

Abstract
Nitric oxide (NO) has earned the reputation of being a signaling mediator with many diverse and often opposing biological activities. The diversity in response to this simple diatomic molecule comes from the enormous variety of chemical reactions and biological properties associated with it. In the last few years, the importance of steady state NO concentrations have emerged as a key determinant of its biological function. Precise cellular responses are differentially regulated by specific NO concentration. We propose 5 basic distinct concentration levels of NO activity; cGMP mediated processes ([NO] <1–30 nM; Akt phosphorylation ([NO] = 30–100 nM); stabilization of HIF-1α ([NO] = 100–300 nM); phosphorylation of p53 ([NO] > 400 nM) and nitrosative stress (1 µM). In general, lower NO concentrations promote cell survival and proliferation, while higher levels favor cell cycle arrest, apoptosis, and senescence. Free radical interactions will also influence NO signaling. One of the consequences of reactive oxygen species (ROS) generation is to reduce NO concentrations. This antagonizes the signaling of nitric oxide and in some cases results in converting a cell cycle arrest profile to a cell survival one. The resulting reactive nitrogen species (RNS) that are generated from these reactions can also have biological effects and increase oxidative and nitrosative stress responses. A number of factors determine the formation of NO and its concentration, such as diffusion, consumption, and substrate availability which are referred to as Kinetic Determinants for Molecular Target Interactions. These are the chemical and biochemical parameters that shape cellular responses to NO. Herein we discuss signal transduction and the chemical biology of NO in terms of the direct and indirect reactions.
Keywords: nitric oxide oxidative nitrosative stress
Introduction
Over the last few decades, free radicals and other reactive small molecules have emerged as important players in a multitude of physiologic and pathologic conditions. From the beginning of this field of research, many of the short lived reactive molecules being studied were found to have opposing effects under seemingly similar circumstances 1. Initially, reactive oxygen species (ROS), reactive nitrogen species (RNS), carbon monoxide (CO), and hydrogen sulfide (H2S) were thought to be primarily cytotoxic species that increased tissue injury 2–6. After the discovery that they were endogenously produced, their role in pathophysiology was re-evaluated. We now know that these “toxic” species are not only endogenously generated but that they are an essential part of the immune response and many physiological signal transduction pathways 7– 10. Although their chemical reactivity can lead to toxicity, the biological properties of these same reactive species can be beneficial and explain their apparently dichotomous actions.
The free radical nitric oxide (NO) is the best example of a reactive molecule demonstrating both cytotoxic and cytoprotective properties 10. Two lines of research led to its discovery. NO was identified in the 1980’s as Endothelium-Derived Relaxation Factor (EDRF), a substance generated by the endothelium that caused vascular relaxation and also the active component of nitrovasodilators 7, 11, 12 13. Conversely, NO was found to be generated by macrophages participating in the anti-tumor and anti-pathogen response 14–16. These few initial observations sparked a new field of investigation and led to an explosion of NO research, which has revealed the importance of this diatomic molecule in nearly every tissue in the body.
However, contradictory results soon began to emerge regarding the participation of NO in pathophysiological responses 17. While a number of studies implied that endogenous NO was toxic, others showed that NO was protective 18–20. The NO mediated toxicity was attributed to the generation of reactive nitrogen oxide species (RNS) that mediated cell death, while the protective effects were proposed to be through antioxidant mechanisms 19. Over the last couple of decades, there was much debate as to the mechanism of this dichotomy and whether NO is a deleterious or beneficial agent.
Unlike most small signaling molecules, the biological effects of nitric oxide are determined by their chemical reactions such as binding to the regulatory heme in soluble guanylate cyclase (sGC) rather than traditional protein receptor ligand interactions. The unique chemistry of NO allows it to participate in numerous reactions 19. During the mid 1990’s, the concept of the chemical biology of NO was introduced to help explain this complexity in the context of biological conditions. The purpose of this thesis was to discern the physiologically relevant chemical reactivity of NO. For instance, various reactions of nitrogen oxides occur over many days at elevated temperature and pressure, which makes them kinetically and thermodynamically unlikely and incompatible with human physiology. On the other hand, some reactions are sufficiently fast to occur under achievable biological conditions.
The chemical biology of NO divides these potential reactions into two categories: direct and indirect 18. The direct effects of NO are those chemical reactions that occur fast enough to allow NO to directly react with a biological target molecule. In contrast, the indirect effects require that NO reacts with oxygen or superoxide to generate RNS, which subsequently react with the biological targets. One advantage of dividing the chemistry of NO in these two categories is that direct effects generally occur at low concentrations while indirect effects occur at much higher concentrations.
Indirect effects can be further subdivided into two categories based on RNS chemistry: nitrosative and oxidative stress 21. Oxidative chemistry refers to a process where the oxidation state of the target molecule is increased. There are several main types of oxidative reactions, electron transfer (i.e. radical formation), hydrogen atom abstraction, and oxygen atom transfer (oxygen atom insertion, addition, transfer, or hydroxylation reactions). Nitrosative stress implies the addition of a nitrosonium [NO+] equivalent to a thiol or secondary amine or hydroxy groups (although this reaction also represents a formal oxidation of a thiol or amine, we make a distinction here since the modifications occur via a nitrosation reaction). Reactive oxygen species (OH radical, O2−) such as those produced by the Fenton reaction are most often associated with oxidative stress. However, peroxynitrite (ONOO−) and nitrogen dioxide (NO2), which can be formed from the reaction of NO with superoxide (O2−), are also potent oxidants (> 1.0 V NHE) 22. In contrast, N2O3 formed from the reaction of NO with O2 (autoxidation), as well as the NO/O2− reaction is a mild oxidant and prefers to nitrosate nucleophiles such as amines and thiols 23–25. The balance between oxidation and nitrosation chemistry as it was found depends largely on the flux of NO (Fig. 1). In the case of the autoxidation in hydrophobic environments, NO2 is first generated but as NO levels increase there is rapid formation of N2O3 (eq. 1 and eq. 2) ultimately forming nitrite in water (eq 3)
| 1) |
| 2) |
| 3) |
Figure 1.
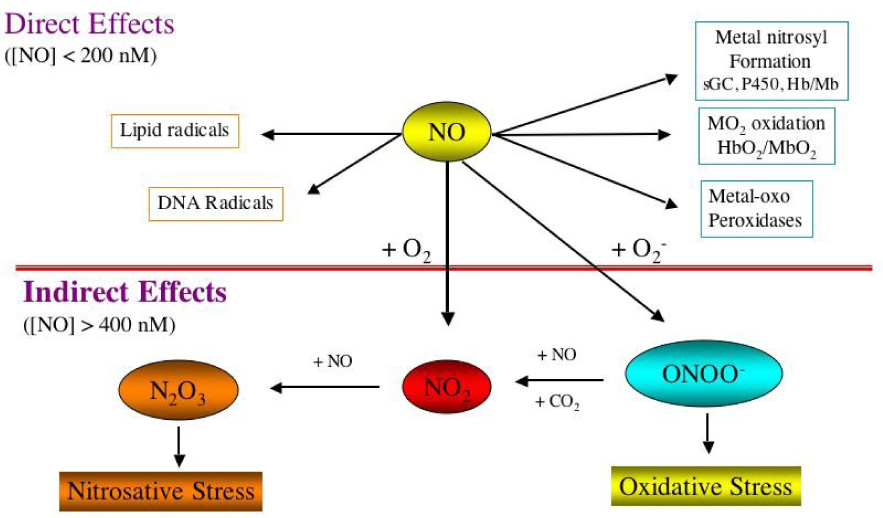
The Chemical Biology of NO. The direct versus the indirect effects.
An equilibrium is formed between NO and NO2 and the proportion of oxidation vs. nitrosation is determined by the amount of substrate present. The same applies to ONOO− formation from the NO/O2− reaction. When ONOO− decomposes in the presence or absence of CO2, powerful oxidants are formed 26, 27. However, the oxidative balance can be tipped towards nitrosation when these oxidizing intermediates are converted to N2O3 through further reaction with NO 28. Neucleophiles are preferentially nitrosated rather than oxidized under these conditions.
| 4) |
| 5) |
| 6) |
The reaction rates of NO with these RNS are nearly diffusion controlled, facilitating the rapid conversion from an oxidative to a nitrosation profile (eq 5 and 6). Therefore, a balance is formed between nitrosative and oxidative chemistry depending on the relative concentration of NO. At low NO fluxes, these reactions would tend to lead to oxidation of substrate, while at higher levels of NO they will preferentially nitrosate. In mechanisms of toxicity, there is a critical balance between the stochiometric amount of RNS generated to participate in oxidative chemistry, which would tend to be low, and the conversion of these intermediates to nitrosating species. This indicates that high NO levels where the indirect effects are predicted, in particular those that can be measured in biological system will favor a nitrosative stress.
The chemical biology of NO gives us a framework to understand how this simple diatomic molecule could have numerous biological properties based simply on its concentration in terms of the chemical toxicology. Low concentrations of NO such as those that occur in vascular and stromal cells (i.e. from eNOS and nNOS) regulate normal physiological processes and the high levels as those expected in activated macrophages (via iNOS) are thought to serve a cytotoxic/cytostatic function 17, 29, 30. However at these higher concentrations, it is not always clear that cell death is the ultimate outcome. Nitrosative stress has a protective side where nitrosation of caspase 3 and 8 as well as PARP leads to protection against apoptosis 31–33. Nitrosation and other oxidants closed NMDA channels preventing calcium influx 34–36, 36–38. Oxidative mechanisms such as nitration also have been shown to have biological signals of protection against apoptosis 39. Nitration of the transferrin receptor leads to proteosomal degradation, which limits iron uptake reducing apoptosis in endothelial cells. These examples suggest that tissues have adapted to conditions of inflammation and the biological mechanisms use this chemistry to mediate protective signals
Concentration Dependence of NO response
Processes ranging from apoptosis,, senescence angiogenesis, inflammation, immunological responses, vascular tone control, cardiac contractility and relaxation, to neuronal death all show seemingly contradictory behavior in response to NO. To better understand this phenomenon, we and others have quantified the effects of NO on different signal transduction mechanisms 40, 41. These observations provide a new perspective of the mechanism of NO signaling under various biological conditions.
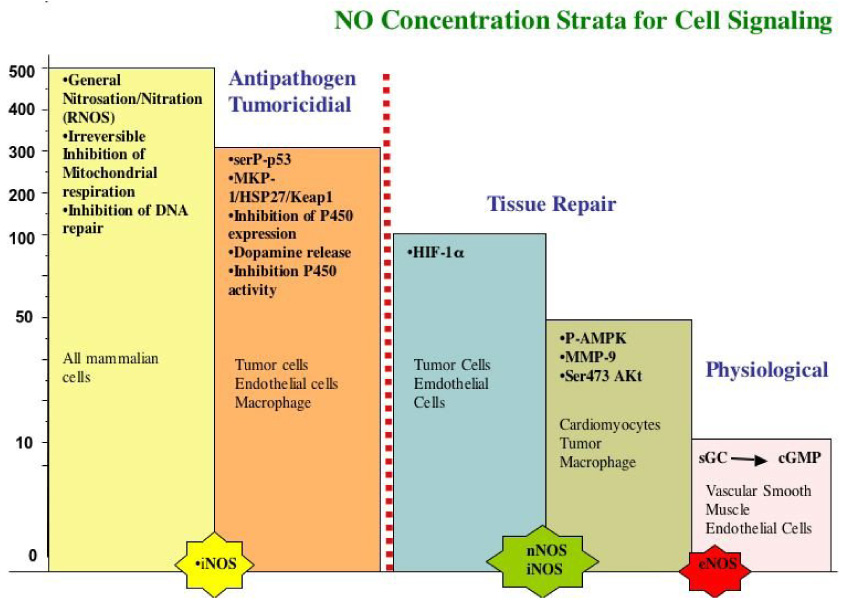
Much of the initial research on NO biology focused on chemical and biochemical modifications of likely biological targets. But as the understanding of the molecular biology of NO and its targets and the physiological context of NO generation increased, it became apparent that the chemistry by which NO elicited specific biological responses was highly dependent on, among other things, its concentration. We surveyed a variety of proteins that are known to be post-translationally modified in response to NO to determine if the redox chemistry differentiates those responses 41. When MCF7 cells were exposed to NO at different concentrations for defined periods of time, various proteins were found to be sensitive to distinct concentrations of NO (Fig. 2) 41. At sustained NO levels between 10–30 nM, phosphorylation of ERK occurs through a cGMP dependent mechanism in MCF7 and endothelial cells. At 30–60 nM NO Akt was phosphorylated 40, 42, 43. When NO reaches a threshold concentration of about 100 nM, HIF-1α is stabilized 40. At 400 nM NO, p53 is phosphorylated and acetylated 40. It is above 1 µM NO that nitrosation of critical proteins such as PARP, caspase and others occurs (summarized in 44). In the Petri dish, this upper level is where NO inhibits the mitochondria respiration 45, 46.
Figure 2.
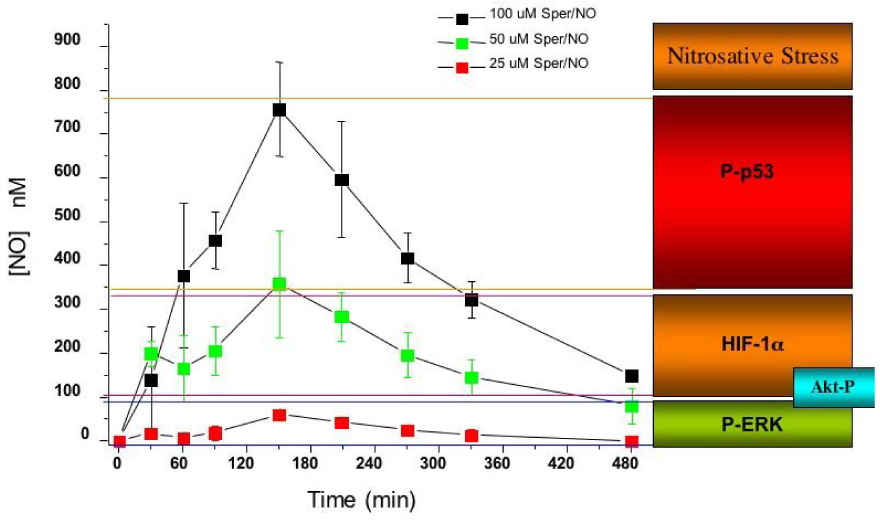
Concentration dependent for the release of NO from SPER/NO and the correlation with different cellular responses. Data from ref 40.
The results of these and other studies provide insight regarding specific mechanisms of action of NO as well as highlight its dichotomous nature of NO under various biological conditions. This has suggested that NO responses are context dependent and highly dependent on the levels of NO. The first level of signaling is cGMP dependent where low doses (<1–30 nM) NO can mediate proliferative and protective effects. These levels are lower in endothelial cells where NO fluxes less than 1 nM can induce proliferation 47. As NO levels increase, Akt becomes phosphorylated at serine 472 42, 43, 48, 49 an event known to be protective against apoptosis by inducing the phosphorylation of Bad and caspase-6 50. When HIF-1α is stabilized at greater NO concentrations this can be a proliferative response and thought to confer protection against tissue injury 51, 52. When NO levels are sufficiently high to induce p53 phosphorylation, there can be a cytostatic and even apoptotic response 53. To make a broad generalization, we conclude that low relative concentrations of NO tend to favor pro-growth and anti-apoptotic responses where higher levels of NO favor pathways that induce cell cycle arrest, senescence, or apoptosis.
In addition to concentration, temporal aspects of NO exposure are equally important. Certain proteins respond immediately to NO exposure, while others require hours or even days to be activated. HIF-1α for example responds immediately to NO (Fig. 2). As long as NO is present at concentrations above the minimal threshold amount necessary to induce its accumulation, HIF-1α is stabilized. Furthermore, when NO levels fall below the minimum activation concentration for HIF-1α, the protein disappears. HIF1α is therefore a real-time dosimeter of NO concentration 40, 41, 54. In contrast, phosphorylation of p53 by NO takes several hours yet sustains long after NO exposure. Again, as long as NO levels peak above the minimum amount necessary for p53 phosphorylation (> 400 nM) these products are observed. We therefore have proteins that are “immediate responders” and those that are “delayed responders”. These can be further classified into those proteins that are “transient responders” or “sustained responders”. HIF-1α falls into this category of an immediate-transient responding protein whereas in the case of p53, it is a delayed-sustained responder. Phosphorylation of ERK via cGMP would qualify as mixed immediate responder but is sustained after the loss of NO. Although these classifications are somewhat subjective they are useful descriptive terms to apply when deciphering the mechanisms of protein regulation by NO (Table 1).
Table 1.
Classifications of NO-regulated proteins in MCF7 cells.
| Immediate | Delayed | |
|---|---|---|
| Sustained | HIF-1α ERK p44/p42 sGC |
HO-1 Bcl-2 |
| Transient | pAMPK Akt |
p53-P-Ser15 MMP-9 Tsp-1 |
Although the biological responses to NO are multifaceted and appear contradictory at times, the concentration dependent responses clarify discrepancies and point to a simpler model. Therefore, many of the dichotomous responses to NO can be explained in terms of its concentration and duration profile. Many of the differential cellular responses may be simply a function of the concentration and duration of exposure, so that distinct biological responses result from specific incremental NO levels. NO is a highly diffusible molecule whose biological concentration is determined partially by its distance from the point of synthesis and partly by the cellular redox environment as discussed below. This suggests that perhaps the rate of NO production is a critical determinant of the cellular responses. Most of the current data has focused on whether NO can regulate specific signal transduction pathways, cause chemical modifications, or inhibit specific enzymes. Until now little was known regarding NO concentration dependence and cellular response mechanisms.
Superoxide (O2−) and the subsequent formation of peroxide (hydro- or alkyl-) are important components of many diseases 1, 55. Superoxide alone can induce signal transduction mechanisms and is generated under a variety of normal and pathological cellular conditions 56. It can activate metal complexes and promote damage through Fenton chemistry and redox cycling. Peroxide can similarly activate phosphorylation cascades and react with metals. Nitric oxide has been shown to have antioxidant properties by its diffusion-controlled reaction with O2− 57. This both prevents the reductive chemistry of O2− and inhibits H2O2 formation. One of the interesting relationships we have recently investigated is the effect of O2− and H2O2 on NO signaling. Just as NO inhibits the effects of O2−, it appears that O2− will ablate NO signaling 58. We have shown that generation of O2− during NO exposure can regulate the concentration of NO and thus affect the response of concentration-dependent, down-stream targets. Since certain regulatory proteins respond to distinct threshold concentrations of NO, the consequences of the introduction of O2− can convert a static-state phenotype to a pro-growth one.
For instance, when MCF7 cells were exposed to high levels of NO (> 500 nM), the presence of superoxide attenuated p53 phosphorylation and but maintained HIF-1α stabilization 41. Conversely, if superoxide fluxes were held constant and NO levels were increased, phosphorylation of p53 and HIF-1α signaling was again attenuated. Since NO is freely diffusible, intercellular or extracellular generation of superoxide had the same effect. These results demonstrated how ROS and NO mutually regulate each other’s signaling mechanism. Consequently, superoxide dismutase (SOD) can reverse these responses by increasing the bioavailability of NO when superoxide is present. Examination of soluble guanylyl cyclase activation and cGMP production by NO demonstrated an increase in the EC50 for nitric oxide as measured in the presence of superoxide. Interestingly, SOD affected the O2−-untreated control cells by shifting the EC50 for cGMP to considerably lower NO concentrations 41. These results suggest that tumor cell NADPH oxidase (NOX) affects NO signaling. Here it was found that superoxide generated extracellular by MCF7 cells dictated the threshold for NO signaling. Thus, there is a strong relationship between ROS generation and NO signaling in which they mutually regulate each others’ behavior.
During the interaction of ROS and NO a number RNS are generated that can affect different biological pathways. However the dominant outcome of the reaction between these species is the scavenging of NO by its simple conversion of to nitrite/nitrate. Although the initial result of NO reacting with O2− is ONOO−, the presence of CO2 catalyzes its rapid conversion to other species. When the flux of NO exceeds O2−, NO2 and N2O3 are predominantly formed. Conversely, when O2− is in excess peroxynitrite mediated oxidation and nitration are quenched suggesting that direct peroxynitrite chemistry has a limited role. It has been shown however that NO2 generated from this reaction can oxidize, nitrate, and nitrosate proteins via N2O3 59, 60 suggesting that this molecule may be involved in cellular signaling. Drapiers and coworkers showed that NO2 and N2O3 could induce HO-1 and IL-8 in monocytes 61 implying that RNS may have both anti-inflammatory and pro-growth properties. Other studies have shown that RNS activate latent TGFβ 62 thereby down-regulating Th1 (t-helper cell 1) type inflammation. Therefore, despite the potential for deleterious effects of RNS, biological signaling has adapted to these reactive intermediates to utilize a feedback mechanism which turns down the inflammatory response and promotes wound healing and tissue restoration. The RNS mediated cellular responses indicate that there are biological alarm systems as well as feedback loops that are important in understanding NO response in vivo.
The Concentration Range of Endogenously Generated Nitric oxide
Although NO elicits many unique cellular responses in vitro, the question remains: are these NO concentrations and conditions actually achievable in vivo? Several studies using different methods examined in vivo concentrations of NO under various biological conditions 63, 64. One method to address this question is to compare the cellular response to NO generated from NO-donor compounds with responses from NO generated from NOS. Experience with NO donor compounds has shown that each NO-specific target has a different concentration sensitivity to NO. By measuring biological effects of NO and correlating them with measured NO concentrations in vitro, we can approximate the in vivo NO concentrations necessary to elicit specific cellular responses. Using this “molecular signature” approach for various concentration levels of NO the redox environment can be determined under specific biological conditions. For example, when MCF7 cells were co-culture with activated NO-producing ANA-1 murine macrophages, an increase in the phosphorylation of Akt and p53 was observed in the MCF7 cells as well as stabilization of HIF-1α 40, 42, 65. When the number of ANA-1 cells was increased, there was a proportional increase in the amount of NO measured in bulk solution and the number of proteins regulated 40, 42. The measured NO concentration from NO-donors in the media necessary to induce p53 phosphroylation was > 400 nM but only > 160 nM when the source was activated ANA-1 macrophages 40. This discrepancy can be rectified by considering that NO generated from NO-donors is uniformly distributed throughout the media, whereas, the enzymatic production of NO from ANA-1 cells generates a concentration gradient emanating from the point of origin. Due to the close proximity of the macrophages to the MCF7 cells in this example, the actual local concentration of NO is substantially greater than 160 nM and at least as much as 400 nM 40, 66, 67, which is the minimum amount of NO necessary to cause p53 phosphorylation 40.
Macrophages are not only an important part of the immune system but can be a major source of NO. They perform a wide variety of functions from fighting bacteria and suppressing tumor growth to coordinating the resolution of tissue repair and restoration processes. Macrophages can generate a number of different levels of NO that serve different functions 68. For instance, eNOS stimulation of guanylyl cyclase in non-activated macrophages is required to fully activate LPS induce iNOS in activated macrophages 69. Interestingly the amount of NO produced from activated macrophages is dependent upon the manner by which they are stimulated. When cultured macrophages are pretreated with IFNγ followed by activation with TNFα or IL-1β the amount of NO measured in solution is approximately 10 times less that when they are stimulated with IFNγ+ LPS (PIPC and Listeria) even though there is only a modest difference in NOS activity 68, 70. In addition, the amount of NO-mediated nitrosation from TNFα or IL-1β stimulation was 30 times less than those stimulated with LPS or PIPC 68. This suggests that NO generated from cytokine stimulation leads to considerably lower NO concentrations in the surrounding environment than those agents that activate through the Toll-like receptors. These examples demonstrate the wide range of potential NO concentrations murine macrophages are capable of producing depending on the various stimulus.
Matrix metalloproteinases (MMP’s) are important mediators of inflammation, angiogenesis, wound response, and cancer 71–74. MMP’s are tightly regulated, both at the levels of transcription and post-translation and are targeted by reactive oxygen (ROS) and reactive nitrogen species (RNS) at both of these levels 75–77. While contradictory results have been reported with respect to redox regulation of MMPs, the ability of ROS and RNS to biphasically regulate MMP activity has been reported and suggests of a critical role of these species in the spatial and temporal regulation of MMPs during matrix remodeling 78–80. Toward this end, the biphasic and dose dependent regulation of matrilysin (MMP-7) by HOCl, initially demonstrated by Peppin and Weiss 78 involved oxidation of the cysteine switch at low HOCl concentrations that mediated activation of the zymogen 79. Higher HOCl concentrations inactivated the enzyme by chlorination/oxidation of adjacent tryptophan-glycine residues within the catalytic domain, leading to structural modifications that impeded substrate binding at the active site cleft 81, 82. The physiologic relevance of oxidant-mediated MMP inactivation was later demonstrated using gp91phox knockouts in a murine model of emphysema 83. Macrophage derived NO has also been shown to biphasically modulate the activity of MMPs. A recent report demonstrated both biological and chemical mechanisms of MMP activation by macrophage derived NO 80. MMP activity (MMP-9, −1, and −13) secreted from macrophages increased in response to low steady state NO levels (~50 nM) via cGMP-mediated suppression of the endogenous tissue inhibitor of metalloproteinase-1 (TIMP-1) 80. Exposure of purified latent MMP-9 to exogenous NO demonstrated a concentration-dependent activation (≥ 500 nM) and inactivation (> 1 µM) of the enzyme, which occurred at higher NO flux. These chemical reactions occurred at concentrations similar to that of activated macrophages. Interestingly, inactivation of TIMP-1 by HOCl has also been reported, again suggesting multiple mechanisms for oxidant regulation of MMPs during matrix remodeling 84. In an epithelial wound model, low NO levels similarly increased MMP-9 mRNA, which translated to enhanced enzymatic activity 85. Together, these results suggest that NO regulation of MMP-9 secreted from macrophages may occur chemically by RNS-mediated protein modification or biologically through cGMP-dependent modulation of the MMP-9/TIMP-1 balance.
One of the first observations that established that macrophages and hepatocytes were a source of NO was the formation of N-nitrosamines from the nitrosation of amines 86, 87. This provided evidence that nitrosative stress can be locally generated from iNOS. Other studies showed that NO and RNS inhibit Dopamine β–hydroxylase and that the endogenous NO generated from macrophages co-cultured with neuroblastoma cells was only mimicked by mM NO donor that generated > µM NO 88. When macrophages were co-cultured with A549 cells latent TGFβ was converted to active TGFβ 62. Again, this could only be recapitulated with NO-donors that gave µM fluxes of NO. Yet another example is the cystostatic effect of NO on Listeria 70. This pathogen propagates in the macrophage. When the thiol dependent protease becomes nitrosated by NO generated from iNOS, the organism is unable to escape the macrophage. Similarly this NO microenvironment effect could only be replicated by mM amounts of the NO donor DETA/NO which gave > µM NO for a prolonged period of time. In a study of astrocytes co-cultured with human microglial cells with HIV, iNOS activity was essential to inhibit the reverse transcriptase activity 89. To mimic this, mM amounts of an NO donor was required giving > µM NO flux. Taken together these results indicate that in the Petri dish NO fluxes can be sustained at µM levels for prolonged periods of time in this microenvironment.
Though these levels can be achieved in a Petri dish, the next question is whether µM levels of NO and indirect chemistry occur in vivo. Several studies have indicated that in murine and human models these upper level fluxes of NO can be achieved in tissue. Inhibition of P450 and COX-2 have provided evidence that locally, high levels of NO can be achieved by inflamed liver cells 90–92. Nitric oxide mediated inhibition of P450 may have important consequence under inflammatory conditions 93, 94. It was found that under sepsis or endotoxmia P450 inhibition correlate with the presence of NOS 95–98. Inhibition of expression and activity requires >100 nM to µM levels of NO to effectively inhibit these proteins. For example in HePG2 cells, 500 µM of DETA/NO with fluxes generating >500 nM NO is required to inhibit P450 1A1 (data not shown). Though oxygen tension in vivo may change this value, this does suggest that high levels of NO can be achieved under inflammatory conditions.
There are some examples in humans that µM NO levels may be achieved in vivo. Human tissue samples from ulcerative colitis found that serine 15 P-p53 co-localized with iNOS. Since relatively high levels of NO are required to phosphorolate p53 in vitro, this indirectly suggests that these levels of NO can be achieved in human. In colorectal cancer, there is a correlation of iNOS with HIF-1α indicating that the levels of NO are > 100 nM 99. Though not definitive they are suggestive that NO can regulate protein function beyond the activity of sGC in vivo.
For instance N-nitrosamines, such as nitrosoproline are generated in vivo under septic conditions 100, 101. Nitrosamine formation can be achieved by two distinct mechanisms, nitrosation and oxidative nitrosylation. Oxidative nitrosylation is where the substrate is first oxidized to a radical which then is followed by radical-radical coupling with NO to form the nitrosated product. Although this is chemically plausible, it remains to be demonstrated under biological conditions. Nitrosation is the direct addition of a nitrosonium equivalent (NO+) from a species such as N2O3. Oxidative nitrosylation occurs from NO under very limited conditions with amines that have low oxidation potentials. Proline and other secondary amines have high oxidation potentials (>1.5 V) and therefore undergo exclusively nitrosative chemistry. Other studies have found increased nitrosated thiols (RSNO) in pathological conditions in human lungs indicating that RNS are generated in vivo. 102 The observation of amine and thiol nitrosation under inflammatory conditions indicates that nitrosative stress mediated from N2O3 species does occur in vivo.
Another indirect indicator of RNS formation is the detection of 3-nitrotyrosine (3-NT). The immunohistochemical staining for this species has been used as a footprint for NO and RNS in tissue. Though peroxynitrite had long been assumed to be the sole source of 3-NT, further investigation revealed that that this species could not be not responsible for all the 3-NT observed in vivo 103, 104. Oxidation of nitrite by one electron to form NO2 through Fenton type or peroxidase chemistry is an alternate mechanism for generation of 3-NT 105, 106. NO2 is unique in that if generated in the absence of NO it can migrate through cells 107. This radical is thiophilic and may have its own signal transduction mechanism such as activation ARE (antioxidant-responsive element) 108. Unlike O2− which is an anion, and OH radical which reacts at its point of generation, NO2 can readily diffuse to adjacent cells and tissue similar to NO, providing a mechanism for signal transduction in vivo. Thus, 3-NT may represent the formation of NO2 from nitrite rather than high levels of NO. Taken together, there is evidence that indirect effects do occur and that the upper end of the NO concentration spectrum can be achieved in vivo.
Nitric oxide is responsible for a surprising array of responses in the nervous system. cGMP levels can be increased endogenously with nM levels of NO 109, 110. Nitric oxide also mediates catecholamine (such as dopamine) release in the stratium 111. Interestingly when this response was compared to synaptosomes and isolated cells, it required a surprisingly high amount of NO (near 1 µM of NO) to replicate this effect. This suggests that the threshold for NO generated in the postsynaptic neurons mediating dopamine release has to be > 200 nM over a short period of time. This is a surprising concentration of NO but isolate the response to a very localize area. The diffusion from this area would result in rapid dilution and not regulate other processes in the region. More importantly the high threshold for this response indicate NO derived at lower levels from other sources will not randomly activate this response. Thus, nNOS in the microenvironment can achieve NO levels 100 times higher than required to stimulate vasorelaxation of vascular smooth muscle cells. Therefore, NO at higher concentration can be used to mediate specific physiological responses in a very confined area over short periods of time.
Burke and coworkers have used microelectrodes successfully to measure these same levels (300 nM) in NMDA stimulated activity in the cerebral cortex. This could indicate that NOS translocated to Ca2+ channels such as that in NMDA may be regulated under inflammatory conditions via the generation of RNS. This mechanism would serve to protect neurons from Ca2+ overload due to increase glutamate release. It also suggests that nitrosation of thiols within this channel via N2O3 is possible in isolated regions of the cell. One interesting aspect of NO regulation of the NMDA channel is that low doses of NO under oxygen-glucose-deprivation (OGD) conditions results in increase opening of the channel. This suggests that under hypoxia, lower concentrations of NO are needed to increase vasodilation to the area. Moreover, it is plausible that direct effects of NO may modulate an increase in channel function while indirect effects close the channel.
The examples cited above demonstrate that there is an upper range of NO concentrations in vivo and that this range can be mimicked by mM concentrations of NO donors. However, what are the lower limits of NO concentration that mediate biological effects? The relaxation of vascular smooth muscle cells (VSMC) requires between 10–30 nM levels of NO to facilitate vasodilation. This was thought to be the lower limit of response to NO. However, several studies using very low fluxes of NO showed that there are different biological effects. Using DETANO, a NO releasing compound with a 24 hr half-life, cellular effects in endothelial cells were observed at NO fluxes less than 1 nM 112. At this level, the NO concentrations escape the detection range of our current methodologies. Bogdan and coworkers showed that these levels of NO occur in immune cell response in NK cells 113. These observations indicate that the concentration range of NO “physiological” responses can occur in sub nM range.
There is evidence of a wide range of NO concentrations that mediate discrete biological responses which depend on both the concentration and temporal profile of NO. The range from sub nM to > µM indicates that NO can elicit different responses over a 1000-fold concentration range. These simple concepts can explain much of the complexity of the biological response. However, how can these different ranges of NO be achieved and what are the factors that enable these different environments to occur. In addition, the NO signaling can be antagonized by ROS. This implies that NOX and other ROS generating enzymes can readily influence the redox environment, fine-tuning the biological response. Ultimately, it is NO's wide range of responses, reactivity, and rapid chemistry make it an ideal and versatile signal transduction agent.
Kinetic Determinants for Molecular Target Interactions
From the above discussion, endogenously generated nitric oxide provides a spectrum of biological responses based on concentration and duration of NO exposure. There are a number of different factors that influence the concentration of NO in vivo and therefore indirectly determine which signaling cascades are activated. Since endogenously generated NO ranges from basal levels in endothelial cells (<1 nM) to that generated by fully activated macrophages (>1 µM), this wide range of concentrations combined with its unique chemical reactivity makes NO a remarkably versatile signal transduction agent. The cellular and biochemical factors that shape these responses are important in understanding the complete mechanism of NO signaling. These factors, referred here as Kinetic Determinants for Molecular Target Interaction are: rates of NO production, diffusion distance, rates of consumption, and reactivity of the nitrogen oxide with the target (Fig. 3). This section discusses the interplay that goes into creating the NO responses in physiology and pathophysiology with respect to the direct and indirect effects.
Figure 3.
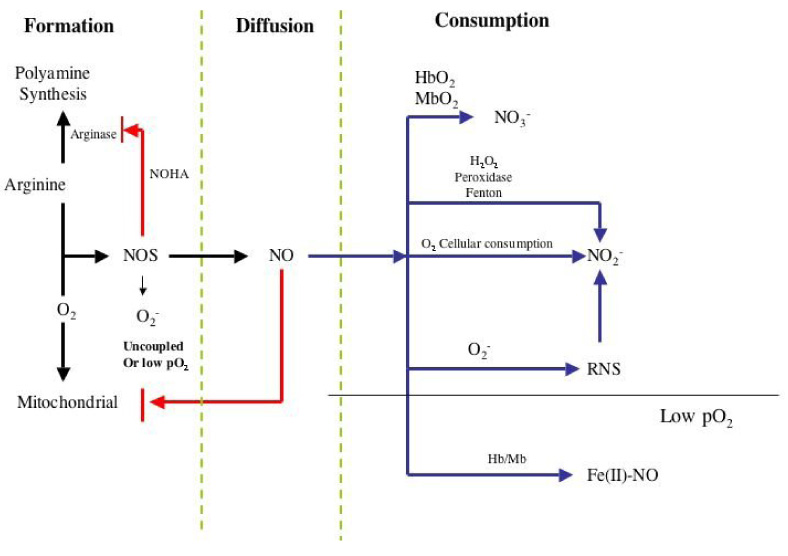
Formation and consumption processes that influence NO Concentration
Formation
Although there are alternative mechanisms to generate NO, such as acidification or reduction of nitrite, the vast majority of mammalian NO is derived enzymatically from nitric oxide synthase (NOS). This family of enzymes converts arginine to citrulline and NO in an NADPH- and O2-dependent process. There are three NOS isoforms that provide a wide range of concentration and temporal NO profiles. Two of these are constitutive (NOS-1; nNOS neuronal and NOS-3 eNOS endothelial) and one is inducible (iNOS; NOS-2) 29. These isoforms are differentially regulated at numerous levels including transcription, translation, post-transcriptional, and biochemical 114–116. There are two major differences between these isoforms, the duration of NO generation, and the local concentration of NO that can be produced. Calmodulin dependency is important for the constitutive forms of the enzyme where calcium fluxes regulate their activity providing short burst of NO. In contrast, iNOS has calmodulin as a subunit and is therefore permanently activated and capable of generating NO for long periods of time. Post-transcriptional modification such as phosphorylation of eNOS can convert this constitutive isoform from generating NO in short bursts to prolonged fluxes 117.
The enzymatic activity of NOS is tightly controlled depending on substrate and cofactor availability, as well as the rate of electron transfer 29, 118. The amount of enzyme and location within the cell determines the concentration of NO in the microenvironment of the molecular target. The substrate and cofactors that control NOS activity are also involved in other major metabolic pathways in the cell, thereby connecting NOS activity with other metabolic pathways.
In the presence of sufficient cofactors (NADPH, FMN, BH4, FAD), the NOS activity is dependent on the arginine and oxygen availability. There are three major arginine pathways that influence the activity of NOS: 1) competition by arginase, 2) arginine uptake and methylation, and 3) proteolysis to form asymmetric dimethylarginine (ADMA) 119. Arginase, the first enzyme in polyamine synthesis, can have significant influence on NOS activity by competing for the cellular arginine pool 120. Though the Km for arginine is higher for arginase as compared to NOS, the cellular concentration of arginase is considerably higher making the portioning of arginine consumption between arginase and iNOS nearly equivalent. In the case of nNOS and eNOS, arginase would then be expected to consume most of the arginine.
Several studies suggest that NOS activity is controlled by arginine availability. Cellular arginine uptake is facilitated by the cationic amino acid transporter (CAT). There are several factors that regulate this transporter. Lysine directly competes for arginine uptake 120, 121. Interestingly the RNS, nitroxyl (HNO), and not NO, decreases the activity of this transporter which may be a feedback mechanism for NOS activity 122. One of the consequences in reducing arginine availability is that NOS becomes a superoxide rather than NO generator 123–125, therefore switching from NO-mediated regulation to ROS generation. This may explain why NOS is beneficial under some conditions while deleterious under others.
Another important aspect of arginine metabolism is the generation of ADMA a known inhibitor of NOS, which is derived from arginine methylation of proteins and subsequent proteolysis 126. One study has shown that ADMA not only inhibits nNOS but converts this enzyme from an NO to superoxide generator 127, which may have major implications in various diseases states. Therefore, NO production and cellular signaling can be influenced by arginine uptake, competitive metabolic pathways and the generation of endogenous NOS inhibitors. Additionally, changes in arginine metabolism may not only affect the rate of NO production but may also be important in the production of superoxide.
Oxygen is another major substrate that regulates the activity of NOS. Changes in pO2 have a profound influence on NOS activity. The oxygen dependency is one of the major differences between the NOS isoforms and is the factor that may provide the most insight into the role each isoform plays in vivo. The Km (i.e. the concentration of oxygen at which the activity of the enzyme is half maximal) for O2 towards each NOS isoform is considerably different. When the O2 concentration in the Km range or lower, the enzyme activity is highly dependent on its concentration, resulting in a direct correlation between NO production and oxygen concentration. However, when O2 concentration is significantly greater than the Km value, the dependency on O2 is lessened as the activity is less affected by increases in oxygen. This becomes important when comparing the Km values for the different NOS isoforms. The Km for eNOS is 23 µM, iNOS is 135 µM, and nNOS is 350 µM 29. These vast differences in Km values indicate that the rate of NO synthesis from nNOS, for example, will be substantially more affected by oxygen fluctuations over a greater range of O2 than eNOS. In other words, nNOS can provide a direct O2 detection system that perhaps is linked to a different subcellular compartimentalization for nNOS vs eNOS (i.e. mitochondria). In contrast, eNOS could be at near full activity even with low oxygen tension allowing regulation of critical functions of the cardiovascular system such vascular tone under these conditions.
Normal tissue oxygen tension range from 5–15 torr (10–20 µM) to as high as 50 torr (60 µM) 128. Therefore the isoforms will have different activity depending on the specific tissue bed. Further complicating this relationship is that NO inhibits the primary mechanism of O2 consumption in tissue, mitochondrial respiration 128, 129. Increases in NO will thus increase O2, which further increases NOS activity. It should be noted that at low O2, NOS generates superoxide rather than NO 130. Though these cellular mechanisms may imply that NO production could spiral out of control, there are two feedback mechanisms that limit this augmentation cycle. The first is that high NO levels retard the enzymatic activity of NOS thereby limiting its production 29. The second is that cellular NO consumption is O2 dependent, and proportional to the oxygen concentration 130. As O2 levels increase, so does the rate of NO metabolism 131. Taken together we see that there is an intimate relationship between these two diatomic gases, which is critical in the regulation of tissue oxygenation and perfusion.
Another important factor of NOS regulation is its location within the cell. Hydrophobic regions of a cell such as membranes have about 10 times higher concentrations of gases (NO, O2, CO) compared to aqueous regions of a cell simply based on their solubility 132. The activity of eNOS can be increased 10 times by simply translocating to the membrane, presumably because of increased oxygen availability 116. Translocation of iNOS in activated macrophages may affect the activity and ultimate NO released from macrophages. The amount of NO detected from cytokine activated and LPS activated is very different despite similar amount of NOS. It has been shown that this increase in NO release from the cell is accompanied with increased membrane iNOS 133. Translocation of nNOS to glutamate recpetor may also increase its NO activity. NOS can exist in both membrane and cytosol. Translocation to a different O2 environment changes its activity and can have a significant effect.
Diffusion
Another important physical characteristic of NO and an important determinant of its concentration is diffusion. Nitric oxide has a low molecular weight, it is uncharged, and soluble in both aqueous and hydrophobic environments. This makes it highly diffusible. The diffusion coefficient for O2 is 2800 µm2 s−1 and NO is 3300 µm2 s−1 134. In tissue, cells are usually within 50–300 µm, or between 1 and 30 cell lengths, from a blood vessels where NO is rapidly consumed. Since NO diffuses as much as 5–10 cell lengths in 1 second, the diffusion to the blood vessel becomes a kinetic barrier in determining which reactions are viable. It suggests that in tissue there is a significant lifetime compared to the lumen of the blood vessel. Thus, compartmentalization between the vessel and tissue allows different gradients to occur.
Simulations (low tissue NO consumptive rate) have demonstrated that large increases in NO concentration can be achieved in tissue which only decreases near blood vessel barriers 134, 135. Therefore it is theoretically feasible to generate high levels of NO. Since p53 is only phosphorylated > 400 nM 40 and some studies have shown that iNOS and P-p53 colocalized in inflamed tissue 65, these concentrations of NO are achieved.
Another important aspect of diffusion is dilution. Under three dimensional conditions, such as tissue, random diffusion of NO away from a single cell producing it ultimately results in rapid dilution. Since the volume of a sphere increases by the cube of its radius, when NO moves a distance of one cell length there is an 8 fold dilution, at 2 lengths a 27 fold dilution, at 4 lengths away there is a 125 fold dilution, and so forth. Within 1 second NO concentration from a single cell has been diluted over 200 times. NO production can be confined to specific areas in the cell resulting in high local concentrations. However short distances away its concentration can diminish substantially. For example, NOS is confined to a specific location such as a synaptic region which is less than a µm across. The NO concentration required to stimulate catecholamine release can be estimated to be 1 µM, which is very high. However, at the equivalent of one cell length away (10 µm) there would be a 1000 fold dilution to <1 nM. This diffusion and subsequent dilution of NO allows for specific sites to be exposed to higher and lower fluxes of NO even within the same cell. In cases such as the vascular endothelium, NOS is located in the junction between the endothelial layer and the vascular smooth muscle cells (VSMC). Since this is juxtaposed to the smooth muscle, its proximity allows NO to migrate to sGC in the VSMC without the interference of the scavenging effects of the blood vessel. Computer simulations of the vessel wall have shown that fluxes of NO in the endothelial VSMC is about 100 nM enough to cause relaxation 134. Microelectrode studies suggest that > 200 nM NO can be generated in this perivascular space 136.
Leukocytes are engineered to fight a bacterium, virus, or tumor cells. For example, if there are four leukocytes surrounding a target pathogen, then NO levels can be additive at the region of NO flux intersection focusing high levels where it is most effective. As discussed above, in the case of Listeria which propagates in macrophages, high NO levels (< 1 µM) induces a cytostatic effect by nitrosateing the protease required for liberation from the leukocyte 70. However, NO dilutes rapidly from the macrophage where 4 cell length away µM levels are diminished to lower than 1 nM. Thus, orientation of the iNOS can determine the shape of the NO concentration under a variety of conditions.
Consumption
Though NO synthesis and diffusion are partial determinants of NO concentrations in vivo, there is another important factor that needs to be considered; consumption. There are several important general mechanisms for NO consumption including RBC reactions, cellular metabolism, and reaction with ROS. An important and fundamental characteristic of NO consumption is that, similar to NO synthesis, most consumptive mechanisms of NO are O2 dependent. This indicates that O2 will be a key factor in determining rate of NO consumption and the scope of its reaction with target molecules. Cellular consumption of NO albeit much slower than its reaction with RBC’s is an important determinant in establishing NO concentration gradients extravascularly 131. Reported values for cellular consumption of NO vary from a rate of 0.01 s−1 at the low end 137, 138 to as high as 7.5 s−1 131. By choosing the upper limit value for NO consumption, simulations have shown that consumptive processes are competitive with diffusion and significantly influence the shape of the NO profile 139. Models using the lower limit demonstrate diffusion is the primary determinate 139. Models and measurements in different vascular beds suggest regulatory NO for vasodilation can reach > 300 nM for brief periods of time 134. However the rate of NO consumption in different tissue beds and the biochemical mechanisms remain to be determined.
The rate of NO consumption by cells is directly dependent on, and proportional to, the oxygen concentration 131 suggesting an important regulatory relationship between NO signaling and oxygen concentration in tissue. While increased oxygen levels will increase NO consumption the converse is that NO regulates oxygen consumption via inhibition of mitochondrial respiration. This important interdependent relationship between NO and O2 provides a direct feedback mechanism to regulate their respective concentrations 140. The respiratory inhibition by NO depends on a number of factors such as the rate of mitochondrial respiration129. At high respiratory rates the IC50 for NO is about ~100 nM at 150 µM O2 which compared to µM levels of NO at lower turnover rates of mitochondria. Thus inhibition of mitochondria in cells with low respiratory rate will require levels of NO that mediate nitrosative stress while higher levels would mediate Akt-P. In tissue, models suggest that at low respiratory rates 200–300 nM NO is required to competitively inhibit by 50% mitochondrial O2 consumption 129. This suggests that the interdependent relationship between NO and O2 depends on the metabolic state of the cell.
It should be noted that despite intense investigation by numerous investigators, the dominant mechanism for general cellular NO consumption has yet to be elucidated. Here, cellular NO consumption refers to the disappearance of NO in the presence of non-erythrocytic cells which represents the sum of various independent cellular NO-consumptive processes. A major mechanism for NO consumption, and a subset of cellular consumption, is via reaction with ROS. The reaction of NO with ROS results in scavenging NO as well as producing intermediates associated with indirect effects 41. This interaction alters NO signaling by lowering its concentration while creating potentially new signaling mechanisms through RNS generation. There are two primary mechanisms for ROS mediated NO consumption; reaction with superoxide and peroxidase/Fenton chemistry. As discussed above, NO reacts with superoxide at near diffusion control rate resulting in peroxynitrite (ONOO−), which is converted in the presence of excess NO to NO2 and N2O3. These species are particularly thiophilic and may provide thiol mediated signaling 21.
Another mechanism for ROS mediated consumption of NO is through interaction with peroxidases and Fenton type reactions 54, 141. It has also been shown that H2O2 through reaction with heme proteins like peroxidases can decrease NO concentrations 142. This reaction leads to nitrite, which in the absence of NO can be oxidized by the same metal catalyst to NO2. Like peroxynitrite, NO2 can nitrate tyrosine residues and oxidize other substances 59, 105, 106. It is possible that the observation of nitrotyrosine indicates lower NO levels despite high NOS output. Thus, the decrease in NO via O2− scavenging, increases NO2 and may serve to change the cellular signaling paradigm.
Two important sources of non-mitochondrial ROS are NADPH oxidase (NOX) and xanthine oxidase (XO). It should be kept in mind that hydrogen peroxide (H2O2) generated from these enzymes has its own signal transduction mechanisms 143. When the Km for O2 for XO (~70 µM) is compared to the NOS isoforms only iNOS falls in this range, while eNOS is far below and nNOS is far above. For this reason translocation of O2−-producing NOX can play role in controlling NO-elicited signaling. Translocation of NOS to the proximity of oxidases or peroxidases may therefore become important. Recently, it has been shown that COX-2 forms a complex with iNOS providing an interesting possibility for regulation of NO and ecosinoid products like PGE2 144.
It has been calculated that the lifetime of NO in vivo is between 10 – 100 ms. Since the reaction rate constant is 1010M−1 s−1 for the NO/O2− reaction, ablation (95% reduced) by superoxide of the NO signaling occurs when the effective NO concentration is above 10–20 nM. Therefore NO concentration less than 1 nM, such as those found in some endothelial responses 112, 145, will be relatively unaffected by this reaction. However, with NO mediated processes that lower blood pressure or suppress MCP-1 and ICAM-1 expression 146–148, superoxide antagonizes these mechanisms.
Another factor to consider is that O2− is primarily generated in the aqueous environments and by virtue of being an anion does not readily migrate into or through membranes. Therefore based on their respective solubility’s, the reaction between NO and O2− occurs mainly in the aqueous layer. The concentration of NO in the aqueous layer will be the determining factor for the magnitude of this reaction. One of the important consequences of this is that translocation of eNOS to the membrane in the proximity of sGC decreases the EC50 while limiting the reaction with superoxide, which is in the aqueous compartment 149.
In endothelial cells, NO mediated angiogenic processes occur below 1 nM, therefore, O2− should have a negligible chemical effect on these mechanisms at sufficient levels of NO production. The more sensitive the target is to NO, the less the effect O2− will have on diminishing its response to NO. However, as the requirement for NO concentration begins to increase such as to levels that stimulate sGC in VSMC, superoxide can have a significant role. The cellular junction between the endothelial cell and VSMC provides an isolated area where superoxide can regulate NO migration to VSMC. In this region, increase NOX or other superoxide producing enzymes would be an important control mechanism for vascular tone. The same may be true for nNOS in the synaptic region may be controlled by O2−. This spatial relationship may be important in shaping the NO mediated signaling.
Another issue to consider when talking about the regulatory effects of O2− on NO signaling is the influence of superoxide dismutase (SOD). This enzyme is responsible for removing O2− by converting it to H2O2 and O2. This becomes important because, like the reaction of NO with O2−, SOD reacts with O2− extremely rapidly and will therefore compete against NO for O2−. Since the rate of reaction of SOD with O2− is about half as fast as the rate of NO with O2−, there must be twice as much SOD than NO for each to scavenge 50% of the O2−.
Where k1 is the rate constant for the reaction of NO with O2− and k2 is the rate constant for the reaction of SOD with O2−. Under biological conditions, SOD at sufficient concentrations, 3−15 µM, has the ability to restore the bioactivity of NO when O2− is present. When SOD removes O2− before it reacts with NO, NO is free to diffuse and perform its signaling functions. Therefore even when O2− is generated concomitant with NO synthesis, if there is sufficient SOD present NO signaling will be unaffected41.
Factors for RNS signaling
As discussed above, NO and derived species are important cell signaling agents. A variety of targets for these NO-derived species (i.e. NO2, ONOO−, N2O3, etc.) exist within cells and include thiols, lipids and aromatic amino acids.44 Clearly, if the normal signaling biochemistry associated with the chemical modification of these targets by nitrogen oxides occurs to an extreme, then this has the potential to have a severe toxicological outcome. Thus, at high NO concentrations, where this chemistry may occur indiscriminantly or to an extreme, toxicity, rather than normal cell signaling may result. However, with normal signaling processes and pathways, the amplification of signals allows reactions to occur at low levels and yet be important in signal transduction.
The formation of the RNS occurs by both the NO/O2− and NO/O2 reaction, which occur at distinct sites in the cell depending on hydrophobicity or hydrophilicity. There are some important factors that dictate the signal transduction mechanism for RNS. These reactive intermediates are electrophilic, thus pKa of the substrate such as thiols will be an important determinate in the reaction dynamics. The three primary NO derived RNS are N2O3 NO2 and peroxynitrite 19, 44. As we discussed above, the later is an important product of the NO consumptive mechanism, while N2O3 and NO2 can be derived from both NO autoxidation and the NO/O2− reaction. As with NO, formation, diffusion, consumption/scavenging and rate of reactivity with the molecular target are important kinetic determinates.
The reaction between NO and O2− to form peroxynitrite (ONOO−) is one of the fastest reactions known in chemistry 150. Since O2− is short-lived and is produced in the aqueous layer, this reaction occurs in the aqueous compartments of tissues. In biological media, peroxynitrite rapidly reacts with CO2 converting it in milliseconds to the anionic species ONOOCO2− (eq. 7). This species decomposes within 15 ms to a radical complex NO2/CO3− (possibly a caged solvent complex) (eq. 8) 26.
| 7) |
| 8) |
| 9) |
The short lifetime and anionic character of these species limits the chemistry of peroxynitrite to a finite distant from the source of O2− in the aqueous media. Under conditions where excess of O2− and NO is produced, scavenging of NO2 and CO3− occur with near diffusion controlled rate constants resulting in abatement of peroxynitrite nitration and oxidation (eq. 9) 151. While the overall reaction between superoxide and ONOOCO2− yields nitrite and carbonate, the reactions with NO form nitrite, carbonate and NO2 (eq 3). The latter then further reacts with NO to give N2O3 (eq. 2) 44. Thus this reaction provides a RNS shunt which can convert NO from a directly acting species to predominantly nitrosative chemistry. This may provide the ability to target oxidation and nitrosative mechanism to specific locations within the cell.
The autoxidation reaction is third order overall with a second order dependency on NO 152 This second order dependency on NO dictates that the relevance of this reaction is highly dependent on the concentration of NO. Since the lifetime of NO is approximately 0.1 second (depending on consumption rate in that tissue bed) mM concentrations would be required for this reaction to be the primary pathway of NO. However, the rate is increased dramatically in membranes and other hydrophobic regions where both O2 and NO are present at a 10 times higher concentration 132. Thus, a fraction of the NO chemistry in cells may occur through the autoxidation reaction in the membrane 153. In the nM range, this reaction in the membrane would be predicted to account for only 0.001 % of the consumptive mechanism of NO. However, a 1 µM NO concentration in the microenvironment of activated macrophage indicates that membrane concentrations may be as high as 10 µM. This reduces the half-life of NO to 50–80 seconds of this reaction suggesting that 1–5% of NO in cells may react through the autoxidation reaction. After prolonged periods of time accumulation of RNS will occur which can react with targets that are thermodynamicly favorable. Since it would take considerable time, due to their low yield, this mechanism serves as a temporal switch directing NO towards specific targets during prolonged NO exposure at defined concentration. Therefore, RNS formation from this reaction is confined to specific areas that are hydrophobic and may serve as sources of unique signaling species after prolonged NO exposure.
The diffusion of these species is an important consideration. The species ONOOCO2− cannot migrate appreciably or cross membranes due to is anionic character and short lifetime. N2O3 is constrained by the hydrolysis reaction to nitrite which has a 1 ms halftime 154. In the cytoplasm the lifetime of N2O3 is further reduced from the reaction with GSH (1 –10 mM) to form GSNO 24. In the membrane where there are fewer nucleophiles and limited water, this species has a longer lifetime, which may increase its probability to interact with specific molecular targets.
In contrast, NO2 is not hydrolyzed in aqueous medium directly. In order to degrade in water, this radical needs to first react with NO to form N2O3 (eq 2) or dimerize to form N2O4, which subsequently hydrolyzes. At high NO2 concentrations, the dimerization is a major reaction, however, these concentrations are unlikely to occur in vivo. Thus, NO determines the NO2 lifetime in water.
The rate constant for the NO-dependent degradation of NO2 and subsequent nitrite formation is 6 ×106 M−1 s−1 (eq 2, 3) indicating that at near µM NO concentrations its lifetime would be limited in aqueous media. Therefore at sub µM NO concentrations NO2 would be able to diffuse between cells 154. It has been found that low fluxes of NO2 readily diffuse into cells such that cells serve as an important "sink" for NO2. Therefore, unlike peroxynitrite and N2O3, which have limited diffusion, NO2 can readily diffuse several cell lengths. There are several studies that show that NO2 can diffuse readily from the extracellular space into cells 59, 60. Since it formed from either the autoxidation or the NO/O2− reaction, this may be an alternative quintessential signal transduction intermediate.
NO2 can be generated in the absence of NO through the oxidation of nitrite from peroxidase reactions or Fenton type reactions. The oxidation of nitrite leads to an increase in nitrotyrosine formation, consistent with the idea that nitrotyrosine is an indicator of the presence of NO2 106. The signal transduction mechanism of NO2 may therefore be very important 155.
The scavenging of these intermediates is an important kinetic determinant of the potential relevance of the signaling processes associated with these species. Peroxynitrite anion is scavenged by GSH but rather slowly 156. In the presence of CO2 the formation of ONOOCO2− is predominant even in the presence of GSH. The decomposition of ONOOCO2−, oxidation of nitrite, and the autoxidation of NO all generate NO2 as a common product and it can be argued that NO2 is an important reactive species associated with the indirect effects of NO. This radical reacts with urate, ascorbate and GSH at >107 M−1 s−1 108. Therefore, in the cytoplasmim where GSH > mM, there is very little probability of these radicals react with specific targets.
In the plasma and extracellular fluids, urate and ascorbate are 100–200 µM, which limits the lifetime of these radicals to less than milliseconds. Thus, the movement of NO2 is limited by the amount of these ubiquitous antioxidants. In the case of N2O3, mM ascorbate and 100 µM GSH but not urate is competitive. Thus, GSH is the more important limiting agent for N2O3 as well as the reaction with water.
The above discussion suggests that the RNS reactivity is limited to the vicinity of its formation (Fig. 4). Thus, site-specific generation of these RNS provides a logical possibility for accomplishing the chemical modification of specific biological targets. It also suggests the proximity of the molecular target (or possibly translocation of the target to the vicinity of the NO source) will be a very important factor in the signaling function of NO. Another important consideration is the concentration of O2, which would have to be high for much of the discussed chemistry to be relevant. Thus, the level of mitochondrial activity, which consumes O2 under certain conditions, generation of O2−, is critical to predicting the likelihood of much of the chemistry discussed here.
Figure 4.
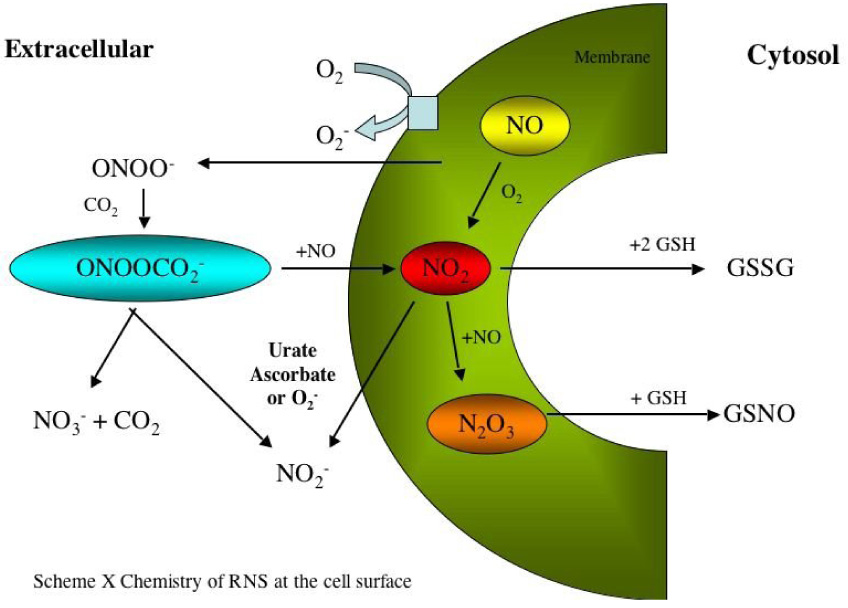
The reactive nitrogen species processes of formation and scavenging.
Summary
The multitude of possibilities in biological responses to NO makes it a fascinating molecule for study. Unlike other simple signaling molecules, there are numerous layers of regulation to consider when assessing possible outcomes of NO exposure. A complex relationship exists between the fundamental chemistry of NO and the important influences of the cellular microenvironment. The importance of concentration when talking about NO-signaling can be appreciated when one considers the distinct concentration-dependence of NO-regulated proteins. Since all biological responses to NO are ultimately a function of its concentration, dynamic fluxes in NOS substrate availability in addition to changes in rates of NO metabolism will have dramatic influences on NO signaling (Fig. 5). The concentration of NO will determine its chemistry (direct vs. indirect), the distance it diffuses, and the type of signaling targets it interacts with. The combination of these factors will establish the cellular phenotype. Many potentially different cellular responses and vastly diverse outcomes are possible through a range of NO concentrations. The presence of other radical species is also a key regulator of NO concentration and will partially dictate its influence on target molecules. By viewing NO in this context it enables us to understand why there can be a seemingly infinite number of biochemical responses to a single signaling molecule. Additionally these principals illustrates how deregulation in NO catabolism or metabolism will be import determinants in the natural course of various disease states.
Figure 5.
Discrete level of NO cellular responses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 1999:71. doi: 10.1016/0748-5514(85)90028-5. [DOI] [PubMed] [Google Scholar]
- 2.Penney DG. Acute carbon monoxide poisoning: animal models: a review. Toxicology. 1990;62:123–160. doi: 10.1016/0300-483x(90)90106-q. [DOI] [PubMed] [Google Scholar]
- 3.Cadenas E, Sies H. Oxidative stress: excited oxygen species and enzyme activity. Adv Enzyme Regul. 1985;23:217–237. doi: 10.1016/0065-2571(85)90049-4. [DOI] [PubMed] [Google Scholar]
- 4.Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- 5.Severs RK. Air pollution and health. Tex Rep Biol Med. 1975;33:45–83. [PubMed] [Google Scholar]
- 6.Mason RP, Chignell CF. Free radicals in pharmacology and toxicology--selected topics. Pharmacol Rev. 1981;33:189–211. [PubMed] [Google Scholar]
- 7.Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu. Rev. Pharmacol. Toxicol. 1990;30:535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]
- 8.Free Radicals in Biology and Medicine (Life Chemistry Reports Series) 1985. [Google Scholar]
- 9.Signal Transduction by Reactive Oxygen and Nitrogen Species: Pathways and Chemical Principles. 2003. [Google Scholar]
- 10.Wink DA, Feelisch M, Fukuto J, Chistodoulou D, Jourd'heuil D, Grisham MB, Vodovotz Y, Cook JA, Krishna M, DeGraff WG, Kim S, Gamson J, Mitchell JB. The cytotoxicity of nitroxyl: possible implications for the pathophysiological role of NO. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 11.Furchgott RF, Vanhoute PM. Endolthelium-derived relaxing and contracting factors. FASEB, J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 12.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 13.Murad F. The nitric oxide-cyclic GMP signal transduction system for intracellular and intercellular communication. Recent Progress in Hormone Research. 1994;49:239–248. doi: 10.1016/b978-0-12-571149-4.50016-7. [DOI] [PubMed] [Google Scholar]
- 14.Granger DL, Hibbs JBJ. High-output nitric oxide: weapon against infection? Trends Microbiol. 1996;4:46–47. doi: 10.1016/0966-842X(96)81506-X. [DOI] [PubMed] [Google Scholar]
- 15.Stuehr DJ, Nathan CF. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathan CF, Hibbs JBJ. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991;3:65–70. doi: 10.1016/0952-7915(91)90079-g. [DOI] [PubMed] [Google Scholar]
- 17.Ignarro LJ. Physiology and pathophysiology of nitric oxide. Kidney Int Suppl. 1996;55:S2–S5. [PubMed] [Google Scholar]
- 18.Wink DA, Hanbauer I, Grisham MB, Laval F, Nims RW, Laval J, Cook JC, Pacelli R, Liebmann J, Krishna MC, Ford MC, JB M. The Chemical Biology of NO. Insights into Regulation, Protective and Toxic Mechanisms of Nitric Oxide. Current Topics in Cellular Regulation. 1996;34:159–187. doi: 10.1016/s0070-2137(96)80006-9. [DOI] [PubMed] [Google Scholar]
- 19.Wink DA, Mitchell JB. The Chemical Biology of Nitric Oxide: Insights into Regulatory, Cytotoxic and Cytoprotective Mechanisms of Nitric Oxide. Free Rad. Biol. Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 20.Grisham MB, Jourd'Heuil D, Wink DA. Nitric oxide. I. Physiological chemistry of nitric oxide and its metabolites:implications in inflammation. Am. J. Physiol. 1999;276:G315–G321. doi: 10.1152/ajpgi.1999.276.2.G315. [DOI] [PubMed] [Google Scholar]
- 21.Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 22.Pryor WA, Squadrito CL. The chemistry of peroxtynitrite and peroxynitrous acid: Products from the reaction of nitric oxide with superoxide. Am. J. Phys. 1996;268:L699–L721. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 23.Wink DA, Darbyshire JF, Nims RW, Saveedra JE, Ford PC. Reactions of the bioregulatory agent nitric oxide in oxygenated aquious media: determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1993;6:23–27. doi: 10.1021/tx00031a003. [DOI] [PubMed] [Google Scholar]
- 24.Wink DA, Nims RW, Darbyshire JF, Christodoulou D, Hanbauer I, Cox GW, Laval F, Laval J, Cook JA, Krishna MC, DeGraff W, Mitchell JB. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 25.Wink DA, Cook JA, Kim S, Vodovotz Y, Pacelli R, Kirshna MC, Russo A, Mitchell JB, Jourd'heuil D, Miles AM, Grisham MB. Superoxide modulates the oxidation and nitrosation of thiols by nitric oxide derived reactive intermediates. J. Biol. Chem. 1997;272:11147–11151. doi: 10.1074/jbc.272.17.11147. [DOI] [PubMed] [Google Scholar]
- 26.Lymar SV, Hurst K. Rapid reaction between peroxynitrite ion and cabon dioxide: implications for biological activity. J. Am. Chem. Soc. 1995;290:52–57. [Google Scholar]
- 27.Uppu RM, Squadrito GL, Pryor WA. Acceleration of peroxynitrite oxidants by carbon dioxide. Arch. Biochem. Biophys. 1996;327:335–343. doi: 10.1006/abbi.1996.0131. [DOI] [PubMed] [Google Scholar]
- 28.Jourd'heuil D, Miranda KM, Kim SM, Espey MG, Vodovotz Y, Laroux S, Mai CT, Miles AM, Grisham MB, Wink DA. The oxidative and nitrosative chemistry of the nitric oxide/superoxide reaction in the presence of bicarbonate. Arch Biochem Biophys. 1999;365:92–100. doi: 10.1006/abbi.1999.1143. [DOI] [PubMed] [Google Scholar]
- 29.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 30.Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YM, Bombeck CA, Billiar TR. Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 1999;84:253–256. doi: 10.1161/01.res.84.3.253. [DOI] [PubMed] [Google Scholar]
- 32.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. doi: 10.1074/jbc.272.49.31138. 2721997. [DOI] [PubMed] [Google Scholar]
- 33.Sidorkina O, Espey MG, Miranda KM, Wink DA, Laval J. Inhibition of poly(ADPRIBOSE) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic Biol Med. 2003;35:1431–1438. doi: 10.1016/j.freeradbiomed.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Gbadegesin M, Vicini S, Hewett SJ, Wink DA, Espey M, Pluta RM, Colton CA. Hypoxia modulates nitric oxide-induced regulation of NMDA receptor currents and neuronal cell death. Am. J. Physiol. 1999;277:C673–C683. doi: 10.1152/ajpcell.1999.277.4.C673. [DOI] [PubMed] [Google Scholar]
- 35.Colton CA, Gbadegesin M, Wink DA, Miranda KM, Espey MG, Vicini S. Nitroxyl anion regulation of the NMDA receptor. J Neurochem. 2001;78:1126–1134. doi: 10.1046/j.1471-4159.2001.00509.x. [DOI] [PubMed] [Google Scholar]
- 36.Lipton SA, Stamler JS. Actions of redox-related congeners of nitric oxide at the NMDA receptor. Neuropharmacology. 1994;33:1229–1233. doi: 10.1016/0028-3908(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 37.Kim WK, Choi YB, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO- Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 38.Vidwans AS, Kim S, Coffin DO, Wink DA, Hewett SJ. Analysis of the neuroprotective effects of various nitric oxide donor compounds in murine mixed cortical cell culture. J. Neurochem. 1999;72:1843–1852. doi: 10.1046/j.1471-4159.1999.0721843.x. [DOI] [PubMed] [Google Scholar]
- 39.Kotamraju S, Tampo Y, Keszler A, Chitambar CR, Joseph J, Haas AL, Kalyanaraman B. Nitric oxide inhibits H2O2-induced transferrin receptor-dependent apoptosis in endothelial cells: Role of ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 2003;100:10653–10658. doi: 10.1073/pnas.1933581100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1 alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006;281:25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 42.Prueitt RL, Boersma BJ, Howe TM, Goodman JE, Thomas DD, Ying L, Pfiester CM, Yfantis HG, Cottrell JR, Lee DH, Remaley AT, Hofseth LJ, Wink DA, Ambs S. Inflammation and IGF-I activate the Akt pathway in breast cancer. Int J Cancer. 2007;120:796–805. doi: 10.1002/ijc.22336. [DOI] [PubMed] [Google Scholar]
- 43.Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007;67:289–299. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 44.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 45.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 46.Borutaite V, Brown GC. S-nitrosothiol inhibition of mitochondrial complex I causes a reversible increase in mitochondrial hydrogen peroxide production. Biochim Biophys Acta. 2006;1757:562–566. doi: 10.1016/j.bbabio.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 47.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102:13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pervin S, Singh R, Freije WA, Chaudhuri G. MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: implications in breast cancer. Cancer Res. 2003;63:8853–8860. [PubMed] [Google Scholar]
- 49.Pervin S, Singh R, Chaudhuri G. Nitric-oxide-induced Bax integration into the mitochondrial membrane commits MDA-MB-468 cells to apoptosis: essential role of Akt. Cancer Res. 2003;63:5470–5479. [PubMed] [Google Scholar]
- 50.Chong ZZ, Li F, Maiese K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005;20:299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brahimi-Horn MC, Pouyssegur J. Harnessing the hypoxia-inducible factor in cancer and ischemic disease. Biochem Pharmacol. 2007;73:450–457. doi: 10.1016/j.bcp.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Paul SA, Simons JW, Mabjeesh NJ. HIF at the crossroads between ischemia and carcinogenesis. J Cell Physiol. 2004;200:20–30. doi: 10.1002/jcp.10479. [DOI] [PubMed] [Google Scholar]
- 53.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 54.Thomas DD, Espey MG, Pociask DA, Ridnour LA, Donzelli S, Wink DA. Asbestos redirects nitric oxide signaling through rapid catalytic conversion to nitrite. Cancer Res. 2006;66:11600–11604. doi: 10.1158/0008-5472.CAN-06-1140. [DOI] [PubMed] [Google Scholar]
- 55.DiGuiseppi J, Fridovich I. The toxicology of molecular oxygen. CRC Crit. Rev. Toxicol. 1984;12:315–342. doi: 10.3109/10408448409044213. [DOI] [PubMed] [Google Scholar]
- 56.Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 57.Wink DA, Miranda KM, MG E, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, Grisham MB. Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal. 2001;3:203–213. doi: 10.1089/152308601300185179. [DOI] [PubMed] [Google Scholar]
- 58.Brune B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid Redox Signal. 2005;7:497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 59.Espey MG, Xavier S, Thomas DD, Miranda KM, Wink DA. Direct real-time evaluation of nitration with green fluorescent protein in solution and within human cells reveals the impact of nitrogen dioxide vs. peroxynitrite mechanisms. Proc Natl Acad Sci U S A. 2002;99:3481–3486. doi: 10.1073/pnas.062604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Espey MG, Thomas DD, Miranda KM, Wink DA. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc Natl Acad Sci U S A. 2002;99:11127–11132. doi: 10.1073/pnas.152157599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turpaev K, Bouton C, Drapier JC. Nitric oxide-derived nitrosating species and gene expression in human monocytic cells. Biochemistry. 2004;43:10844–10850. doi: 10.1021/bi049831y. [DOI] [PubMed] [Google Scholar]
- 62.Vodovotz Y, Chesler L, Chong H, Kim SJ, Simpson JT, DeGraff W, Cox GW, Roberts AB, Wink DA, Barcellos-Hoff MH. Regulation of transforming growth factor beta 1 by nitric oxide. Cancer Res. 1999;59:2142–2149. [PubMed] [Google Scholar]
- 63.Zhang X. Real time and in vivo monitoring of nitric oxide by electrochemical sensors--from dream to reality. Front Biosci. 2004;9:3434–3446. doi: 10.2741/1492. [DOI] [PubMed] [Google Scholar]
- 64.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 65.Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, Quezado MM, Zurer I, Rotter V, Wink DA, Appella E, Harris CC. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc Natl Acad Sci U S A. 2003;100:143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laurent M, Lepoivre M, Tenu JP. Kinetic modelling of the nitric oxide gradient generated in vitro by adherent cells expressing inducible nitric oxide synthase. Biochem. J. 1996;314:109–113. doi: 10.1042/bj3140109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C, Trudel LJ, Wogan GN, Deen WM. Thresholds of nitric oxide-mediated toxicity in human lymphoblastoid cells. Chem Res Toxicol. 2003;16:1004–1013. doi: 10.1021/tx0340448. [DOI] [PubMed] [Google Scholar]
- 68.Espey MG, Miranda KM, Pluta RM, Wink DA. Nitrosative capacity of macrophages is dependent on nitric-oxide synthase induction signals. J. Biol. Chem. 2000;275:11341–11347. doi: 10.1074/jbc.275.15.11341. [DOI] [PubMed] [Google Scholar]
- 69.Connelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J Biol Chem. 2003;278:26480–26487. doi: 10.1074/jbc.M302238200. [DOI] [PubMed] [Google Scholar]
- 70.Ogawa R, Pacelli R, Espey MG, Miranda KM, Friedman N, Kim SM, Cox G, Mitchell JB, Wink DA, Russo A. Comparison of control of Listeria by nitric oxide redox chemistry from murine macrophages and NO donors: insights into listeriocidal activity of oxidative and nitrosative stress. Free Radic Biol Med. 2001;30:268–276. doi: 10.1016/s0891-5849(00)00470-6. [DOI] [PubMed] [Google Scholar]
- 71.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang C, Werb Z. The many faces of metalloproteases: cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001;11:S37–S43. doi: 10.1016/s0962-8924(01)02122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parks WC. Matrix metalloproteinases in repair. Wound Repair Regen. 1999;7:423–432. doi: 10.1046/j.1524-475x.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- 75.Okamoto T, Akuta T, Tamura F, van DVA, Akaike T. Molecular mechanism for activation and regulation of matrix metalloproteinases during bacterial infections and respiratory inflammation. Biol Chem. 2004;385:997–1006. doi: 10.1515/BC.2004.130. [DOI] [PubMed] [Google Scholar]
- 76.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med. 2004;37:768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 77.Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. 2004;9:43–51. doi: 10.1023/B:HREV.0000011393.40674.13. [DOI] [PubMed] [Google Scholar]
- 78.Peppin GJ, Weiss SJ. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc Natl Acad Sci U S A. 1986;83:4322–4326. doi: 10.1073/pnas.83.12.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 80.Ridnour LA, Windhausen AN, Isenberg JS, Yeung N, Thomas DD, Vitek MP, Roberts DD, Wink DA. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2007;104:16898–16903. doi: 10.1073/pnas.0702761104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J Biol Chem. 2003;278:28403–28409. doi: 10.1074/jbc.M304739200. [DOI] [PubMed] [Google Scholar]
- 82.Fu X, Kao JL, Bergt C, Kassim SY, Huq NP, d'Avignon A, Parks WC, Mecham RP, Heinecke JW. Oxidative cross-linking of tryptophan to glycine restrains matrix metalloproteinase activity: specific structural motifs control protein oxidation. J Biol Chem. 2004;279:6209–6212. doi: 10.1074/jbc.C300506200. [DOI] [PubMed] [Google Scholar]
- 83.Kassim SY, Fu X, Liles WC, Shapiro SD, Parks WC, Heinecke JW. NADPH oxidase restrains the matrix metalloproteinase activity of macrophages. J Biol Chem. 2005;280:30201–30205. doi: 10.1074/jbc.M503292200. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Rosen H, Madtes DK, Shao B, Martin TR, Heinecke J, Fu X. Myeloperoxidase Inactivates TIMP-1 by Oxidizing Its N-terminal Cysteine Residue: AN OXIDATIVE MECHANISM FOR REGULATING PROTEOLYSIS DURING INFLAMMATION. J Biol Chem. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van dVA. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marletta MA. A. Mammalian synthesis of nitrite, nitrate, nitric oxide and N-nitrosating agents. Chem. Res. Toxicol. 1988;1:249–257. doi: 10.1021/tx00005a001. [DOI] [PubMed] [Google Scholar]
- 87.Liu RH, Hotchkiss JH. Potential genotoxicity of chronically elevated nitric oxide: a review. Mutat. Res. 1995;339:73–89. doi: 10.1016/0165-1110(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 88.Zhou X, Espey MG, Chen JX, Hofseth LJ, Miranda KM, Hussain SP, Wink DA, Harris CC. Inhibitory effects of nitric oxide and nitrosative stress on dopamine-beta-hydroxylase. J. Biol. Chem. 2000;275:21241–21246. doi: 10.1074/jbc.M904498199. [DOI] [PubMed] [Google Scholar]
- 89.Hori K, Burd PR, Furuke K, Kutza J, Weih KA, Clouse KA. Human immunodeficiency virus-1-infected macrophages induce inducible nitric oxide synthase and nitric oxide (NO) production in astrocytes: astrocytic NO as a possible mediator of neural damage in acquired immunodeficiency syndrome. Blood. 1999;93:1843–1850. [PubMed] [Google Scholar]
- 90.Stadler J, Harbrecht BG, Di Silvio M, Curran RD, Jordan ML, Simmons RL, Billiar TR. Endogenous nitric oxide inhibits the synthesis of cyclooxygenase products and interleukin-6 by rat Kupffer cells. J. Leukoc. Biol. 1993;53:165–172. doi: 10.1002/jlb.53.2.165. [DOI] [PubMed] [Google Scholar]
- 91.Stadler J, Trockfeld J, Schmalix WA, Brill T, Siewert JR, Greim H, Doehmer J. Inhibition of cytochromes P4501A by nitric oxide. Proc Natl Acad Sci U S A. 1994;91:3559–3563. doi: 10.1073/pnas.91.9.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Snyder GD, Holmes RW, Bates JN, Van VBJ. Nitric oxide inhibits aromatase activity: mechanisms of action. J Steroid Biochem Mol Biol. 1996;58:63–69. doi: 10.1016/0960-0760(96)00008-8. [DOI] [PubMed] [Google Scholar]
- 93.Tunctan B, Yaghini FA, Estes A, Malik KU. Inhibition by nitric oxide of cytochrome P450 4A activity contributes to endotoxin-induced hypotension in rats. Nitric Oxide. 2006;14:51–57. doi: 10.1016/j.niox.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 94.Yaghi A, Bend JR, Webb CD, Zeldin DC, Weicker S, Mehta S, McCormack DG. Excess nitric oxide decreases cytochrome P-450 2J4 content and P-450-dependent arachidonic acid metabolism in lungs of rats with acute pneumonia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1260–L1267. doi: 10.1152/ajplung.00273.2003. [DOI] [PubMed] [Google Scholar]
- 95.Lee SH, Lee SM. Suppression of hepatic cytochrome p450-mediated drug metabolism during the late stage of sepsis in rats. Shock. 2005;23:144–149. doi: 10.1097/01.shk.0000150778.39484.54. [DOI] [PubMed] [Google Scholar]
- 96.Carcillo JA, Doughty L, Kofos D, Frye RF, Kaplan SS, Sasser H, Burckart GJ. Cytochrome P450 mediated-drug metabolism is reduced in children with sepsis-induced multiple organ failure. Intensive Care Med. 2003;29:980–984. doi: 10.1007/s00134-003-1758-3. [DOI] [PubMed] [Google Scholar]
- 97.Takemura S, Minamiyama Y, Imaoka S, Funae Y, Hirohashi K, Inoue M, Kinoshita H. Hepatic cytochrome P450 is directly inactivated by nitric oxide, not by inflammatory cytokines, in the early phase of endotoxemia. J Hepatol. 1999;30:1035–1044. doi: 10.1016/s0168-8278(99)80257-8. [DOI] [PubMed] [Google Scholar]
- 98.Muller CM, Scierka A, Stiller RL, Kim YM, Cook DR, Lancaster JRJ, Buffington CW, Watkins WD. Nitric oxide mediates hepatic cytochrome P450 dysfunction induced by endotoxin. Anesthesiology. 1996;84:1435–1442. doi: 10.1097/00000542-199606000-00020. [DOI] [PubMed] [Google Scholar]
- 99.Yu JX, Cui L, Zhang QY, Chen H, Ji P, Wei HJ, Ma HY. Expression of NOS and HIF-1 alpha in human colorectal carcinoma and implication in tumor angiogenesis. World J Gastroenterol. 2006;12:4660–4664. doi: 10.3748/wjg.v12.i29.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 1995;93:17–48. doi: 10.1016/0304-3835(95)03786-V. [DOI] [PubMed] [Google Scholar]
- 101.Wishnok JS, Tannenbaum SR, Stillwell WG, Glogowski JA, Leaf CD. Urinary markers for exposures to alkylating or nitrosating agents. Environ Health Perspect. 1993;99:155–159. doi: 10.1289/ehp.9399155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, Loscalzo J, Stamler JS. Endogenous nitrogen oxides and bronchodilator S-nitosothiols in human airways. Proc. Natl. Acad. Sci. USA. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pfeiffer S, Lass A, Schmidt K, Mayer B. Protein tyrosine nitration in mouse peritoneal macrophages activated in vitro and in vivo: evidence against an essential role of peroxynitrite. FASEB J. 2001;15:2355–2364. doi: 10.1096/fj.01-0295com. [DOI] [PubMed] [Google Scholar]
- 104.Pfeiffer S, Lass A, Schmidt K, Mayer B. Protein tyrosine nitration in cytokine-activated murine macrophages. Involvement of a peroxidase/nitrite pathway rather than peroxynitrite. J Biol Chem. 2001;276:34051–34058. doi: 10.1074/jbc.M100585200. [DOI] [PubMed] [Google Scholar]
- 105.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 106.Thomas DD, Espey MG, Vitek MP, Miranda KM, Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2- reaction. Proc Natl Acad Sci U S A. 2002;99:12691–12696. doi: 10.1073/pnas.202312699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Espey MG, Miranda KM, Thomas DD, Wink DA. Distinction between Nitrosating Mechanisms within Human Cells and Aqueous Solution. J Biol Chem. 2001;276:30085–30091. doi: 10.1074/jbc.M101723200. [DOI] [PubMed] [Google Scholar]
- 108.Ford E, Hughes MN, Wardman P. Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic Biol Med. 2002;32:1314–1323. doi: 10.1016/s0891-5849(02)00850-x. [DOI] [PubMed] [Google Scholar]
- 109.Garthwaite J. Neural nitric oxide signalling. Trends Neurosci. 1995;18:51–52. [PubMed] [Google Scholar]
- 110.Dawson TM, Dawson VL, Synder SH. A novel neuronal messenger molecule in brain: the free radical, nitric oxide. Ann. Neurol. 1992;32:297–311. doi: 10.1002/ana.410320302. [DOI] [PubMed] [Google Scholar]
- 111.Hanbauer I, Wink D, Osawa Y, Edelman GM, Gally JA. Role of nitric oxide in NMDA-evoked release of [3H]-dopamine from striatal slices. Neuroreports. 1992;3:409–412. doi: 10.1097/00001756-199205000-00008. [DOI] [PubMed] [Google Scholar]
- 112.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schindler H, Lutz MB, Rollinghoff M, Bogdan C. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J Immunol. 2001;166:3075–3082. doi: 10.4049/jimmunol.166.5.3075. [DOI] [PubMed] [Google Scholar]
- 114.Stueh rDJ. Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- 115.Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 116.Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- 117.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ghosh DK, Salerno JC. Nitric oxide synthases: domain structure and alignment in enzyme function and control. Front Biosci. 2003;8:d193–d209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- 119.Morris SMJ. Arginine metabolism: boundaries of our knowledge. J Nutr. 2007;137:1602S–1609S. doi: 10.1093/jn/137.6.1602S. [DOI] [PubMed] [Google Scholar]
- 120.Morris SMJ. Arginine metabolism in vascular biology and disease. Vasc Med. 2005;10 Suppl 1:S83–S87. doi: 10.1177/1358836X0501000112. [DOI] [PubMed] [Google Scholar]
- 121.Morris SMJ. Recent advances in arginine metabolism. Curr Opin Clin Nutr Metab Care. 2004;7:45–51. doi: 10.1097/00075197-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 122.Saenz DA, Bari SE, Salido E, Chianelli M, Rosenstein RE. Effect of nitroxyl on the hamster retinal nitridergic pathway. Neurochem Int. 2007;51:424–432. doi: 10.1016/j.neuint.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 123.Vasquez-Vivar J, Hogg N, Martasek P, Karoui H, Pritchard KAJ, Kalyanaraman B. Tetrahydrobiopterin-dependent inhibition of superoxide generation from neuronal nitric oxide synthase. J Biol Chem. 1999;274:26736–26742. doi: 10.1074/jbc.274.38.26736. [DOI] [PubMed] [Google Scholar]
- 124.Pou S, Keaton L, Surichamorn W, Rosen GW. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem. 1999;274:9573–9580. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 125.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KAJ. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl. 2003;4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 127.Cardounel AJ, Xia Y, Zweier JL. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetrahydrobiopterin. J Biol Chem. 2005;280:7540–7549. doi: 10.1074/jbc.M410241200. [DOI] [PubMed] [Google Scholar]
- 128.Buerk DG. Nitric oxide regulation of microvascular oxygen. Antioxid Redox Signal. 2007;9:829–843. doi: 10.1089/ars.2007.1551. [DOI] [PubMed] [Google Scholar]
- 129.Mason MG, Nicholls P, Wilson MT, Cooper CE. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:708–713. doi: 10.1073/pnas.0506562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abu-Soud HM, Rousseau DL, Stuehr DJ. Nitric oxide binding to the heme of neuronal nitric-oxide synthase links its activity to changes in oxygen tension. J. Biol. Chem. 1996;271:32515–32518. doi: 10.1074/jbc.271.51.32515. [DOI] [PubMed] [Google Scholar]
- 131.Thomas DD, Liu X, Kantrow SP, Lancaster JR. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA. 2001;98:355–360. doi: 10.1073/pnas.011379598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu X, Miller MJS, Joshi MS, Thomas DD, Lancaster JRJ. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vodovotz Y, Kwon N, Popischil M, Manning J, Paik J, Nathan C. Inactivation of nitric oxide synthase following prolonged incubation of mouse macrophages with interferon-gamma and bacterial lipopolysaccharide. J. Immunol. 1994;152:4110–4118. [PubMed] [Google Scholar]
- 134.Chen X, Buerk DG, Barbee KA, Jaron DA. A model of NO/O2 transport in capillary-perfused tissue containing an arteriole and venule pair. Ann Biomed Eng. 2007;35:517–529. doi: 10.1007/s10439-006-9236-z. [DOI] [PubMed] [Google Scholar]
- 135.Lancaster J. Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc. Natl. Acad. Sci. USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vukosavljevic N, Jaron D, Barbee KA, Buerk DG. Quantifying the L-arginine paradox in vivo. Microvasc Res. 2006;71:48–54. doi: 10.1016/j.mvr.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 137.Vaughn MW, Kuo L, Liao JC. Effective diffusion distance of nitric oxide in the microcirculation. Am J Physiol. 1998;274:H1705–H1714. doi: 10.1152/ajpheart.1998.274.5.H1705. [DOI] [PubMed] [Google Scholar]
- 138.Vaughn MW, Kuo L, Liao JC. Estimation of nitric oxide production and reaction rates in tissue by use of a mathematical model. Am J Physiol. 1998;274:H2163–H2176. doi: 10.1152/ajpheart.1998.274.6.H2163. [DOI] [PubMed] [Google Scholar]
- 139.Lamkin-Kennard KA, Buerk DG, Jaron D. Interactions between NO and O2 in the microcirculation: a mathematical analysis. Microvasc Res. 2004;68:38–50. doi: 10.1016/j.mvr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 140.Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, Bailey SM, Darley-Usmar VM. Nitroxia: the pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med. 2005;38:297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 141.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 142.Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci U S A. 2003;100:14766–14771. doi: 10.1073/pnas.2435008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 145.Isenberg JS, Wink DA, Roberts DD. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc Res. 2006;71:785–793. doi: 10.1016/j.cardiores.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 146.Kubes P, Kanwar S, Niu XF, Gaboury JP. Nitric oxide inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB, J. 1993;264:G143–G149. doi: 10.1096/fasebj.7.13.8405815. [DOI] [PubMed] [Google Scholar]
- 147.Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain-induced reactive oxygen species involved in ICAM-1 gene induction in endothelial cells. Hypertension. 1998;31:125–130. doi: 10.1161/01.hyp.31.1.125. [DOI] [PubMed] [Google Scholar]
- 148.Tsao PS, Wang B, Buitrago R, Shyy JY, Cooke JP. nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96:934–940. doi: 10.1161/01.cir.96.3.934. [DOI] [PubMed] [Google Scholar]
- 149.Koesling D, Russwurm M, Mergia E, Mullershausen F, Friebe A. Nitric oxide-sensitive guanylyl cyclase: structure and regulation. Neurochem Int. 2004;45:813–819. doi: 10.1016/j.neuint.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 150.Koppenol WH. Chemistry of peroxynitrite and its relevance to biological systems. Met Ions Biol Syst. 597-6191999. [PubMed] [Google Scholar]
- 151.Lancaster JRJ. Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chem Res Toxicol. 2006;19:1160–1174. doi: 10.1021/tx060061w. [DOI] [PubMed] [Google Scholar]
- 152.Ford PC, Wink DA, Stanbury DM. Autooxidation kinetics of aqueous nitric oxide. FEBS Lett. 1993;326:1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- 153.Moller MN, Li Q, Vitturi DA, Robinson JM, Lancaster JRJ, Denicola A. Membrane "lens" effect: focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem Res Toxicol. 2007;20:709–714. doi: 10.1021/tx700010h. [DOI] [PubMed] [Google Scholar]
- 154.Schwartz SE, White WH. Trace Atmospheric Constituents. Properties, Transformation and Fates. 1983. pp. 1–117. [Google Scholar]
- 155.Wink DA. Ion implicated in blood pact. Nat Med. 2003;9:1460–1461. doi: 10.1038/nm1203-1460. [DOI] [PubMed] [Google Scholar]
- 156.Radi R, Beckman JS, Bush KM, Freeman BA. Peoxynitrite oxidation of sulfhydryls: The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]