enrolment of the overall tripartite complex on fully mature organelles—imply that regulation of melanosome transport might represent an independent role played by OA1 in pigment cells. Nevertheless, given the lack of morphogenetic defects at immature melanosomal stages and the variability of the macromelanosomal phenotype, it is also possible that the primary defect in ocular albinism lies in organelle motility, which secondarily leads to aberrant biogenesis. For instance, as a melanosomal GPCR, OA1 might be sensitive to the maturation stage of the organelles and transduce this information to the cytosol by triggering a signalling cascade to retain pigmented, yet incompletely developed, organelles on MTs for full maturation. In the absence of OA1, melanosomes might be prematurely transported to the cell periphery and become susceptible to abnormal fusion events, giving rise to macromelanosomes, especially in cells highly active in membrane remodelling and disposal such as the RPE (46).
OA1 shares genomic localization and functional features with SHROOM2/APXL
A combined defect in melanosome distribution and maturation, somewhat reminiscent of OA1 loss-of-function, has been recently reported in the RPE of Xenopus embryos lacking SHROOM2/APXL function (47), supporting the idea that melanosome biogenesis and transport are tightly bound processes. Interestingly, the human ortholog of the SHROOM2/APXL gene, like the OA1 gene, was originally isolated by our group from the ocular albinism type 1 critical region on the distal short arm of the X-chromosome (48). The involvement of SHROOM2/APXL in ocular albinism appears unlikely, given that most patients carry mutations within the OA1 gene (34,49), while none was identified in SHROOM2/APXL (48). It is nevertheless surprising that a gene possibly implicated in the same pathways as OA1 localizes close to the OA1 gene not only in humans (where the two genes lie 20 kb apart and are transcribed in opposite directions; MVS, unpublished observations), but also mice (50) and even frogs (47). Whether a related function in RPE physiology is also paralleled by a common transcriptional regulation will be an interesting matter for future investigations.
MATERIALS AND METHODS
Melanocyte line generation and culture
Melanocyte lines were established from Oa1+/+, Oa1+/− and Oa1−/− female mice of the 129/Sv strain (27) and named melan-A (designating the agouti A/A genotype), melan-Oa1+/− and melan-Oa1−/−, respectively. Trunk skins from neonatal mice and mitomycin C-treated XB2 feeder cells were used for establishment of melanocyte cell lines, as described previously (51). Melanocytes were cultured in RPMI 1640 (Invitrogen) supplemented with 10% foetal bovine serum (Sigma), 2 mm glutamine (Invitrogen), 100 U/ml penicillin G (Invitrogen), 1000 U/ml streptomycin (Invitrogen), 200 nm tetradecanoyl phorbol-13-acetate (Sigma) and 200 pm cholera toxin (Sigma), at 37°C with 10% CO2. Cells were used for experiments up to passage 30 (10 for subclones). Relative to Oa1+/− and Oa1+/+ cells, Oa1−/− melanocytes displayed a quite dendritic morphology, which became particularly evident in the presence of cholera toxin. This phenotype was not corrected by transduction with wild-type OA1 (the cell morphology of transduced Oa1−/−LOA1SN and Oa1−/−LOA1Δ18SN melanocytes was comparable with parental Oa1−/− cells, see below), therefore a role of OA1 in dendrite formation remains to be established. However, to reduce morphological differences, cholera toxin was used exclusively during generation of the lines, whereas it was subsequently omitted for their analysis. Oa1+/+ and Oa1+/− melanocytes were indistinguishable based on melanosome size and distribution. Nevertheless, since Oa1, like human OA1, is located on the X-chromosome (52–54) and might be subjected to X-inactivation, we looked for the presence of mosaic Oa1 expression within the Oa1+/− cell line [due to the immortalization procedure, see (51), these melanocyte lines are expected to originate from multiple clones]. Subclones generated by limiting dilution were tested for melanosomal abnormalities and Oa1 expression by RT–PCR (see below). All nine clones analyzed were comparable to the parental line for cell morphology and melanosomal phenotype (Supplementary Material, Fig. S1A) and were able to express full-length Oa1 (Supplementary Material, Fig. S1B), suggesting that even if Oa1 is subjected to X-inactivation, the Oa1+/− cell line derives from one or more precursors carrying the mutant X-chromosome inactivated. Thus, we routinely used the Oa1+/− line as wild-type control, since in our hands these cells were morphologically more homogeneous and showed a more stable pigmentation through passages and freezing–thawing cycles than Oa1+/+ cells. However, experiments were confirmed with Oa1+/+ cells and procedures involving a limited number of cells (as melanosome tracking) were performed on the PCR-tested Oa1+/− subclone #5 (Supplementary Material, Fig. S1). Cytoskeletal drug treatments were performed as follows: 1 µm cytochalasin D (Sigma) for 30–60 min at 37°C (after 15 min of treatment, the reorganization of AFs in short patches was already visible and melanosomes began to redistribute; over 90 min, cells often showed morphological changes, becoming smaller and dendritic); 0.5 µm latrunculin A (Sigma) for 10–40 min at 37°C (even after 10 min, cells showed morphological changes, becoming smaller and dendritic); 30 µm nocodazole (Sigma) for 30–60 min at 37°C. Disruption of cytoskeletal networks was tested by anti-tubulin antibodies and phalloidin staining. In all treatments, only the target cytoskeletal system was affected (Supplementary Material, Fig. S2 and not shown).
Reverse transcription polymerase chain reaction
Cytoplasmic RNA was isolated with RNeasy Mini kit (Qiagen), and reverse transcribed with oligo(dT) primers using the SuperScript First-Strand Synthesis System for RT–PCR kit (Invitrogen), according to manufacturers' instructions. The resulting cDNA was used as template for conventional RT–PCR reactions with primers FowmOa1-1: 5′-CCACTGCCTGTGACTTGCTTG-3′ and RevmOa1-9: 5′-CCTGGGCTTGGGAAATGGAGT-3′, which amplify a 986-bp product spanning all nine exons of the Oa1 gene. PCR conditions were 1 cycle at 94°C for 5 min, 60°C for 2 min and 72°C for 5 min, followed by 40 cycles at 94°C for 1 min, 62°C for 1 min and 72°C for 5 min in the presence of 5% DMSO.
Expression vectors, transfection and retroviral infection
Constructs pR/OA1wt, pR/OA1T232K, LOA1SN and LOA1Δ18SN, containing the wild-type and mutant OA1 cDNAs, were previously described (22,35). Plasmids pEGFP-MC-LT and pEGFP-BR-LT were kindly provided by Dr J.A. Hammer III (31,32) and plasmid pEGFP-Rab27a was kindly provided by Dr M.C. Seabra (55). For transient transfections, melanocytes were plated on 13-mm glass coverslips, transfected using FuGENE 6 according to the manufacturer's instructions (Roche) and analysed 24–72 h post-transfection. For stable transduction, retroviruses were produced in Phoenix Eco packaging cells, following the Nolan Lab protocol (http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html). Phoenix cells were transfected at 70% of confluence with LOA1SN and LOA1Δ18SN retroviral vectors using FuGENE 6 in a 10-cm plate. Twenty-four hours after transfection, the medium was changed and the retroviral supernatant collected after additional 24 h. After centrifugation and filtration to eliminate cell debris, the retroviral supernatant was either frozen at −80°C or supplemented with polybrene (Sigma) at 8 μg/ml and used for melanocyte infection. The day before infection, melanocytes were seeded in 6-cm plates at 30–40% of confluence and the following day incubated with 4 ml of retroviral supernatant/polybrene mix for 4 h. Seventy-two hours post-infection, cells were selected as a polyclonal population with 700 µg/ml of G418 (Sigma), which was maintained thereafter in order to avoid loss of retroviral integration.
Immunofluorescence analysis
Immunofluorescence analysis was performed as previously described (26), using the following primary antibodies: W7 anti-OA1, rabbit anti-human OA1 polyclonal antiserum (19); anti-β-tubulin, mouse anti-β-tubulin monoclonal antibody (Boheringer); anti-giantin, rabbit anti-giantin polyclonal antiserum (a gift of Dr A. De Matteis). Secondary antibodies were Cy3/Cy2-conjugated donkey anti-rabbit IgG and donkey anti-mouse IgG (Jackons ImmunoResearch Laboratories). For staining of AFs, rhodamine (TRITC)-conjugated phalloidin (Sigma) was used. For nuclear staining, Hoechst 33342 (Sigma) at 100 ng/ml was used for 5 min prior to mounting. Coverslips were mounted using Mowiol 4–88 reagent (Calbiochem) and viewed with a PlanApochromat 63× n.a. 1.4 oil objective on an epifluorescence Axiophot microscope (Zeiss). Images were acquired with a C4742-95 CCD camera (Hamamatsu Photonics) using HiPic32 software (Hamamatsu Photonics) and processed with Photoshop (Adobe). pEGFP-MC-LT-transfected cells typically showed a punctuated and ring-like staining superimposed to a diffuse signal. At short expression times (24 h), the ring-like staining was often spread all over the cell and was clearly surrounding black melanosomes, whereas at longer expression times most of the staining was concentrated in the perinuclear region together with black melanosomes. Conversely, pEGFP-BR-LT-transfected cells displayed at any time a punctuated staining superimposed to a diffuse signal, with an evident accumulation of fluorescence at the MT organizing centre and only occasional co-localization with black melanosomes, as described previously (32). Quantification of melanosome redistribution following pEGFP-MC-LT transfection was performed by calculating in each experiment the percent of transfected cells (visualized by means of the EGFP fluorescence), showing a dilute-like phenotype (pronounced accumulation of melanosomes in the central cytoplasm). A small percent of cells with an abnormally round or dendritic morphology, indicating cell suffering, was excluded from the counts. Since melanosome aggregation can sometimes result from transfection-dependent cell suffering (∼5% of Oa1+/− cells transfected with pEGFP-BR-LT or pEGFP alone showed a dilute-like phenotype), normalization for pEGFP-MC-LT-specific effects was obtained by subtracting in each experiment the percent of aggregation observed in twin coverslips transfected with the brain-specific pEGFP-BR-LT construct. Quantitative immunofluorescence analysis of the co-localization between the EGFP-MC-LT fusion protein and pigment granules was performed on digital micrographs of cells displaying a bright ring-like pattern of the fusion protein, before the appearance of a full dilute-like phenotype (typically 24 h of expression). Distinct black melanosomes (15–30) were randomly marked throughout the cytoplasm of individual cells, with the exception of areas overcrowded with organelles (perinuclear areas, tips of dendrites, areas under the plasma membrane), and their co-localization with EGFP-MC-LT was assessed. EGFP-MC-LT staining was considered as co-localizing with melanosomes when (i) forming a complete or partial ring surrounding single melanosomes, and (ii) appearing as a clear and distinct spot superimposed to isolated melanosomes. More selective co-localization criteria, i.e. considering only the ring-like pattern of EGFP-MC-LT staining, led to a reduction in the co-localization percentage (45–50%), however, no differences were found between wild-type and Oa1-deficient cells. In addition, although in these counts both giant and normal size melanosomes were considered, exclusion of the giant organelles did not lead to differences between wild-type and Oa1-deficient cells. Statistical analyses were performed using Microsoft Excel software.
Live cell imaging
Mouse melanocytes grown on 24-mm glass coverslips were transferred to round adaptors (OKO-lab) with 2 ml of medium supplemented with 25 mm HEPES, pH 7.4, and placed on a heated stage at 37°C, to be observed in bright field either with a PlanApochromat 63× n.a. 1.4 oil objective (Zeiss) on an Axiovert S100 TV2 microscope (Zeiss), or with a 60× n.a. 1.4 oil objective (Olympus) on a Olympus IX70 microscope (DeltaVision System, Applied Precision). Each coverslip was analysed for a total time not exceeding 1 h to ensure properly buffered conditions (33). Cells treated with cytochalasin D and nocodazole were incubated with the drugs 30 min prior to observations and recorded up to 1 h from the beginning of the treatment. Cells expressing pEGFP-MC-LT were analyzed 24 h post-transfection by choosing among the EGFP-positive cells those showing an already evident dilute-like phenotype. Images were recorded with a ORCA II CCD camera (Hamamatsu Photonics), using Image-Pro Plus software (Media Cybernetics), or with a CoolSNAP CCD camera (Roper Scientific, Photometrics), using softWoRx 3.5 software (Applied Precision). Each wild-type and Oa1-deficient line to be compared were acquired in the same conditions on the same day. ImageJ (plugin ‘Manual tracking’) from NIH Image (http://rsb.info.nih.gov/ij/) was used to track the melanosome paths. All data collected were transferred and elaborated in an Excel worksheet (Microsoft). A number of 50–100 distinct melanosomes were chosen randomly in comparable areas of the cytoplasm of 5–12 different Oa1+/− or Oa1−/− cells from two independent experiments, and individual movements (or steps) were calculated as the displacement between successive frames (resulting in 179 steps per organelle). Similarly, 40–50 distinct melanosomes were analyzed from five different Oa1+/+ or Oa1−/− cells, or Oa1−/−LOA1SN or Oa1−/−LOA1Δ18SN cells. In untreated conditions, melanosomes were selected based on their position in the first frame within or outside of the perinuclear area, defined as the cytoplasm within a 15-µm-radius circle around the nucleus and corresponding to the MT-enriched region in most cells (Fig. 5A, white circles). In other conditions, melanosome motility was quite uniform throughout the cytoplasm and organelles were chosen mostly out of the perinuclear area, which is inconveniently thick and crowded, particularly upon cytochalasin D treatment or pEGFP-MC-LT expression. Movement direction was calculated relative to the centroid position of the nucleus, or of the melanosome aggregate in pEGFP-MC-LT-transfected cells. Some heterogeneity in melanosome size was present both in wild-type and Oa1-deficient lines, however, this variability was definitively more pronounced in the latter, with melanosomes showing various dimensions from normal (∼1 µm or less) to clearly giant (2–3 µm). In initial trials, despite the tracking of giant melanosomes was always avoided, random sampling of organelles of normal and apparently similar size resulted in the selection of melanosomes with slightly, yet significantly, bigger average diameters in Oa1-deficient lines compared with their wild-type counterparts (wild-type organelles were in average 1.5–2 pixels smaller, corresponding to ∼0.15–0.2 µm). Since this physical variable had an effect on the frequency of fast movements (data not shown), we established a size range for melanosomes to be analysed, corresponding to 7–9 ± 1 pixels in the digital movie acquisitions (corresponding to ∼0.7–0.9 ± 0.1 µm), for both major and minor diameters. The sizes of all tracked melanosomes, chosen within the allowed range, were precisely determined by two independent investigators by considering as the melanosome margins the more external series of pixels clearly distinct from the surrounding background and extending for at least half of the organelle shape (Supplementary Material, Fig. S4). The dimensions of organelles obtained from each wild-type cell line and its Oa1-deficient counterpart were compared to exclude statistically significant differences. The average major and minor diameters and Student's t-test values of melanosomes analysed for each wild-type and Oa1-deficient line in the different conditions are reported in Supplementary Material, Table S1.
Electron microscopy
Cell culture monolayers were fixed for 15 min at 4°C with 4% paraformaldehyde and 2.5% glutaraldehyde in 125 mm phosphate buffer. The monolayers were detached by rubber and centrifuged at high speed. The pellet was post-fixed for 1 h with OsO4 in 125 mM phosphate buffer, washed, and embedded in Epon. Conventional thin sections were collected on uncoated grids, stained with uranyl and lead citrate, and acquired by a CCD camera-equipped Leo912ab electron microscope and AnalySIS software (Soft Imaging System). Electron microscopy analysis of mouse retinas was performed on comparable regions of the eyes from wild-type and Oa1-KO mice belonging to the same litter and sacrificed at the same time, as previously described (27,29). For quantification of melanosome distribution, rectangular areas of RPE, corresponding to few cells and delimited by the apical and basal membranes, were divided transversely into apical zone (corresponding to one-third of the total area) and cell body (corresponding to the remaining two thirds), and the organelles contained within were counted. In case of slight irregularities of the apical membrane, as in the Oa1-KO section shown in Figure 1A, the external limit of the apical region was placed at an intermediate position and the RPE cytoplasm that remained excluded was calculated to compensate gaps in the area of the apical zone under consideration. All quantitative analyses on organelle size, density and distribution were performed considering mature melanosomes.
SUPPLEMENTARY MATERIAL
FUNDING
The financial support of the Vision of Children Foundation, San Diego-CA (to M.V.S.), the National Institutes of Health/National Eye Institute (EY014540 to M.V.S.); Telethon-Italy (F.3 to M.V.S.) and Wellcome Trust (078327/Z/05 to DCB) is gratefully acknowledged.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr E.C. Dell'Angelica for helpful suggestions and critical reading of the manuscript and Drs C. Covino and M.C. Panzeri for help with optical and electron microscopy. Part of this work was carried out in Alembic, an advanced microscopy laboratory established by the San Raffaele Institute and the Vita-Salute San Raffaele University, Milan, Italy.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Quevedo W.C., Fitzpatrick T.B., Szabo G., Jimbow K. Biology of melanocytes. In: Fitzpatrick T.B., Eisen A.Z., Wolff K., Freedberg I.M., Austen K.F., editors. Dermatology in General Medicine. Vol. 1. New York: McGraw-Hill; 1987. pp. 224–251. [Google Scholar]
- 2.Orlow S.J. Melanosomes are specialized members of the lysosomal lineage of organelles. J. Invest. Dermatol. 1995;105:3–7. doi: 10.1111/1523-1747.ep12312291. [DOI] [PubMed] [Google Scholar]
- 3.Dell'Angelica E.C., Mullins C., Caplan S., Bonifacino J.S. Lysosome-related organelles. FASEB J. 2000;14:1265–1278. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 4.Raposo G., Marks M.S. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–248. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- 5.Raposo G., Tenza D., Murphy D.M., Berson J.F., Marks M.S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 2001;152:809–824. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seiji M., Shimao K., Birbeck M.S., Fitzpatrick T.B. Subcellular localization of melanin biosynthesis. Ann. N. Y. Acad. Sci. 1963;100:497–533. [PubMed] [Google Scholar]
- 7.Marks M.S., Seabra M.C. The melanosome: membrane dynamics in black and white. Nat. Rev. Mol. Cell Biol. 2001;2:738–748. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- 8.Barral D.C., Seabra M.C. The melanosome as a model to study organelle motility in mammals. Pigment Cell Res. 2004;17:111–118. doi: 10.1111/j.1600-0749.2004.00138.x. [DOI] [PubMed] [Google Scholar]
- 9.Lopes V.S., Ramalho J.S., Owen D.M., Karl M.O., Strauss O., Futter C.E., Seabra M.C. The ternary Rab27a-Myrip-Myosin VIIa complex regulates melanosome motility in the retinal pigment epithelium. Traffic. 2007;8:486–499. doi: 10.1111/j.1600-0854.2007.00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klomp A.E., Teofilo K., Legacki E., Williams D.S. Analysis of the linkage of MYRIP and MYO7A to melanosomes by RAB27A in retinal pigment epithelial cells. Cell Motil. Cytoskeleton. 2007;64:474–487. doi: 10.1002/cm.20198. [DOI] [PubMed] [Google Scholar]
- 11.Gross S.P., Tuma M.C., Deacon S.W., Serpinskaya A.S., Reilein A.R., Gelfand V.I. Interactions and regulation of molecular motors in Xenopus melanophores. J. Cell Biol. 2002;156:855–865. doi: 10.1083/jcb.200105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers S.L., Gelfand V.I. Myosin cooperates with microtubule motors during organelle transport in melanophores. Curr. Biol. 1998;8:161–164. doi: 10.1016/s0960-9822(98)70063-6. [DOI] [PubMed] [Google Scholar]
- 13.Byers H.R., Yaar M., Eller M.S., Jalbert N.L., Gilchrest B.A. Role of cytoplasmic dynein in melanosome transport in human melanocytes. J. Invest. Dermatol. 2000;114:990–997. doi: 10.1046/j.1523-1747.2000.00957.x. [DOI] [PubMed] [Google Scholar]
- 14.Nascimento A.A., Roland J.T., Gelfand V.I. Pigment cells: a model for the study of organelle transport. Annu. Rev. Cell Dev. Biol. 2003;19:469–491. doi: 10.1146/annurev.cellbio.19.111401.092937. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs D., Azarian S.M., Lillo C., Kitamoto J., Klomp A.E., Steel K.P., Libby R.T., Williams D.S. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J. Cell Sci. 2004;117:6473–6483. doi: 10.1242/jcs.01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Futter C.E., Ramalho J.S., Jaissle G.B., Seeliger M.W., Seabra M.C. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Mol. Biol. Cell. 2004;15:2264–2275. doi: 10.1091/mbc.E03-10-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King R.A., Hearing V.J., Creel D.J., Oetting W.S. Albinism. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. 7th edn. III. New York: McGraw-Hill, Inc; 1995. pp. 4353–4392. Chapter 147. [Google Scholar]
- 18.O'Donnell F.E., Jr, Hambrick G.W., Jr, Green W.R., Iliff W.J., Stone D.L. X-linked ocular albinism. An oculocutaneous macromelanosomal disorder. Arch. Ophthalmol. 1976;94:1883–1892. doi: 10.1001/archopht.1976.03910040593001. [DOI] [PubMed] [Google Scholar]
- 19.Schiaffino M.V., Baschirotto C., Pellegrini G., Montalti S., Tacchetti C., De Luca M., Ballabio A. The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc. Natl. Acad. Sci. USA. 1996;93:9055–9060. doi: 10.1073/pnas.93.17.9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samaraweera P., Shen B., Newton J.M., Barsh G.S., Orlow S.J. The mouse ocular albinism 1 gene product is an endolysosomal protein. Exp. Eye Res. 2001;72:319–329. doi: 10.1006/exer.2000.0962. [DOI] [PubMed] [Google Scholar]
- 21.Schiaffino M.V., d'Addio M., Alloni A., Baschirotto C., Valetti C., Cortese K., Puri C., Bassi M.T., Colla C., De Luca M., et al. Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat. Genet. 1999;23:108–112. doi: 10.1038/12715. [DOI] [PubMed] [Google Scholar]
- 22.d'Addio M., Pizzigoni A., Bassi M.T., Baschirotto C., Valetti C., Incerti B., Clementi M., De Luca M., Ballabio A., Schiaffino M.V. Defective intracellular transport and processing of OA1 is a major cause of ocular albinism type 1. Hum. Mol. Genet. 2000;9:3011–3018. doi: 10.1093/hmg/9.20.3011. [DOI] [PubMed] [Google Scholar]
- 23.Sone M., Orlow S.J. The ocular albinism type 1 gene product, OA1, spans intracellular membranes 7 times. Exp. Eye Res. 2007;85:806–816. doi: 10.1016/j.exer.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staleva L., Orlow S.J. Ocular albinism 1 protein: trafficking and function when expressed in Saccharomyces cerevisiae. Exp. Eye Res. 2006;82:311–318. doi: 10.1016/j.exer.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Innamorati G., Piccirillo R., Bagnato P., Palmisano I., Schiaffino M.V. The melanosomal/lysosomal protein OA1 has properties of a G protein-coupled receptor. Pigment Cell Res. 2006;19:125–135. doi: 10.1111/j.1600-0749.2006.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piccirillo R., Palmisano I., Innamorati G., Bagnato P., Altimare D., Schiaffino M.V. An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J. Cell Sci. 2006;119:2003–2014. doi: 10.1242/jcs.02930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Incerti B., Cortese K., Pizzigoni A., Surace E.M., Varani S., Coppola M., Jeffery G., Seeliger M., Jaissle G., Bennett D.C., et al. Oa1 knock-out: new insights on the pathogenesis of ocular albinism type 1. Hum. Mol. Genet. 2000;9:2781–2788. doi: 10.1093/hmg/9.19.2781. [DOI] [PubMed] [Google Scholar]
- 28.Lopes V.S., Wasmeier C., Seabra M.C., Futter C.E. Melanosome maturation defect in Rab38-deficient retinal pigment epithelium results in instability of immature melanosomes during transient melanogenesis. Mol. Biol. Cell. 2007;18:3914–3927. doi: 10.1091/mbc.E07-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortese K., Giordano F., Surace E.M., Venturi C., Ballabio A., Tacchetti C., Marigo V. The ocular albinism type 1 (OA1) gene controls melanosome maturation and size. Invest. Ophthalmol. Vis. Sci. 2005;46:4358–4364. doi: 10.1167/iovs.05-0834. [DOI] [PubMed] [Google Scholar]
- 30.Rachel R.A., Dolen G., Hayes N.L., Lu A., Erskine L., Nowakowski R.S., Mason C.A. Spatiotemporal features of early neuronogenesis differ in wild-type and albino mouse retina. J. Neurosci. 2002;22:4249–4263. doi: 10.1523/JNEUROSCI.22-11-04249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X., Bowers B., Rao K., Wei Q., Hammer J.A., 3rd Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function In vivo. J. Cell Biol. 1998;143:1899–1918. doi: 10.1083/jcb.143.7.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X., Wang F., Rao K., Sellers J.R., Hammer J.A., 3rd Rab27a is an essential component of melanosome receptor for myosin Va. Mol. Biol. Cell. 2002;13:1735–1749. doi: 10.1091/mbc.01-12-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu X., Hammer J.A., III Making sense of melanosome dynamics in mouse melanocytes. Pigment Cell Res. 2000;13:241–247. doi: 10.1034/j.1600-0749.2000.130405.x. [DOI] [PubMed] [Google Scholar]
- 34.Schnur R.E., Gao M., Wick P.A., Keller M., Benke P.J., Edwards M.J., Grix A.W., Hockey A., Jung J.H., Kidd K.K., et al. OA1 mutations and deletions in X-linked ocular albinism. Am. J. Hum. Genet. 1998;62:800–809. doi: 10.1086/301776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiaffino M.V., Dellambra E., Cortese K., Baschirotto C., Bondanza S., Clementi M., Nucci P., Ballabio A., Tacchetti C., De Luca M. Effective retroviral-mediated gene transfer in normal and mutant human melanocytes. Hum. Gene Ther. 2002;13:947–957. doi: 10.1089/10430340252939050. [DOI] [PubMed] [Google Scholar]
- 36.Aspengren S., Hedberg D., Wallin M. Melanophores: a model system for neuronal transport and exocytosis? J. Neurosci. Res. 2007;85:2591–2600. doi: 10.1002/jnr.21132. [DOI] [PubMed] [Google Scholar]
- 37.Deacon S.W., Serpinskaya A.S., Vaughan P.S., Lopez Fanarraga M., Vernos I., Vaughan K.T., Gelfand V.I. Dynactin is required for bidirectional organelle transport. J. Cell Biol. 2003;160:297–301. doi: 10.1083/jcb.200210066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sammak P.J., Adams S.R., Harootunian A.T., Schliwa M., Tsien R.Y. Intracellular cyclic AMP not calcium, determines the direction of vesicle movement in melanophores: direct measurement by fluorescence ratio imaging. J. Cell Biol. 1992;117:57–72. doi: 10.1083/jcb.117.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodionov V., Yi J., Kashina A., Oladipo A., Gross S.P. Switching between microtubule- and actin-based transport systems in melanophores is controlled by cAMP levels. Curr. Biol. 2003;13:1837–1847. doi: 10.1016/j.cub.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 40.Deacon S.W., Nascimento A., Serpinskaya A.S., Gelfand V.I. Regulation of bidirectional melanosome transport by organelle bound MAP kinase. Curr. Biol. 2005;15:459–463. doi: 10.1016/j.cub.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 41.Caviston J.P., Holzbaur E.L. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Soldati T., Schliwa M. Powering membrane traffic in endocytosis and recycling. Nat. Rev. Mol. Cell Biol. 2006;7:897–908. doi: 10.1038/nrm2060. [DOI] [PubMed] [Google Scholar]
- 43.Jordens I., Marsman M., Kuijl C., Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6:1070–1077. doi: 10.1111/j.1600-0854.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 44.Seabra M.C., Coudrier E. Rab GTPases and myosin motors in organelle motility. Traffic. 2004;5:393–399. doi: 10.1111/j.1398-9219.2004.00190.x. [DOI] [PubMed] [Google Scholar]
- 45.Nurnberg B., Ahnert-Hilger G. Potential roles of heterotrimeric G proteins of the endomembrane system. FEBS Lett. 1996;389:61–65. doi: 10.1016/0014-5793(96)00584-4. [DOI] [PubMed] [Google Scholar]
- 46.Futter C.E. The molecular regulation of organelle transport in mammalian retinal pigment epithelial cells. Pigment Cell Res. 2006;19:104–111. doi: 10.1111/j.1600-0749.2006.00303.x. [DOI] [PubMed] [Google Scholar]
- 47.Fairbank P.D., Lee C., Ellis A., Hildebrand J.D., Gross J.M., Wallingford J.B. Shroom2 (APXL) regulates melanosome biogenesis and localization in the retinal pigment epithelium. Development. 2006;133:4109–4118. doi: 10.1242/dev.02563. [DOI] [PubMed] [Google Scholar]
- 48.Schiaffino M.V., Bassi M.T., Rugarli E.I., Renieri A., Galli L., Ballabio A. Cloning of a human homologue of the Xenopus laevis APX gene from the ocular albinism type 1 critical region. Hum. Mol. Genet. 1995;4:373–382. doi: 10.1093/hmg/4.3.373. [DOI] [PubMed] [Google Scholar]
- 49.Bassi M.T., Bergen A.A., Bitoun P., Charles S.J., Clementi M., Gosselin R., Hurst J., Lewis R.A., Lorenz B., Meitinger T., et al. Diverse prevalence of large deletions within the OA1 gene in ocular albinism type 1 patients from Europe and North America. Hum. Genet. 2001;108:51–54. doi: 10.1007/s004390000440. [DOI] [PubMed] [Google Scholar]
- 50.Disteche C.M., Dinulos M.B., Bassi M.T., Elliott R.W., Rugarli E.I. Mapping of the murine tbl1 gene reveals a new rearrangement between mouse and human X chromosomes. Mamm. Genome. 1998;9:1062–1064. doi: 10.1007/s003359900926. [DOI] [PubMed] [Google Scholar]
- 51.Sviderskaya E.V., Bennett D.C., Ho L., Bailin T., Lee S.T., Spritz R.A. Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J. Invest. Dermatol. 1997;108:30–34. doi: 10.1111/1523-1747.ep12285621. [DOI] [PubMed] [Google Scholar]
- 52.Bassi M.T., Schiaffino M.V., Renieri A., De Nigris F., Galli L., Bruttini M., Gebbia M., Bergen A.A.B., Lewis R.A., Ballabio A. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat. Genet. 1995;10:13–19. doi: 10.1038/ng0595-13. [DOI] [PubMed] [Google Scholar]
- 53.Bassi M.T., Incerti B., Easty D.J., Sviderskaya E.V., Ballabio A. Cloning of the murine homologue of the Ocular Albinism type 1 (OA1) gene: sequence, genomic structure and expression analysis in pigment cells. Genome Res. 1996;6:880–885. doi: 10.1101/gr.6.9.880. [DOI] [PubMed] [Google Scholar]
- 54.Newton J.M., Orlow S.J., Barsh G.S. Isolation and characterization of a mouse homolog of the X-linked ocular albinism (OA1) gene. Genomics. 1996;37:219–225. doi: 10.1006/geno.1996.0545. [DOI] [PubMed] [Google Scholar]
- 55.Hume A.N., Collinson L.M., Rapak A., Gomes A.Q., Hopkins C.R., Seabra M.C. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J. Cell Biol. 2001;152:795–808. doi: 10.1083/jcb.152.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.