

Abstract
Motivation: Identification of functionally important residues in proteins plays a significant role in biological discovery. Here, we present INTREPID—an information–theoretic approach for functional site identification that exploits the information in large diverse multiple sequence alignments (MSAs). INTREPID uses a traversal of the phylogeny in combination with a positional conservation score, based on Jensen–Shannon divergence, to rank positions in an MSA. While knowledge of protein 3D structure can significantly improve the accuracy of functional site identification, since structural information is not available for a majority of proteins, INTREPID relies solely on sequence information. We evaluated INTREPID on two tasks: predicting catalytic residues and predicting specificity determinants.
Results: In catalytic residue prediction, INTREPID provides significant improvements over Evolutionary Trace, ConSurf as well as over a baseline global conservation method on a set of 100 manually curated enzymes from the Catalytic Site Atlas. In particular, INTREPID is able to better predict catalytic positions that are not globally conserved and hence, attains improved sensitivity at high values of specificity. We also investigated the performance of INTREPID as a function of the evolutionary divergence of the protein family. We found that INTREPID is better able to exploit the diversity in such families and that accuracy improves when homologs with very low sequence identity are included in an alignment. In specificity determinant prediction, when subtype information is known, INTREPID-SPEC, a variant of INTREPID, attains accuracies that are competitive with other approaches for this task.
Availability: INTREPID is available for 16919 families in the PhyloFacts resource (http://phylogenomics.berkeley.edu/phylofacts).
Contact: sriram_s@cs.berkeley.edu
Supplementary information: Relevant online supplementary material is available at http://phylogenomics.berkeley.edu/INTREPID.
1 INTRODUCTION
The problem of identifying the positions in a protein critical for its structure or function plays a significant role in biological discovery. These residues (such as the catalytic triad of serine, aspartate and histidine found in proteases) provide valuable clues about the functions of proteins. Since experimental methods to determine the roles of individual positions are time-consuming and expensive, computational methods are widely used for protein functional residue prediction; these provide initial clues that can be followed up by experiments.
Casari et al. (1995) developed one of the first computational approaches to identify positions conferring functional specificity. Another method for functional residue prediction is Evolutionary Trace (ET) (Lichtarge et al., 1996). The original ET method defines progressively more conservative cuts of a phylogeny. The level of the cut at which a column shows a specific pattern of conservation (either family-wide or subfamily-specific) is used to assign a score to each position in a protein. A more recent method, ConSurf (Landau et al., 2005), computes the rate of evolution at each position based on phylogenetic analysis; residues with lower rates of evolution are considered more important. Variants of both ET, one of which uses an entropy-based score, (Aloy et al., 2001; Mihalek et al., 2004) and ConSurf (Glaser et al., 2006; Mayrose et al., 2004; Nimrod et al., 2005) have also been developed. In general, predictive methods have relied on protein surface geometry (Peters et al., 1996), energy considerations (Elcock, 2001; Laurie and Jackson, 2005), chemical properties (Ko et al., 2005; Ondrechen et al., 2001) and sequence conservation (Casari et al., 1995; Landau et al., 2005; Landgraf et al., 2001; Lichtarge et al., 1996) or have attempted to combine different features (Gutteridge et al., 2003; Petrova and Wu, 2006; Youn et al., 2007).
A number of methods focusing exclusively on specificity-determining residues have also been developed (Del Sol Mesa et al., 2003; Donald and Shakhnovich, 2005; Hannenhalli and Russell, 2000; Kalinina et al., 2004; Mirny and Gelfand, 2002; Pei et al., 2006). Capra and Singh (2008) developed a method for scoring the positions in an alignment, termed GroupSim, which was found to be competitive with a number of previous methods. Some of the methods proposed for specificity determinant prediction require the subtypes to be specified (Capra and Singh, 2008; Hannenhalli and Russell, 2000; Kalinina et al., 2004; Mirny and Gelfand, 2002; Pirovano et al., 2006), while others (Del Sol Mesa et al., 2003; Donald and Shakhnovich, 2005; Pei et al., 2006) do not. In practice, subtypes are seldom known for a protein family. Thus, methods which can work without explicit knowledge of subtypes (i.e. from a tabula rasa) are more suitable for general use.
In this article, we present a new method—INTREPID (INformation-theoretic TREe traversal for Protein functional site IDentification). INTREPID takes as input a target protein, a multiple sequence alignment (MSA) and a gene tree of the family containing the target protein; a protein structure can also be included to boost performance but is not required. In this article, we focus on methods that exploit only sequence information, since structural information is not available for a majority of proteins. Methods employing an MSA as an input operate on the assumption that all residues in a column are homologous; this assumption can be violated due to structural and functional variability across specific lineages and errors in alignments. A number of enzyme families exhibit variability in the location of catalytic residues (Todd et al., 2002), while other enzyme families exhibit variation at catalytic positions. The inteins have been known to exhibit variations in their catalytic residues that in turn affect the intein-mediated splicing mechanisms. For instance, functional inteins with an N-terminal alanine instead of the catalytic cysteine or serine have been observed (Johnson et al., 2007; Southworth et al., 2000). INTREPID is designed to be robust to these issues.
The key idea in INTREPID is the use of phylogenetic information by examining the conservation patterns at each node of a phylogenetic tree on a path from the root to the leaf corresponding to the sequence of interest. For instance, catalytic residues tend to be conserved across distant homologs and thus will appear conserved at (or near) the root of a gene tree. In contrast, specificity determinants will not be conserved across all members of a family, but are likely to be conserved within one or more subtypes. Thus, prediction of these two distinct types of positions requires a different approach for each task. Any suitable conservation score can be used within the tree traversal of INTREPID depending on the type of functional residue to be predicted. A number of functions have been developed for determining functional residues by scoring the columns of a MSA, including information–theoretic scores based on Shannon Entropy (Sander and Schneider, 1991; Shenkin et al., 1991), Relative Entropy (Wang and Samudrala, 2006), and Jensen–Shannon (JS) divergence (Capra and Singh, 2007). INTREPID uses the JS divergence as it has been found to be the most accurate conservation-based score for functional residue identification (Capra and Singh, 2007).
In the catalytic residue prediction problem, we apply INTREPID to large protein families for enzymes in the catalytic-site atlas (CSA) (Porter et al., 2004). We compare INTREPID to other sequence-based methods, such as ET, ConSurf and baseline methods based on global conservation scores. We also compare INTREPID to the machine learning methods reported in Petrova and Wu (2006) and in Youn et al. (2007). We also analyze the effect of alignment diversity on the accuracy of catalytic residue prediction. Finally, we apply INTREPID-SPEC, a variant of INTREPID adapted to specificity determinant prediction, to the dataset of putative specificity-determining positions (SDPs) generated by Capra and Singh (2008).
2 METHODS
The input to INTREPID comprises a target protein p whose functional residues are to be predicted, an MSA of proteins homologous to p and an estimated evolutionary tree of these homologs, i.e. the gene tree.
Each residue in p is analyzed independently to derive its predicted importance, based on the conservation patterns at each node on a path from the root to the leaf corresponding to protein p. INTREPID uses a key observation that was first exploited in the context of functional residue identification by Casari et al. (1995) and reinforced since then by numerous studies: residues playing critical roles for protein structure or function are often under strong negative selection. This negative selection enables these residues to be detected due to their strong conservation across a family of related proteins. Catalytic residues in enzyme-active sites are an example of such a class. In predicting catalytic residues based on sequence conservation, the evolutionary context is critical, i.e. the degree of sequence divergence across homologs included in the analysis will have a significant impact on the method performance. In a closely related set of proteins, even positions that are not critical for function may appear well conserved. Thus, truly critical residues may only be revealed against a backdrop of evolutionary divergence.
Unfortunately, conservation patterns in an MSA can be affected by inadvertently included non-homologs, alignment and phylogeny errors and functional divergence in specific lineages e.g. where a residue conserved in one subtree is not conserved in another subtree due to changes in function. INTREPID is designed to detect catalytic residues exhibiting such behavior by combining the conservation patterns observed at different nodes of the tree.
2.1 Computing the positional importance score
INTREPID computes an importance score IMPp(x) for every position x in protein p using a traversal of the phylogenetic tree from the root to the leaf corresponding to p. The tree traversal enables us to exploit the information over the entire tree, instead of requiring us to select a particular cut of a tree into subtrees. It also helps to avoid the contribution of noise from subfamilies or entire lineages that may disagree on the importance of particular positions.
Every node encountered in this traversal corresponds to a subtree containing p and one or more homologs, and provides a different perspective on the potential importance of each position in p. For instance, at the leaf corresponding to p, no homologs are available to highlight which positions are conserved and which are variable, and it is impossible to predict which of the positions in p are likely to be critical for function. At the other extreme, residues that are perfectly conserved across the entire family will be evident when viewed from the root of the tree. As we traverse a path from the root to the leaf, positions formerly appearing to be variable will become fixed in specific lineages; at a leaf, all positions will be perfectly conserved. To enable us to compensate for subtrees with highly correlated or very few sequences, the score IMPp accounts for the evolutionary distance spanned as estimated by the sequence divergence.
We denote by S the subtree corresponding to a node encountered in the tree traversal, cons(S,x) is the conservation of position x within subtree S, and 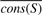 is the average conservation across all columns in subtree S. The importance score at a position x is computed as
is the average conservation across all columns in subtree S. The importance score at a position x is computed as
| (1) |
In this article, we use the JS divergence (Lin and Wong, 1990) between the amino acid distribution and the background [with prior weight = 1/2 as in Capra and Singh (2007)]. The importance score thus assigns a high score to those residues that are conserved over a large subtree of divergent sequences. When subtrees with many highly similar sequences are considered, the average conservation will be high. In this case, even though the positional conservation is also high, the difference between these two numbers will be fairly low. The maximum observed positional conservation on the path from the root to the leaf at each position x is its importance. We finally normalize the score across all the positions in the protein p so that the reported score at position x is  where
where  and σ(IMPp) are the average and SDs of the importance scores across all the columns in the MSA.
and σ(IMPp) are the average and SDs of the importance scores across all the columns in the MSA.
We illustrate INTREPID with an example.
Figure 1 shows six protein sequences of length four each. The target protein is marked with an arrow. The nodes traced by the tree traversal are S1,S2,S3,S4 and S5. We first compute the average JS divergence in each of the subtrees. In subtree S1, the average JS divergence is:  . Repeating this calculation for each of the other subtrees, we get
. Repeating this calculation for each of the other subtrees, we get 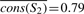 ,
, 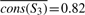 ,
, 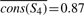 , and
, and 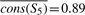 .
.
Fig. 1.

An example of the INTREPID algorithm. This example shows six protein sequences of length four each. The target protein p is marked with an arrow. The nodes visited by the tree traversal are S1, S2, S3, S4 and S5. As explained in the text, INTREPID ranks the positions in the order 2, 1, 4 and 3, while simple global conservation would rank position 4 above position 1.
Now let us look at column 1. In a tree traversal from the root (node S1) to the leaf corresponding to p, we compute the following importance scores: cons(S1,1)=0.73−0.74=−0.1, cons(S2,1)=0.82−0.79=0.03, cons(S3,1)=0.91−0.82=0.09, cons(S4,1)=0.91−0.87=0.04, cons(S5,1)=0.91−0.89=0.02.
The maximum importance score IMPp(1)=0.09, corresponds to the score at node S3 where position 1 is completely conserved. Computing these scores for other positions: IMPp(2)=0.13, IMPp(3)=−0.03, IMPp(4)=0.05. As expected, we see that position 2 has the highest importance score followed by position 1. If simple global conservation had been used (i.e. each position had been ranked based on its conservation across the family), then position 4 would have a higher rank than position 1. INTREPID gives a higher score to position 1 than to position 4 because of the higher conservation in position 1 in the subtree containing p. In other words, position 4 appears to be important for a majority of the family but may have evolved a different role in the lineage corresponding to subtree S4. On the other hand, position 1 appears to be associated with a function that is preserved within the subtree S3 but is lost or modified outside.
Different measures of positional conservation can be used within the tree traversal protocol. We also considered using the log-odds of the frequency of the most frequent amino acid and the relative entropy between the amino acid distribution of position x within subtree S and a background distribution (Wang and Samudrala, 2006). Consistent with the results reported in Capra and Singh (2007), the score based on JS divergence was found to be the most accurate. We use the distribution from the BLOSUM62 alignments (Henikoff and Henikoff, 1992) as the background distribution. See Supplementary Materials for details and experimental results using the different positional conservation scores.
2.2 INTREPID-SPEC
While the scoring functions discussed in the previous section are designed to detect family-defining positions (and catalytic positions in particular), this basic tree traversal protocol can be adapted to detect SDPs as well. Specificity-determining positions tend to be conserved within—but different across—subfamilies. For this problem, we compute the positional conservation score as the relative entropy of the amino acid distributions within and outside a subtree. This variant is termed INTREPID-SPEC. The importance score at position x is computed as
| (2) |
where S is a node on the path from the root to the leaf corresponding to p, pxS denotes the probability distribution of amino acids at position x for the sequences within subtree S, and pxSc denotes the probability distribution of amino acids at position x over the other sequences. In computing the scores in Equation 2, S ranges over all the nodes in the tree traversal except the root. To avoid saturated probabilities (and handle subtrees with very few sequences), we use add-one pseudocounts (Durbin et al., 1998). Such a relative entropy score was used by Hannenhalli and Russell (2000) for specificity-residue prediction when the subtypes are known. Using the score within the tree traversal allows us to predict specificity determinants even when the subtypes are not known.
3 EXPERIMENTS
In this section, we start by describing experiments to assess INTREPID on the prediction of catalytic residues, and examine the effect of protein family divergence on the accuracy of catalytic residue prediction. We then assess the accuracy of INTREPID on specificity determinant prediction.
3.1 Catalytic residue prediction
3.1.1 Preliminaries
We compared INTREPID to two methods that use only sequence information to predict functionally important residues, ET (Lichtarge et al., 1996) and ConSurf (Pupko et al., 2002). We also included in our comparison a baseline method termed Global-JS which applies the JS-divergence score to each column of the alignment as performed by Capra and Singh (2007). We used the results from servers implementing ET and ConSurf to ensure that each of these methods would be run with parameters for which it has been optimized: the ET server from Baylor College of Medicine (http://mammoth.bcm.tmc.edu/traceview/) (BCMET), which implements the improved evolution–entropy hybrid version of ET (Mihalek et al., 2004), and the ConSurf web server at Tel Aviv University (http://consurf.tau.ac.il).
While evaluating these methods, the question of how the reported scores are typically handled by users needs to be addressed. We consider two ways of post-processing the scores reported. In the first case, we use the ranks of the residues instead of the scores. This treatment is more useful under the assumption that every protein should have some predicted residues (if, for instance, the protein is known to be an enzyme). In the second case, we normalize the scores of each method on each protein and then analyze all 100 proteins as a set, sorting the normalized scores for each position. In this approach, for some score cutoff, some proteins may have no predicted positions while others may have several. Normalizing the scores improved the accuracies of both BCMET and ConSurf compared to using unnormalized scores.
We computed the following metrics for comparison (note that although sensitivity and recall are synonymous terms, we follow convention and use each term according to the analysis):
Here, a true positive (TP) is a residue identified by the CSA as catalytic which is selected by a method, a false negative (FN) is a catalytic residue that is missed, a false positive (FP) is a residue erroneously selected by a method (i.e. it is not listed in the CSA), and a true negative (TN) is a non-catalytic residue that is correctly not selected. Specificity measures how well a method rejects non-catalytic residues. Since the ratio of catalytic to non-catalytic residues is low, even apparently high values of specificity can correspond to a large number of false positives. Precision, which measures the fraction of predicted catalytic residues that are correct, is a more relevant measure of performance in this setting. We plot the receiver operating characteristic (ROC) curve (Sensitivity versus 1-Specificity) and the Precision–Recall curve (Precision versus Recall) for each of these methods. The ROC curve has been truncated to the high-specificity region for clarity (Specificity ≥80%) although the trends shown are similar over the entire range of specificities (see Supplementary Material for Precision–Recall curves and ROC curves over the entire range).
3.1.2 INTREPID is significantly more accurate than other sequence-based methods
Figure 2 compares the performance of INTREPID, Global-JS, BCMET and ConSurf on the CSA-100 dataset (see Section 4 for details). We see from the figure that INTREPID has the highest sensitivity over the entire range of specificities and is significantly more accurate than the other methods. Table 1 compares the different methods under various metrics. For example, at 90% specificity, INTREPID attains a sensitivity of 85.03% relative to sensitivities of 70.06% and 73.8% by BCMET and ConSurf, respectively. The baseline method (Global-JS) performs quite well (a sensitivity of 78.66% at a specificity of 90%). At a precision of 10%, INTREPID attains a recall of 75.0% while Global-JS has a recall of 64.0%. ConSurf and BCMET never attain a precision of 10% resulting in 0% recall at this level. When the normalized scores are used in place of the ranks, we see from Table 1 that INTREPID has the highest sensitivity followed by Global-JS, BCMET and ConSurf.
Fig. 2.

Results for catalytic residue prediction on CSA-100 dataset using rank-based scores: ROC curves comparing INTREPID, Global-JS, BCMET and ConSurf. The ROC curve shows INTREPID to have the highest sensitivity over the range of high specificity (≥80%) followed by Global-JS. BCMET performs better as the specificity decreases. Refer to Table 1 for full AUC scores.
Table 1.
Statistics comparing the different algorithms on the CSA-100 dataset
| INTREPID | Global-JS | ConSurf | BCMET | ||
|---|---|---|---|---|---|
| Residue ranks | Sensitivity95 | 70.06 | 64.33 | 49.20 | 40.76 |
| Sensitivity90 | 85.03 | 78.66 | 73.80 | 70.06 | |
| Sensitivity80 | 93.95 | 90.13 | 89.78 | 92.04 | |
| Recall10 | 75.0 | 64.0 | 0.00 | 0.00 | |
| AUC | 0.944 | 0.924 | 0.907 | 0.914 | |
| AUC95 | 0.024 | 0.022 | 0.011 | 0.010 | |
| AUC90 | 0.063 | 0.058 | 0.046 | 0.039 | |
| AUC80 | 0.154 | 0.145 | 0.127 | 0.124 | |
| P-value | – | 3.89 × 10−18 | 1.64 × 10−17 | 1.34 × 10−17 | |
| Normalized scores | Sensitivity95 | 67.83 | 58.28 | 36.74 | 54.46 |
| Sensitivity90 | 85.03 | 75.48 | 59.42 | 74.84 | |
| Sensitivity80 | 92.99 | 89.81 | 87.86 | 91.72 | |
| Recall10 | 71.0 | 56.0 | 3.83 | 31.21 | |
| AUC | 0.935 | 0.910 | 0.884 | 0.919 | |
| AUC95 | 0.022 | 0.018 | 0.011 | 0.016 | |
| AUC90 | 0.060 | 0.053 | 0.036 | 0.048 | |
| AUC80 | 0.149 | 0.137 | 0.111 | 0.134 | |
| P-value | – | 3.89 × 10−18 | 3.89 × 10−18 | 5.27 × 10−18 |
BCMET refers to the ET server from Baylor College of Medicine. In the top panel, the ranks of the residues were used while in the bottom panel, the normalized scores were used. Sensitivity is measured at specificities of 95%, 90%, and 85% respectively and recall at 10% precision. AUCx (x =80,90,95) refers to the area under the ROC curve when specificity is at least x%; AUC is the area under the entire curve. The P-value refers to the Wilcoxon signed rank P-values between the AUC of the INTREPID and each of the other methods. INTREPID improves significantly over the other methods on all metrics. Based on their ranks, ConSurf and BCMET do not reach a precision of 10% and hence have zero recall. The confidence intervals on these statistics are reported in Supplementary Table S-3.
Since the ConSurf server selects a smaller, more closely related set of sequences as input to Rate4Site (the program that computes the site-specific evolutionary rates as part of the ConSurf protocol), we also tested the prediction power of Rate4Site on the CSA-100 dataset that contains a greater level of sequence divergence. Rate4Site failed to complete on 77 of the 100 alignments due to memory allocation problems. By removing sequences with >80% identity, we obtained Rate4Site results on 71 out of the 100 inputs. We refer to these 71 families as the CSA-71 dataset. We also ran INTREPID on these reduced alignments as well as the full alignments for these 71 families. Figure 3 compares the performance of INTREPID, run on alignments made non-redundant at 80% identity and on the original alignments for the CSA-71 dataset, with Rate4Site. INTREPID, when run on the reduced MSA, has a small but statistically significant improvement over Rate4Site (Wilcoxon paired sign-rank test P-value of 1.3 × 10−5). At 90% specificity, INTREPID attains sensitivities of 83.6% on the full MSA and 85.1% on the reduced MSA, while Rate4Site attains a sensitivity of 84.6%. Similarly, at 10% precision, INTREPID on the full MSA, INTREPID on the reduced MSA and Rate4Site have 75%, 80% and 75% recall. See Figure 3 for details. The figure also shows the considerable difference in accuracies between Rate4Site when run on these alignments and when run as part of ConSurf; this difference is likely a result of the different alignments used. Importantly, INTREPID has an average running time of 25.7 s on this dataset compared to Rate4Site which requires 2 h and 52 min on average.
Fig. 3.

Results on the CSA-71 dataset comparing INTREPID, Rate4Site and ConSurf using rank-based scores. Results were obtained on alignments derived from the original dataset by removing sequences with more than 80% sequence identity to one another; the 71 alignments used here were the alignments on which Rate4Site completed successfully. INTREPID was run on the reduced MSA as well as on the full MSAs for these 71 families. INTREPID, when run on both MSAs, and Rate4Site have similar accuracies though INTREPID is slightly more accurate (AUC90 for the methods are 0.061,0.061 and 0.059, respectively; AUC for the methods are 0.941,0.938 and 0.940, respectively; the difference in accuracy between INTREPID, run on the reduced MSA, and Rate4Site is statistically significant with a P-value of 1.3 × 10−5). Rate4Site is considerably more accurate than the ConSurf webserver (which also uses the Rate4Site program)—this difference is likely a result of the different alignments used.
We also evaluated INTREPID on two other datasets consisting of the protein families used by Petrova and Wu (2006) and by Youn et al. (2007), respectively. On the Petrova dataset, INTREPID, with a sensitivity of 90.57% at a false positive rate of 13%, is as accurate as their method which attains a sensitivity of 90% at the same false positive rate (i.e. the results are essentially indistinguishable). This is a very surprising result because INTREPID uses only sequence conservation, while the method reported in Petrova and Wu (2006) uses a learning algorithm to combine sequence and structural features. Youn et al. (2007) present two variants of their method, one employing only sequence information, while the second combines sequence and structural information. They present results for both variants on a dataset based on ASTRAL 40 v1.65 (Brenner et al., 2000) selected to be non-redundant at the SCOP family level. On a similarly constructed dataset, INTREPID attains a recall of 28.13% at a precision of 15% and an area under the curve (AUC) of 0.906. When restricted to sequence features alone, their method attains a sensitivity of about 16% at 15% precision and an AUC of 0.866. Thus, INTREPID improves over the method used in Youn et al. (2007) when restricted to sequence features alone. In contrast, their method that combines sequence and structural information attains a much higher recall of about 65% at about the same precision. Reassuringly, the performance of INTREPID is approximately the same across these different datasets suggesting that these results would generalize well to new protein families.
3.1.3 Greater evolutionary divergence improves the accuracies of INTREPID
To measure the impact of evolutionary divergence on method performance, we controlled the sequence diversity of the alignment used. We created restricted alignments at the x%-level, i.e. sequences were discarded from each of these alignments so that the minimum percent identity from any sequence to the seed was at least x%. We varied x over 10%,15%,20% and 25%, respectively. For comparison, we also included the original alignment which is labeled ‘Unrestricted’. The effect of evolutionary divergence on INTREPID is shown in Figure 4. We see that as the divergence of the family increases, INTREPID accuracy increases. At 90% specificity, INTREPID has 42% sensitivity at 25% identity trimming. INTREPID reaches 85% sensitivity when no sequences are removed. The trends shown here suggest that INTREPID is robust to divergence in protein families. All methods tested for the impact of sequence divergence on catalytic residue prediction—INTREPID, Global-JS and Rate4Site—benefit from increased sequence diversity (see Supplementary Materials).
Fig. 4.

Effect of alignment diversity on catalytic residue prediction: ROC curve for INTREPID on alignments with varying degrees of evolutionary divergence, indicated by the minimum percent identity to the seed. The original alignment with no sequences removed is labeled ‘Unrestricted’. INTREPID performs significantly better with increasing evolutionary divergence. For instance, INTREPID achieves 42% sensitivity at 90% specificity and 25% identity trimming but reaches 85% sensitivity when no sequences are removed.
3.1.4. INTREPID is more robust to catalytic residues that are not conserved across the MSA
The advantage of INTREPID over global conservation analysis can be inferred from the level at which the maximum score is attained in the tree traversal. A little less than 50% of the catalytic residues have their maximum scores at the root. However, for 56 of the catalytic residues (≈18% of all catalytic residues in the dataset), the maximum score is attained at least 5 levels away from the root. In 34 of the 56 residues, INTREPID assigns a better rank than Global-JS while Global-JS assigns a better rank on 15 (see Figure S-8 in Supplementary Materials). Thus, INTREPID is more effective at identifying catalytic residues that are not conserved across the entire protein family. To illustrate this point, we consider two such families.
The first example is the enoyl-[acyl-carrier-protein] reductase from Escherichia coli (PDB id: 1mfp). CSA lists two catalytic residues: K163 and Y156. All methods give high ranks to K163, while Y156 is far more challenging. INTREPID given Y156 a rank of 18 (out of 258 positions), and BCMET, Global-JS and ConSurf give ranks of 31, 58 and 100, respectively. The homologs gathered for this protein family are found to include other short chain dehydrogenases (such as 3-oxoacyl-[acyl-carrier-protein] reductase). In these other families, this position generally contains a glutamine. The catalytic role of this glutamine has been observed in human 15-hydroxyprostaglandin dehydrogenase (Cho et al., 2006). The global frequency of tyrosine at position 156 is only about 25% though it is conserved within a subtree containing 199 sequences in a family with 833 sequences (see Supplementary Figs S-9 and S-10).
Another example is Flavocytochrome b2 from Saccharomyces Cerevisiae (PDB id:1fcB). This protein is part of the flavin mononucleotide (FMN)-dependent oxidoreductases. The poor conservation at the active-site residues in this family has been observed by Todd et al. (2001). This lack of conservation is most evident at the catalytic residues Y143, H373, and R376 (see Supplementary Figure S-11). On H373 INTREPID, ConSurf and BCMET all give ranks of 1, while Global-JS gives a rank of 22. On R376, INTREPID, ConSurf, BCMET and Global-JS give ranks of 7, 23, 4 and 9, respectively, while on Y143, the respective ranks are 23, 66, 56 and 20.
3.2 Specificity determinant prediction
Methods for specificity determinant prediction can be classified as those that require the subtypes to be known a priori (Capra and Singh, 2008; Hannenhalli and Russell, 2000; Kalinina et al., 2004; Mirny and Gelfand, 2002; Pirovano et al., 2006) and those that do not (Del Sol Mesa et al., 2003; Donald and Shakhnovich, 2005; Pei et al., 2006). INTREPID does not require knowledge of the subtypes. For specificity determinant prediction, we use INTREPID-SPEC (described in Section 2.2). We can implicitly provide subtype information to INTREPID-SPEC by building a separate tree for each subtype which are then joined at the root to obtain a tree for the family. We compared INTREPID-SPEC to the GroupSim heuristic that was found to be competitive with other sequence-based methods in Capra and Singh (2008). Note that all the methods that were benchmarked in Capra and Singh (2008), including GroupSim, use subtype information. We used the dataset generated by Capra and Singh (2008) for the evaluation. Following the definitions used in Capra and Singh (2008), residues that pass the SDPO filter (low overlap of residues across subtypes and conserved in at least one subtype) are considered positives and those that do not pass the SDPL filter (low overlap of residues across subtype) are considered negatives. We used the alignments from this original dataset.
We ran INTREPID-SPEC on this dataset by choosing each protein in turn as the target p, computing an importance score and then averaging this score across all the proteins. Since we are interested in SDPs, we ignore the conservation score at the root during the tree traversal.
3.2.1 INTREPID-SPEC is competitive with other sequence-based methods for specificity determinant prediction
INTREPID-SPEC, when subtype information is used, has accuracies similar to GroupSim as seen in Figure 5. [Capra and Singh (2008) have shown that GroupSim is competitive with other sequence-based methods suggesting that INTREPID-SPEC would have similar accuracies to these other methods as well]. Although INTREPID-SPEC does a tree traversal even when subtype information is provided implicitly, our results show that the maximum scores for the specificity determinants are attained at the point in the tree that separates the known subtypes.
Fig. 5.

Comparison of methods for specificity determinant prediction: INTREPID-SPEC run on trees built using subtype information and INTREPID-SPEC run with no subtype information are compared to GroupSim. INTREPID-SPEC (with subtypes provided) attains accuracies competitive with GroupSim. Including subtype information improves INTREPID-SPEC recall by roughly 10% at all levels.
We also ran INTREPID-SPEC on trees constructed without knowledge of subtypes (Fig. 5). INTREPID-SPEC with subtype information has 10% greater precision across the range of recall values than when no subtype information is available. This difference in performance can be attributed to the bias induced by the rooting of the tree on the process of averaging the INTREPID-SPEC scores across all the sequences in the family. In a family with multiple subtypes, this procedure gives higher ranks to those SDPs that differentiate a subtype that is joined to the rest of the family at the root. This bias explains why trees built using subtype information lead to improved accuracy. When the subtype information is not used, the top ranked residues often separate subtrees which do not correspond to the original subtypes. While such predictions are penalized in our present evaluation, these may be biologically interesting.
4 MATERIALS AND METHODS
For catalytic residue prediction, we identified a set of 100 enzymes from the manually curated section of the CSA (Porter et al., 2004) selected to ensure that no pair had detectable homology (i.e. we required a BLAST E-value >1). We term this the CSA-100 dataset. A PSI-BLAST (Altschul et al., 1997) search was performed with each of these 100 enzymes as a seed against the UniProt database (Apweiler et al., 2004). PSI-BLAST was run for four iterations with an E-value inclusion threshold of 1 × 10−4, from which a maximum of 1000 homologs were retrieved. The resulting homologs were realigned using MUSCLE (Edgar, 2004) with MAXITERS set to 2. Identical sequences were discarded. Columns in which the seed had a gap were removed. A neighbor-joining tree was built from this alignment using the PHYLIP package (Felsenstein, 1993). The dataset has alignments with a minimum of 32 sequences, a maximum of 1033 sequences and a median of 843 sequences. The average percent identity of the alignments varies from 6.4% to 31.14% with a median of 14.99%. The dataset contains a total of 314 catalytic residues out of a total of 36 229 residues with a median of three catalytic residues per enzyme.
For the comparison with the Petrova and Wu (2006) dataset, we generated alignments and trees by the protocol described above using the 79 enzymes reported in their paper (Petrova and Wu, 2006). The resulting dataset contains 244 catalytic residues out of a total of 23 332 residues. For the comparison with the Youn et al. (2007) dataset, we picked a random domain from each SCOP family for which we generated alignments and trees as described above. This dataset contains 1172 catalytic residues out of a total of 119 433 residues.
For specificity determinant prediction, we used the alignments from the dataset constructed by Capra and Singh (2008). Neighbor-joining trees were built using the PHYLIP package (Felsenstein, 1993).
5 CONCLUSIONS
In this article, we have presented INTREPID, a novel method to predict functional residues from sequence information only. The primary innovation in INTREPID is its use of the phylogeny of the family to infer the evolutionary pressures on positions within different subgroups. INTREPID infers functionally important positions through a traversal of the phylogeny from the root to the target protein located at a leaf; at each point on this path and for each position independently, INTREPID computes a positional conservation score based on JS divergence between the distribution of amino acids at that position and a background distribution. Positional scores are adjusted to take into consideration the scores of other positions within the same subtree; thus positional scores for a subtree containing highly similar sequences will be small, even though individual positions may be highly conserved. In contrast, a position that is highly conserved within a subtree that is otherwise highly variable will have a high JS divergence. Each position is then assigned the maximal JS score achieved over all nodes on the path. Positions that are conserved across the entire family achieve their maximum score at the root, whereas other positions will achieve their maximum at some distance from the root. Since even catalytic residues are not always perfectly conserved across a family (if, for instance, sequences with divergent functions are included in the analysis, or due to alignment errors), this tree traversal enables INTREPID to exploit the information in highly divergent datasets. In fact, our analysis of CSA-defined catalytic residues shows that 18% of catalytic residues in the dataset have their maximum score at least 5 levels from the root of the tree.
We have presented results comparing INTREPID with two of the leading methods in functional residue prediction that make use of sequence information only—ET and ConSurf and with a simple baseline method that computes the JS divergence between the amino acid distribution at a position and a background distribution (Global-JS). We compared each method on a benchmark dataset of 100 manually curated sequence-divergent enzymes from the CSA. Our results show that INTREPID has significantly superior accuracy than each of these methods, attaining a sensitivity of 85% at 90% specificity (in contrast. ET and ConSurf attain sensitivities of 70% and 74%, respectively at the same specificity) and attaining a recall of about 64% at 10% precision (in contrast neither ET nor ConSurf attain a precision >10%). Since the ConSurf server selects a more conservative set of sequences than those we selected, we also did a separate experiment in which we submitted our larger alignments to the Rate4Site algorithm (the core algorithm within ConSurf). As Rate4Site failed to complete on the full alignments, we filtered the alignments to reduce highly similar sequences. The method performances are very close on the 71 alignments on which Rate4Site completed successfully (ROC analysis shows INTREPID has a small but statistically significant edge over Rate4Site on this dataset).
In addition to these comparisons with methods using sequence information only, we compared INTREPID to the machine learning algorithms reported by Petrova and Wu (2006) and by Youn et al. (2007) which make use of structural information. Surprisingly, on the Petrova dataset, INTREPID is as accurate as their support vector machine (SVM)-based method, even though the latter uses both sequence and structure-based features. On the Youn et al. (2007) dataset, INTREPID is more accurate than the variant of their method that makes use of only sequence features. Reassuringly, the performance of INTREPID is approximately the same across these different datasets suggesting that it would generalize well to new protein families.
To analyze the effect of the evolutionary divergence on prediction accuracy, we created alignments in which the minimum pairwise identity to the seed was restricted. The sensitivity of INTREPID was found to increase as the alignments became more divergent. These results, while in agreement with several previous studies (Aloy et al., 2001; Landgraf et al., 2001; Panchenko et al., 2004), suggest that highly divergent families (with minimum pairwise identity as low as 10%) can significantly improve catalytic residue prediction.
Prediction of active-site residues based on sequence information alone is clearly affected by the quality of the sequence data, in particular, on the effective coverage and extent of the sequence space around the protein of interest. To test the impact on this kind of sequence space coverage, we analyzed the accuracy of INTREPID in predicting catalytic residues for sequences not used as seeds in clustering homologs (i.e. which may be towards the periphery of the sequence space). As expected, accuracy decreases as evolutionary distance to the seed increases. Our limited results suggest that the sequence of interest should have sequence identity >50% to the seed (see Supplementary Materials).
In summary, the utility of INTREPID in catalytic-site prediction can be traced to the following features. First, INTREPID relies solely on sequence information, making it useful when no structural data are available. Second, INTREPID is computationally efficient, making it useful in large-scale application, and allowing it to be used on large datasets. For instance, INTREPID is considerably faster than Rate4Site, with 400-fold lower average runtime. Third, INTREPID can be used on datasets including highly divergent sequences; in fact, its accuracy improves as more divergent sequences are included. While INTREPID is designed to make use of sequence information alone, it can be used as a component in a prediction protocol that attempts to combine sequence information with other types of information.
On the task of specificity determinant prediction, a variant of INTREPID, INTREPID-SPEC, was as accurate as the GroupSim method proposed by Capra and Singh (2008) when both methods were given subtype information. Unlike GroupSim however, INTREPID-SPEC does not require subtype information since the tree traversal provides an implicit grouping of sequences. We found that subtype information results in an improvement in precision of about 10% across the range of recall values.
In this work, we have focused on functional residue prediction in enzymes. In future work, we plan to assess the performance of these methods on non-enzymes as well as on other types of functional residues. Scoring functions that may be better suited to detect other types of conservation signals can be plugged into the INTREPID framework to obtain improved predictions. Finally, all the estimated accuracies of catalytic residue prediction methods depend critically on the characteristics of the dataset used to benchmark method performance. The poor performance of a method on a protein family may simply be the result of insufficient experimental data available for that family.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the anonymous reviewers for insightful and constructive comments. The authors would also like to thank Anthony Capra and Mona Singh for the SDP dataset and the GroupSim code, and Eunseog Youn and Sean Mooney for access to their dataset.
Conflict of Interest: none declared.
REFERENCES
- Aloy P, et al. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J. Mol. Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- Altschul S, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, et al. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- Capra JA, Singh M. Characterization and prediction of residues determining protein functional specificity. Bioinformatics. 2008;24:1473–1480. doi: 10.1093/bioinformatics/btn214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, et al. A method to predict functional residues in proteins. Nat. Struct. Biol. 1995;2:171–178. doi: 10.1038/nsb0295-171. [DOI] [PubMed] [Google Scholar]
- Cho H, et al. Role of glutamine 148 of human 15-hydroxyprostaglandin dehydrogenase in catalytic oxidation of prostaglandin e2. Bioorg. Med. Chem. 2006;14:6486–6491. doi: 10.1016/j.bmc.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Del Sol Mesa A, et al. Automatic methods for predicting functionally important residues. J. Mol. Biol. 2003;326:1289–1302. doi: 10.1016/s0022-2836(02)01451-1. [DOI] [PubMed] [Google Scholar]
- Donald JE, Shakhnovich EI. Determining functional specificity from protein sequences. Bioinformatics. 2005;21:2629–2635. doi: 10.1093/bioinformatics/bti396. [DOI] [PubMed] [Google Scholar]
- Durbin R, et al. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- Edgar RC. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J. Mol. Biol. 2001;312:885–896. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author. Seattle: Department of Genetics, University of Washington; 1993. [Google Scholar]
- Glaser F, et al. A method for localizing ligand binding pockets in protein structures. Proteins. 2006;62:479–488. doi: 10.1002/prot.20769. [DOI] [PubMed] [Google Scholar]
- Gutteridge A, et al. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J. Mol. Biol. 2003;330:719–734. doi: 10.1016/s0022-2836(03)00515-1. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S, Russell R. Analysis and prediction of functional sub-types from protein sequence alignments. J. Mol. Biol. 2000;303:61–76. doi: 10.1006/jmbi.2000.4036. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Nat. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MA, et al. NMR structure of a KlbA intein precursor from Methanococcus jannaschii. Protein Sci. 2007;16:1316–1328. doi: 10.1110/ps.072816707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinina OV, et al. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Sci. 2004;13:443–456. doi: 10.1110/ps.03191704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, et al. Prediction of active sites for protein structures from computed chemical properties. Bioinformatics. 2005;21(Suppl. 1):i258–i265. doi: 10.1093/bioinformatics/bti1039. [DOI] [PubMed] [Google Scholar]
- Landau M, et al. Consurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33(Web Server Issue):W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf R, et al. Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. J. Mol. Biol. 2001;307:1487–1502. doi: 10.1006/jmbi.2001.4540. [DOI] [PubMed] [Google Scholar]
- Laurie AT, Jackson RM. Q-sitefinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- Lichtarge O, et al. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Lin J, Wong SKM. A new directed divergence measure and its characterization. Int. J. Gen. Syst. 1990;17:73–81. [Google Scholar]
- Mayrose I, et al. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol. Biol. Evol. 2004;21:1781–1791. doi: 10.1093/molbev/msh194. [DOI] [PubMed] [Google Scholar]
- Mihalek I, et al. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- Mirny LA, Gelfand MS. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. J. Mol. Biol. 2002;321:7–20. doi: 10.1016/s0022-2836(02)00587-9. [DOI] [PubMed] [Google Scholar]
- Nimrod G, et al. In silico identification of functional regions in proteins. Bioinformatics. 2005;21(Suppl. 1):i328–i337. doi: 10.1093/bioinformatics/bti1023. [DOI] [PubMed] [Google Scholar]
- Ondrechen MJ, et al. Thematics: a simple computational predictor of enzyme function from structure. Proc. Natl Acad. Sci. USA. 2001;98:12473–12478. doi: 10.1073/pnas.211436698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko AR, et al. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 2004;13:884–892. doi: 10.1110/ps.03465504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, et al. Prediction of functional specificity determinants from protein sequences using log-likelihood ratios. Bioinformatics. 2006;22:164–171. doi: 10.1093/bioinformatics/bti766. [DOI] [PubMed] [Google Scholar]
- Peters KP, et al. The automatic search for ligand binding sites in proteins of known three-dimensional structure using only geometric criteria. J. Mol. Biol. 1996;256:201–213. doi: 10.1006/jmbi.1996.0077. [DOI] [PubMed] [Google Scholar]
- Petrova N, Wu C. Prediction of catalytic residues using support vector machine with selected protein sequence and structural properties. BMC Bioinformatics. 2006;7:312. doi: 10.1186/1471-2105-7-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirovano W, et al. Sequence comparison by sequence harmony identifies subtype-specific functional sites. Nucleic Acids Res. 2006;34:6540–6548. doi: 10.1093/nar/gkl901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CT, et al. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32(Database issue):D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupko T, et al. Rate4site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18(Suppl. 1):S71–S77. doi: 10.1093/bioinformatics/18.suppl_1.s71. [DOI] [PubMed] [Google Scholar]
- Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- Shenkin P, et al. Information-theoretical entropy as a measure of sequence variability. Proteins. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]
- Southworth M, et al. An alternative protein splicing mechanism for inteins lacking an N-terminal nucleophile. EMBO J. 2000;19:5019–5026. doi: 10.1093/emboj/19.18.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd A, et al. Plasticity of enzyme active sites. Trends Biochem. Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- Todd AE, et al. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Wang K, Samudrala R. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinformatics. 2006;7:385. doi: 10.1186/1471-2105-7-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn E, et al. Evaluation of features for catalytic residue prediction in novel folds. Protein Sci. 2007;16:216–226. doi: 10.1110/ps.062523907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.