

Abstract
Apoptolidins A−D are microbial secondary metabolites shown to be selectively cytotoxic against several cancer cell lines and noncytotoxic against normal cells. Total syntheses of apoptolidinones A and D are reported. The efficient synthetic strategy leading to the apoptolidinones features construction of the common 20-membered macrolactone by an intramolecular Suzuki reaction and stereocontrolled aldol reactions establishing the C19/C20 and C22/C23 stereocenters. In contrast to apoptolidin A, the aglycones apoptolidinone A and D were shown to be noncytotoxic when evaluated against human lung cancer cells (H292).
Introduction
Small molecule natural products provide opportunities for scientific advances in chemistry and biology through their study. An excellent example of this line of investigation is the discovery and study of cell-specific apoptosis-inducing natural products. In the 1990s Seto and Hayakawa initiated a cell-based screening program aimed at the identification of new genotype-selective cytotoxic agents from microbial extracts.(1) In 1993 the Seto group reported the isolation of apoptolidin, a 20-membered macrolide found to induce apoptosis in rat glia cells transformed with the adenovirus oncogene and produced by Nocardiopsis sp. (ca. 200 mg/2 L of fermentation).(2) Subsequently several minor apoptolidin congeners were isolated from the same microorganism by Wender’s group and given the names apoptolidin B−D with apoptolidin adopting the name apoptolidin A (Figure 1).(3) Apoptolidins A−D were reported to inhibit growth of H292 cancer cells (lung carcinoma) in the submicromolar range, with apoptolidin B showing the greatest activity (GI50 = 7 ± 4 nM).
Figure 1.

Structures of apoptolidins and isoapoptolidins.
Not surprisingly, the reported cell-selective induction of apoptosis by apoptolidin A stimulated investigations on several research fronts in order to better understand the reported cell-selective cytotoxicity. Khosla and co-workers determined that apoptolidin A and structurally related cytotoxic macrolides ossamycin and cytovaricin inhibit mitochondrial F0F1-ATPase, identifying this enzyme as a promising target for the development of cell-selective anticancer agents.(4) The apoptolidins have proven to be rather labile compounds easily subjected to a base-induced acyl migration from the C19 to C20 hydroxyl group to produce isoapoptolidins (Figure 1),(5) compounds possessing diminished activity against mitochondrial F0F1-ATPase. The molecular complexity and instability of apoptolidin A has led Wender and co-workers to search for semisynthetic analogs possessing superior stability and/or pharmacokinetic properties.(6) Comparison of the enzyme inhibition of mitochondrial F0F1-ATPase to antiproliferative activity in E1A-transformed rat fibroblasts of apoptolidin A derived analogs suggested either the existence of a secondary biological target or a more complex mode of action.
The complex molecular architecture and novel cytotoxic profile of apoptolidin A has stimulated considerable interest from the synthetic community.(7) Total syntheses of the fully glycosylated ensemble, apoptolidin A, have been reported by the groups of Nicolaou and Koert.(8) Several groups including our own have described the synthesis of apoptolidinone (A).(9) We describe herein the total synthesis of apoptolidinones A and D and evaluation of their antiproliferative properties.
Results and Discussion
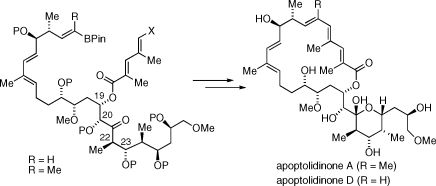
An unsaturated 20-membered macrolactone conjugated to a stereochemically rich pyran ring constitutes the gross structural features of apoptolidinone A/D, reminiscent of polyketides such as the bafilomycin macrolides.(10) We anticipated macrolide I (Scheme 1) would serve as a logical synthetic precursor to apoptolidinone. However, because of the tendency of apoptolidin to ring expand to iso-apoptolidin under basic conditions, proper choice of protecting groups was considered of paramount importance. As we gathered information from published studies and work from our own group, we ultimately identified triethylsily (P = TES) groups as the protecting groups of choice. Disconnection of the macrolide at C5−C6 which relied on introduction of a vinyl boronate by a cross-metathesis reaction allowed, in principle, synthetic access to apoptolidinones A (R = Me) and D (R = H). Disconnection at C11−C12 and application of two stereoselective aldol retrons leads to the identification of fragments II−VI as key building blocks for the assembly of apoptolidinone A/D.
Scheme 1.

The C23−C28 fragment (Scheme 1, IV) was derived by a Crimmins syn aldol reaction between the chlorotitanium enolate derived from oxazolidinethione 9 and aldehyde 4 (Scheme 2).(11) The latter was easily prepared from (R)-glycidyl methyl ether (1) starting with epoxide ring opening with the carbanion derived from 1,3-dithiane followed by alcohol protection and dithiane hydrolysis. Reduction of the TES protected aldol 6 with lithium borohydride led to alcohol 7, which on oxidation under Swern conditions gave aldehyde 8.
Scheme 2. Synthesis of Fragment 8.

Reagents and conditions: (a) n-BuLi, 1,3-dithiane, THF, −40 °C, 95%; (b) TBSCl, ImH, DMAP, DMF, 0 °C, 95%; (c) MeI (xs), K2CO3, MeCN/water (10:1), 40 °C, 90%; (d) 9, TiCl4, 0 °C then (−)-sparteine, −78 to 0 °C, CH2Cl2, 95%; (e) TESCl, ImH, DMF, 0 °C, 95%; (f) LiBH4, MeOH, 0 °C, 90%; (g) (COCl)2, DMSO, Et3N, −78 °C, 88%.
Our synthetic strategy (Scheme 1) projected the equivalent of a C20−C22 fragment (VI) to conjoin aldehydes IV and V through two regio- and stereoselective aldol reactions by one of two coupling orders (VI + IV + V and VI + V + IV). To this end we developed reaction conditions that would allow access to either isomeric silyl enol ethers 12 or 13 starting from TES protected 1-hydroxy-2-butanone 11 (Scheme 3).(12) Deprotonation of 11 under kinetic conditions (LDA, THF, −78 °C) followed by the addition of TMSCl afforded a 15:73:12 mixture of isomers (Z)-12, (E)-12, and (Z)-13 (Scheme 3).13,14 Thermodynamic conditions (TMSOTf, Et3N, THF, 0 °C) afforded (Z)-13 as the exclusive isomer as determined by GC analysis.
Scheme 3. Synthesis of Silyl Enol Ethers 12 and 13.

Reagents and conditions: (a) TESCl, ImH, CH2Cl2, 0 °C, 77%; (b) LDA, THF, −78 °C then TMSCl, 80%; (c) TMSOTf, Et3N, THF, 0 °C, 92%.
The C6−C11 fragment that incorporates two stereocenters (C8 and C9) was to be introduced in the form a vinyl boronate (II, Scheme 1). To this end, we examined the asymmetric crotylation of 3-iodoacrolein (14)(15) and 3-borylacrolein (15)(16) employing either Roush’s (18)(17) or Brown’s (19) syn selective crotylating agent (Scheme 4).(18) As reported earlier, addition of the (Z)-crotylboronate reagent 18 to the pinacol ester of 3-boronoacrolein afforded, following in situ silylation, syn homoallylic ether 17 with modest enantioselectivity (80% ee) and yield (40−43%).(7g) The addition of 18 to 3-iodoacrolein proceeded with poor asymmetric induction (26% ee), whereas addition of the Brown reagent (19) to 14 gave 16 in high enantiomeric excess (90% ee) and good yield (67%). Vinyl iodide 16 was readily converted to vinyl boronate 17 by lithium−halogen exchange followed by a boronate quench.
Scheme 4.

Construction of the C12−C19 fragment (V, Scheme 1) started from lactone 20, readily derived from (S)-malic acid(19) and converted to 3-methoxy-γ-butyrolactone (21) (Scheme 5).(20) Reduction of lactone 21 with DIBAL-H afforded lactol 22, which on condensation with 1,3-propanedithiol afforded dithiane 23. Swern oxidation of primary alcohol 23 provided aldehyde 24 in near quantitative yield. A five-carbon unit was introduced to aldehyde 24 by chelation-controlled addition of the Grignard reagent derived from bromide 25, prepared from dihydrofuran according to Kocienski’s procedure as described by Koert and co-workers in their reported synthesis of apoptolidinone.9b,21 A dichloromethane solution of secondary alcohol 26 was treated sequentially with iodine followed by imidazole and triethylchlorosilane in one pot to provide vinyl iodide 28 in 89−96% yield. Dithiane hydrolysis of 28 was efficiently accomplished using the Fetizon−Jurion procedure to provide aldehyde 29 in 64−74% yield.(22) With fragments II−VI available, we turned our attention to their coupling to complete the synthesis of apoptolidinone A (Scheme 6).
Scheme 5. Synthesis of Fragment 29.

Reagents and conditions: (a) Ag2O, MeI (solvent) 86%; (b) DIBAL-H, THF, −78 °C; (c) 1,3-propanedithiol, BF3·OEt2, CH2Cl2, 28 °C, 86% from lactone 21; (d) (COCl)2, DMSO, i-Pr2NEt, −78 °C, 98%; (e) 25, Mg, 1,2-dibromoethane, Et2O, −78 °C, 85%; (f) TESCl, ImH, CH2Cl2, 28 °C; (g) I2, CH2Cl2, 0 °C, 90% from 26; (h) MeI (xs), K2CO3, MeCN/pH 7 buffer (4:1), 28 °C, 92%.
Scheme 6. Synthesis of Apoptolidinone A.

Reagents and conditions: (a) 17, Pd(Ph3P)4, Tl(OEt), THF/H2O (3:1), 28 °C, 86%; (b) 12, BF3·OEt2, CaH2, CH2Cl2, −94 °C, 72%; (c) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, PhMe, −78 °C, 71%; (d) LHMDS, THF, HMPA, −78 °C, 2 h then 8, THF, 48%; (e) TESOTf, 2,6-lutidine, CH2Cl2, 0 °C, 97%; (f) 34, isopropenyl pinacol boronic ester (5 equiv), PhMe, 50 °C, 46 h, 22%; (g) Pd(Ph3P)4, Tl(OEt), THF/H2O (3:1), 28 °C, 30 min, 84%; (h) HF·pyridine, THF, −10 to 10 °C, 48h, 94%.
Initially, Suzuki−Miyaura cross-coupling of aldehyde 29 and vinyl boronate 17 was accompanied by elimination of the β-methoxy group of aldehyde 30 to afford the corresponding α,β-unsaturated aldehyde.(23) However optimization of coupling conditions [Tl(OEt), Pd(PPh3)4, THF(aq)] and reaction time (15 min) eventually led to an observed 70% yield of 30 without any observed β elimination.(24) We next turned our attention to investigating two key stereocontrolled aldol reactions; leading to the formation of the C(19)−C(20) and C(22)−C(23) carbon−carbon bonds. Mukaiyama aldol reaction between aldehyde 30 and an 8:1 mixture of silyl enol ether 12 and 13 afforded anti,syn-31a as the major isomer (31a−d: 12:3.8:1.6:1) (Figure 2).(25) The assigned C(19)−C(20) syn relative stereochemistry rested on the observed coupling constant of the aldol product (J19,20 = 3.5 Hz) and the proclivity for the formation of syn aldol products in the Mukaiyama aldol reaction of silyl enol ethers (regardless of double bond geometry) with chiral aldehydes.(26) The assigned C(17)−C(19) anti configuration rested on the well-established 1,3-asymmetric induction model proposed by Evans and co-workers in the mid-1990s.(27) The second major isomer was assigned the syn,syn (31b) relative stereochemistry (J19,20 = 3.9 Hz) (Figure 2). The minor aldol products were assigned the structures of anti,anti-31c and syn,anti-31d (J19,20 = 4.7 and 4.2 Hz), although the assignment of the C(17)−C(19) relative configuration remained ambiguous. The carbonyl resonances in the 13C NMR of 31c and 31d were not observed due to the trace amount of material isolated. Yamaguchi esterification of anti,syn-31 with carboxylic acid 32(28) led to isolation of dienoate 33 in 83% yield.(29) We anticipated the matched double stereodifferentiating aldol reaction between a metal enolate derived from ketone 33 and aldehyde 8 to proceed with high stereoselectivity.(30) In the event kinetic deprotonation (LHMDS, HMPA, THF, −78 °C) of 33 followed by aldol condensation with aldehyde 8 afforded syn aldol product 35 as a single isomer in 48% yield. Following silylation of 35 a dichloromethane solution of alkene 36, isopropenyl boronate, and Grubbs second-generation catalyst (34) held at reflux for 5 h afforded a 30% yield of vinylboronate 37 that was judged by 1H NMR spectroscopy analysis to be a single geometric isomer. However, analysis by HPLC revealed a significant quantity of an isomeric byproduct as determined by further 1H NMR analysis.(31) Employing the same catalyst in toluene heated to 50 °C resulted in a 22% yield of 37 and significant reduction of the isomeric byproduct.(32) Vinylboronate 37 derived by the latter procedure subjected to an intramolecular Suzuki−Miyaura reaction delivered macrolactone 38 in a consistent 84% yield. Finally, desilylation of 38 provided apoptolidinone A in 94% yield.
Figure 2.

Aldol stereoisomers derived from Mukaiyama aldol between aldehyde 30 and silylenol ether 12.
A synthesis of apoptolidinone D required only a change in the cross-metathesis partner of alkene 36 from isopropenyl boronic ester to 1-propenyl boronic ester. However, we decided to probe the assembly of alkene 36 by an alternate order of aldol coupling (cf. Scheme 1, VI + V + IV). To this end, we examined the addition of silyl enol ether (Z)-13 to syn substituted aldehyde 8 (Scheme 7). As pointed out by Evans, syn substituted aldols are stereochemically nonreinforcing substrates where the C24 substitutent of 8 favors a Felkin while the C25 a non-Felkin addition.26,33 Literature precedent suggested the stereoselectivity of the addition of (Z)-13 to 8 could not be predicted with confidence. In the event, we were surprised to observe treatment of a solution of 8 and (Z)-13 with borontrifluoride etherate led not to a Mukaiyama aldol reaction but instead silyl enol ether 39 as a single stereoisomer, the product of a heteroene reaction.(34) Stereochemical assignment of C22 and C23 was based on NMR analysis of pyran 40, derived from 39 by exhaustive desilylation (p-TSA, MeOH, 28 °C, 69%). The NOESY spectrum of 40 revealed NOEs from H-23 to 22-Me and 24-Me and H-23 to H-27.(35) This observation combined with the large coupling constants (J22−23 = 9.9 Hz and J23−24 = 9.9 Hz) led to the assigned relative stereochemistry shown, indicating a Felkin addition to 8, the undesired stereoselectivity.
Scheme 7.

Scheme 8. Synthesis of Apoptolidinone D.

Reagents and conditions: (a) 34, 1-propenyl pinacol boronic ester (10 equiv), PhMe, 80 °C, 6 h then 40 °C, 24 h, 53%; (b) Pd(Ph3P)4, Tl(OEt), THF/H2O (3:1), 28 °C, 15 min, 95%; (c) HF·pyridine, THF, −10 to 10 °C, 48 h, 87%.
Apoptolidinone D was completed starting with a cross-metathesis between 36 and 1-propenyl pinacol boronic ester promoted by Grubbs second generation catalyst to deliver vinyl boronate 41 in superior yield and stereoselectivity relative to the same reaction with isopropenyl boronate leading to apoptolidinone A. Macrocyclization of 41 under the previously described Suzuki conditions gave 42 in 95% yield. Desilylation of 42 provided apoptolidinone D in 87% yield.
Wender’s group has reported GI50ʼs for apoptolidin A (32 nM), apoptolidin B (7 nM), apoptolidin C (24 nM), and apoptolidin D (110 nM) against H292 (human lung carcinoma) cells.3,36 The effect reported by Wender’s group was cytostasis observed over a 2−3 day (ca. 72 h) period. We observed that treatment of H292 lung carcinoma cells with apoptolidin A results in growth arrest. In addition, however, extended maintenance of cells in the presence of apoptolidin A results in a delayed toxicity not previously reported. The effect is drastic, resulting in >95% cell death after 7 days in culture with apoptolidin A concentrations as low as 30 nM (Figure 3). In contrast, the synthetic aglycones apoptolidonone A and apoptolidinone D were inactive in the assay, inhibiting neither cell growth nor viability at the concentrations tested.
Figure 3.

Toxicity of apoptolidin A and aglycones in H292 human lung carcinoma cells. (a) Cells were plated at a density of 500/well in 96-well plates and treated for 7 days with apoptolidin A, apoptolidinone A, and apoptolidinone D at concentrations from 3 nM to 10 μM. Viability was measured by loading cells with 2 μM Calcein-AM and reading plates using a Spectramax (Molecular Dynamics) plate reader; λabs = 494, λem = 517. (b) Effective concentration 50 (EC50) values for apoptolidin A, apoptolidinone A, and apoptolidinone D. (c) Bright field photomicrographs of H292 cells cultured for 7 days in the presence of either 10 or 30 nM apoptolidin A.
Conclusions
In conclusion, we have developed a convergent synthesis of apoptolidinones A and D. Evaluation of the cytotoxicity of these aglycones using human lung cancer cells (H292) confirms the importance of the sugars of apoptolidin in expressing overall cytotoxicity as reported by Koert and co-workers.(8e) Current efforts are aimed at the development of synthetic probes aimed at interrogating the cellular target(s) of the apoptolidins.
Experimental Section
Aldol 31
To a solution of diene 30 (0.40 g, 0.76 mmol) and silyl enol ethers (7:1, 12/13; 0.83 g, 3.03 mmol, 4 equiv) in dichloromethane (32 mL) at 0 °C was added CaH2 (64 mg, 1.52 mmol, 2 equiv). After 15 min, the reaction was cooled to −94 °C and BF3·OEt2 (105 μL, 1.52 mmol, 2 equiv) was added. The reaction mixture was stirred for 45 min at −94 °C and quenched with saturated NaHCO3 (25 mL), and the aqueous layer was extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (hexanes/EtOAc, 20:1−15:1) to afford 0.39 g (72%) of alcohol 31a−d as a 12:3.8:1.6:1 mixture of diastereoisomers (by HPLC, 21.4 mm × 25 cm column, 49 min gradient, 0−10% ethyl acetate in hexanes). For synthetic purposes the mixture can be advanced to the next step without further purification, as the resulting mixture of Yamaguchi esterification products can be easily separated to afford the desired ester 33.
Data for 31a
tR = 34.25 min; [α]25D −9.1° (c 2.1, CHCl3); IR (neat) 3505, 2953, 2873, 1724, 1462, 1418, 1244, 1091, 1018 cm−1; 1H NMR (500 MHz C6D6) δ 6.25 (d, J = 16.0 Hz, 1H), 6.05−5.98 (m, 1H), 5.63 (dd, J = 16.0, 7.5 Hz, 1H), 5.55 (t, J = 7.0 Hz, 1H), 5.08−5.03 (m, 2H), 4.24−4.18 (m, 1H), 4.07 (t, J = 7.5 Hz, 1H), 4.04 (d, J = 4.0 Hz, 1H). 3.99−3.95 (m, 1H), 3.66 (ddd. J = 10.0, 4.0, 2.0 Hz, 1H), 3.30 (s, 3H), 2.61 (d, J = 9.0 Hz, 1H), 2.53−2.38 (m, 4H), 2.25−2.16 (m, 1H), 1.95−1.89 (m, 1H), 1.88−1.82 (m, 1H), 1.76 (s, 3H), 1.66−1.60 (m, 1H), 1.60−1.52 (m, 1H), 1.11 (d, J = 6.5 Hz, 3H), 1.05−0.99 (m, 21H), 0.91 (t, J = 8.0 Hz, 9H), 0.68−0.62 (m, 12 H), 0.52 (q, J = 8.0 Hz, 6H); 13C NMR (125 MHz, C6D6) δ 213.3, 141.3, 135.7, 133.7, 132.5, 128.6, 114.2, 81.7, 80.8, 78.1, 71.6, 70.6, 58.2, 45.2, 33.5, 32.2, 31.7, 25.3, 15.0, 12.6, 7.3, 7.1, 7.1, 6.9, 5.4, 5.4, 5.0; HRMS (MALDI) m/z 749.5078 [(M + Na)+ calcd for C39H78O6Si3Na, 749.5004].
Data for 31b
tR = 31.75 min; [α]25D −2.6° (c 0.16, CHCl3); IR (neat) 3498, 2955, 2877, 1716, 1459, 1414, 1239, 1083, 1006, 966, 845, 677 cm-1; 1H NMR (500 MHz C6D6) δ 6.27 (dd, J = 15.5, 4.5 Hz, 1H), 6.07−5.98 (m, 1H), 5.65 (dd, J = 15.5, 7 Hz, 1H), 5.56−5.50 (m, 1H), 5.10−5.04 (m, 2H), 4.25−4.19 (m, 1H), 4.17 (d, J = 4 Hz, 1H), 4.09 (t, J = 6.2 Hz, 1H), 3.93−3.88 (m, 1H), 3.44−3.39 (m, 1H), 3.30 (d, J = 4.5 Hz, 1H), 3.09 (s, 3H), 2.73−2−58 (m, 2H), 2.45−2.32 (m, 2H), 2.24−2.14 (m, 1H), 2.04 (d, J = 15 Hz, 1H), 1.83−1.73 (m, 2H), 1.76 (s, 3H), 1.59−1.52 (m, 1H), 1.15−1.09 (m, 6H), 1.07−0.94 (m, 27H), 0.69−0.56 (m, 18H); 13C NMR (125 MHz, C6D6) δ 211.9, 141.4, 135.7, 133.8, 132.4, 128.8, 114.3, 84.5, 81.9, 78.1, 73.8, 71.6, 57.1, 45.2, 32.9, 31.9, 31.8, 25.2, 15.1, 12.7, 7.4, 7.2, 7.0, 5.5, 5.4, 5.2; HRMS (MALDI) m/z 749.5020 [(M + Na)+ calcd for C39H78O6Si3Na, 749.5004].
Data for 31c
tR = 32.80 min; [α]25D −9.3° (c 0.08, CHCl3); IR (neat) 3470, 3400, 2955, 2913, 2878, 2361, 2337, 1714, 1459, 1414, 1377, 1238, 1096, 1008, 968, 912, 844, 741, 671, 558, 522 cm−1; 1H NMR (500 MHz C6D6) δ 6.26 (d, J = 19.5 Hz, 1H), 6.08−5.97 (m, 1H), 5.65 (dd, J = 19.5, 9.2 Hz, 1H), 5.58−5.52 (m, 1H), 5.10−5.04 (m, 2H), 4.25−4.20 (m, 1H), 4.14 (d, J = 5.8 Hz, 1H), 4.09 (t, J = 7.9 Hz, 1H), 4.00−3.95 (m, 1H), 3.60−3.57 (m, 1H), 3.27 (s, 3H), 3.01 (d, J = 5 Hz, 1H), 2.62−2.47 (m, 2H), 2.45−2.32 (m, 2H), 2.27−2.15 (m, 1H), 2.00−1.92 (m, 1H), 1.77 (s, 3H), 1.65−1.55 (m, 1H), 1.13 (d, J = 8.6 Hz, 3H), 1.08−1.00 (m, 18H), 0.95 (t, J = 10.0 Hz, 9H), 0.66 (q, J = 9.8 Hz, 12H), 0.58 (q, J = 9.9 Hz, 6H);13C NMR (150 MHz, C6D6) δ 141.4, 135.8, 133.8, 132.5, 128.7, 114.2, 84.5, 82.4, 81.9, 78.1, 73.9, 72.4, 71.6, 71.1, 58.0, 57.1, 45.3, 32.9, 32.5, 32.0, 31.9, 25.2, 15.1, 12.7, 7.4, 7.2, 7.0, 6.9, 5.6, 5.5, 5.4, 5.2, 5.1; HRMS (MALDI) m/z 749.5038 [(M + Na)+ calcd for C39H78O6Si3Na, 749.5004].
Data for 31d
tR = 36.80 min; [α]25D −50.0° (c 0.01, CHCl3); IR (neat) 2955, 2878, 1713, 1459, 1414, 1379, 1239, 1105, 1007, 968, 912, 845, 741, 583, 553, 525 cm−1; 1H NMR (500 MHz C6D6) δ 6.30 (d, J = 19.6 Hz, 1H), 6.08−5.98 (m, 1H), 5.67 (dd, J = 19.5, 9.1 Hz, 1H), 5.56−5.50 (m, 1H), 5.11−5.04 (m, 2H), 4.30−4.17 (m, 1H), 4.11 (t, J = 7.9 Hz, 1H), 4.07 (d, J = 5.2 Hz, 1H), 3.94−3.89 (m, 1H), 3.48 (d, J = 12.6 Hz, 1H), 3.29 (s, 3H), 2.55−2.32 (m, 4H), 2.22−2.12 (m, 1H), 1.91−1.85 (m, 1H), 1.78 (s, 3H), 1.71−1.59 (m, 2H), 1.51−1.42 (m, 1H), 1.13 (d, J = 8.5 Hz, 3H), 1.10−1.00 (m, 18H), 0.94 (t, J = 9.9 Hz, 9H), 0.75−0.62 (m, 12H), 0.56 (q, J = 9.9 Hz, 6H); 13C NMR (150 MHz, C6D6) δ 141.4, 135.7, 133.8, 132.2, 128.9, 114.3, 82.1, 81.5, 78.1, 72.9, 70.7, 57.9, 45.3, 34.2, 34.0, 32.1, 25.3, 15.1, 12.7, 7.3, 7.2, 7.1, 7.0, 5.6, 5.5, 5.0; HRMS (MALDI) m/z 749.5030 [(M + Na)+ calcd for C39H78O6Si3Na, 749.5004].
Macrolactone 38
To a solution of vinyl boronate 37 (5 mg, 3.1 μmol) in THF/H2O (4 mL, 3:1, degassed) was added Pd(Ph3P)4 (0.7 mg, 0.62 μmol, 0.2 equiv). The resulting yellow solution was stirred for 5 min before TlOEt (0.3 μL, 4.6 μmol, 1.5 equiv) was added. The solution was stirred for 15 min (color turned from yellow to gray). The reaction was quenched with saturated NaHCO3 (5 mL), and the aqueous layer was extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. The residue was purified by flash chromatography (hexanes/EtOAc, 25:1) to afford 3.5 mg (84%) of lactone 38 as a colorless oil: [α]25D +17.5° (c 0.32, CHCl3); IR (neat) 3497, 3382, 2954, 2915, 2877, 1700, 1459, 1241, 1111, 1081, 968, 835, 781, 741; 1H NMR (500 MHz, C6D6) δ 7.49 (s, 1H), 6.26 (s, 1H), 6.06 (d, J = 16.0 Hz, 1H), 5.71−5.67 (m, 1H), 5.57 (t, J = 8.0 Hz, 1H), 5.38 (dd, J = 16.0, 9.0 Hz, 1H), 5.16 (d, J = 10.0 Hz, 1H), 5.00 (d, J = 4.0 Hz, 1H), 4.31−4.28 (m, 1H), 4.24 (dd, J = 9.5, 1.0 Hz, 1H), 3.94−3.89 (m, 1H), 3.82 (t, J = 8.5 Hz, 1H), 3.60 (dt, J = 8.5, 2.5 Hz, 1H), 3.52−3.44 (m, 1H), 2.48 (s, 3H), 3.39−3.36 (m, 2H), 3.16 (s, 3H), 3.02 (t, J = 8.5 Hz, 1H), 2.61−2.56 (m, 1H), 2.53−2.46 (m, 1H), 2.16−1.90 (m, 4H), 2.07 (s, 3H), 1.82 (s, 3H), 1.80−1.74 (m, 1H), 1.63 (d, J = 1.0 Hz, 3H), 1.59 (s, 3H), 1.41 (d, J = 7.0 Hz, 3H), 1.36−1.26 (m, 6H), 1.21 (d, J = 7.0 Hz, 3H), 1.17−1.13 (m, 21H), 1.10 (d, J = 7.0 Hz, 3H), 1.08−1.02 (m, 18 H), 1.00 (s, 9H), 0.97−0.80 (m, 18 H), 0.75−0.64 (m, 15 H), 0.18 (s, 3H), 0.16 (s, 3H); 13C NMR (125 MHz, C6D6) δ 209.8, 168.4, 146.2, 145.5, 141.5, 136.4, 133.3, 132.3, 132.2, 131.8, 129.3, 123.4, 82.1, 81.1, 77.1, 76.2, 73.9, 73.1, 70.4, 70.0, 61.0, 58.7, 47.3, 43.6, 41.0, 40.8, 36.2, 34.9, 30.1, 26.1, 24.8, 18.3, 17.9, 17.4, 16.3, 14.0, 11.9, 11.1, 10.3, 7.5, 7.4, 7.3, 7.2, 7.1, 6.6, 5.9, 5.8, 5.6, 5.4, −4.0, −4.5; HRMS (ESI) m/z 1371.9600 [(M + Li)+ calcd for C73H144O11Si6Li, 1371.9484].
Apoptolidinone A
To solution of 6.4 mg (4.7 μMol) macrolactone 38 in 2 mL of THF at −10 °C was added HF-Py (0.1 mL) dropwise. After 1 h the temperature was raised to 0 °C and after an additional 12 h to 10 °C. After 36 h the reaction mixture was diluted with Et2O, washed with water, and concentrated in vacuo, leaving ca. 2 mL of Et2O. (It was noticed that drying over MgSO4 significantly decreased the amount of product in the solution.) The residue was purified by flash chromatography (CH2Cl2/MeOH, 15:1) to afford 3 mg (94%) of apoptolidinone A as a white solid: [α]25D +68°(c 0.15, CHCl3); IR (neat) 3395, 2925, 1664, 1596, 1390, 1258, 1095, 1023, 968, 754, 710 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.37 (s, 1H), 6.19 (s, 1H), 6.10 (d, J = 16.0 Hz, 1H), 5.64 (dd, J = 9.0, 7.0 Hz, 1H), 5.33 (dd, J = 15.5, 8.5 Hz, 1H), 5.32−5.29 (m, 1H), 5.22 (d, J = 10.5 Hz, 1H), 4.09 (ddd, J = 9.0, 3.5, 2.5 Hz, 1H), 3.77 (app t, J = 9.5 Hz, 1H), 3.74 (dd, J = 11.0, 5.0 Hz, 1H), 3.53−3.58 (m, 2H), 3.45−3.42 (m, 1H), 3.36 (s, 3H), 3.30 (s, 3H), 3.23−3.15 (m, 2H), 2.73 (dd, J = 10.0, 4.5 Hz, 1H), 2.52−2.41 (m, 2H), 2.19 (s, 3H), 2.18−2.13 (m, 1H), 2.11 (s, 3H), 2.09−2.02 (m, 2H), 1.92 (d, J = 1.0 Hz, 3H), 1.78−1.72 (m, 2H), 1.67 (s, 3H), 1.60−1.51 (m, 2H), 1.44−1.37 (m, 1H), 1.32−1.26 (m, 1H), 1.13 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 172.7, 149.1, 147.3, 143.7, 137.7, 134.9, 133.1, 133.0, 132.6, 129.6, 123.8, 101.3, 83.8, 80.6, 78.6, 75.5, 74.6, 73.7, 72.3, 69.2, 68.1, 61.4, 59.4, 41.0, 40.8, 38.6, 38.4, 36.6, 36.4, 24.5, 17.8, 17.8, 16.4, 14.0, 12.2, 12.1, 5.3; HRMS (ESI) m/z 687.4298 [(M + Li)+ calculated for C37H60LiO11, 687.4296].
Apoptolidinone D
To solution of 19.9 mg (14.7 μMol) macrolactone 42 in 4 mL of THF at −10 °C was added HF-Py (0.2 mL) dropwise. After 1 h the temperature was raised to 0 °C and after an additional 12 h to 10 °C. After 36 h the reaction mixture was diluted with Et2O, washed with water, and concentrated in vacuo leaving ca. 4 mL of Et2O. (It was noticed that drying over MgSO4 significantly decreased the amount of product in the solution.) The residue was purified by flash chromatography (CH2Cl2/MeOH, 15:1) to afford 8.5 mg (87%) of apoptolidinone D as a off-white solid: [α]25D +67.5°; IR (neat) 3382, 2927, 2362, 1666, 1599, 1457, 1390, 1255, 1100, 1024, 965, 754 cm−1; 1H NMR (600 MHz, CD3OD) δ 7.39 (s, 1H), 6.27−6.22 (m, 2H), 5.99 (d, J = 15.6 Hz, 1H), 5.57 (t, J = 8.1 Hz, 1H), 5.48−5.44 (m, 1H), 5.32−5.28 (m, 2H), 4.08 (dt, J = 8.4, 2.4 Hz, 1H), 3.78−3.73 (m, 2H), 3.55−3.50 (m, 1H), 3.53 (d, J = 1.2 Hz, 1H), 3.42−3.35 (m, 2H), 3.37 (s, 3H), 3.26 (s, 3H), 3−18−3.12 (m, 2H), 2.66 (dd, J = 9.6, 5.4 Hz, 1H), 2.49−2.42 (m, 1H), 2.30−2.23 (m, 1H), 2.17−1−95 (m, 5H), 2.13 (s, 3H), 2.08 (s, 3H), 1.79−1.72 (m, 2H), 1.72−1.53 (m, 3H), 1.64 (s, 3H), 1.43−1.22 (m, 4H), 1.18 (d, J = 6.6 Hz, 3H), 1.02 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CD3OD) δ 172.6, 147.5, 146.0, 143.0, 137.4, 135.0, 133.5, 132.2, 130.9, 126.6, 124.2, 101.3, 84.0, 80.0, 78.5, 75.4, 74.9, 73.8, 72.5, 69.2, 68.1, 61.4, 59.4, 46.7, 40.9, 38.5, 38.3, 36.3, 36.1, 24.8, 18.6, 15.6, 13.9, 12.2, 12.1, 5.3; HRMS (MALDI) m/z 689.3881 [(M + Na)+ calcd for C36H58NaO11, 689.3877].
Acknowledgments
This work was supported by the National Institutes of Health (CA 059515) and the Vanderbilt Institute of Chemical Biology.
Supporting Information Available
Experimental procedures and characterization data and NMR spectra of apoptolidinones A/D and all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Hayakawa Y.; Adachi K.; Komeshima N. J. Antibiot. 1987, 40, 1349–1352. [DOI] [PubMed] [Google Scholar]; b Hayakawa Y.; Sohda K.; Shinya K.; Hidaka T.; Seto H. J. Antibiot. 1995, 48, 954–961. [DOI] [PubMed] [Google Scholar]; c Hayakawa Y.; Sohda K. Y.; Furihata K.; Kuzuyama T.; ShinYa K.; Seto H. J. Antibiot. 1996, 49, 974–979. [DOI] [PubMed] [Google Scholar]; d Hayakawa Y.; Sohda K. Y.; Seto H. J. Antibiot. 1996, 49, 980–984. [DOI] [PubMed] [Google Scholar]
- a Kim J. W.; Adachi H.; ShinYa K.; Hayakawa Y.; Seto H. J. Antibiot. 1997, 50, 628–630. [DOI] [PubMed] [Google Scholar]; b Hayakawa Y.; Kim J. W.; Adachi H.; Shin-ya K.; Fujita K.; Seto H. J. Am. Chem. Soc. 1998, 120, 3524–3525. [Google Scholar]
- a Wender P. A.; Longcore K. E. Org. Lett. 2007, 9, 691–694. [DOI] [PubMed] [Google Scholar]; b Wender P. A.; Sukopp M.; Longcore K. Org. Lett. 2005, 7, 3025–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Salomon A. R.; Voehringer D. W.; Herzenberg L. A.; Khosla C. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 14766–14771. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Salomon A. R.; Voehringer D. W.; Herzenberg L. A.; Khosla C. Chem. Biol. 2001, 8, 71–80. [DOI] [PubMed] [Google Scholar]
- a Pennington J. D.; Williams H. J.; Salomon A. R.; Sulikowski G. A. Org. Lett. 2002, 4, 3823–3825. [DOI] [PubMed] [Google Scholar]; b Wender P. A.; Gulledge A. V.; Jankowski O. D.; Seto H. Org. Lett. 2002, 4, 3819–3822. [DOI] [PubMed] [Google Scholar]
- a Wender P. A.; Jankowski O. D.; Longcore K.; Tabet E. A.; Seto H.; Tomikawa T. Org. Lett. 2006, 8, 589–592. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wender P. A.; Jankowski O. D.; Tabet E. A.; Seto H. Org. Lett. 2003, 5, 2299–2302. [DOI] [PubMed] [Google Scholar]; c Wender P. A.; Jankowski O. D.; Tabet E. A.; Seto H. Org. Lett. 2003, 5, 487–490. [DOI] [PubMed] [Google Scholar]
- a Abe K.; Kato K.; Arai T.; Rahim M. A.; Sultana I.; Matsumura S.; Toshima K. Tetrahedron Lett. 2004, 45, 8849–8853. [Google Scholar]; b Bouchez L. C.; Vogel P. Chem. Eur. J. 2005, 11, 4609–4620. [DOI] [PubMed] [Google Scholar]; c Chng S. S.; Xu J.; Loh T. P. Tetrahedron Lett. 2003, 44, 4997–5000. [Google Scholar]; d Craita C.; Didier C.; Vogel P. J. Chem. Soc., Chem. Commun. 2007, 2411–2413. [DOI] [PubMed] [Google Scholar]; e Crimmins M. T.; Long A. Org. Lett. 2005, 7, 4157–4160. [DOI] [PubMed] [Google Scholar]; f Daniel P. T.; Koert U.; Schuppan J. Angew. Chem., Int. Ed. 2006, 45, 872–893. [DOI] [PubMed] [Google Scholar]; g Jin B. H.; Liu Q. S.; Sulikowski G. A. Tetrahedron 2005, 61, 401–408. [Google Scholar]; h Nicolaou K. C.; Li Y. W.; Weyershausen B.; Wei H. X. J. Chem. Soc., Chem. Commun. 2000, 307–308. [Google Scholar]; i Paquette W. D.; Taylor R. E. Org. Lett. 2004, 6, 103–106. [DOI] [PubMed] [Google Scholar]; j Schuppan J.; Ziemer B.; Koert U. Tetrahedron Lett. 2000, 41, 621–624. [Google Scholar]; k Sulikowski G. A.; Lee W. M.; Jin B.; Wu B. Org. Lett. 2000, 2, 1439–1442. [DOI] [PubMed] [Google Scholar]; l Toshima K.; Arita T.; Kato K.; Tanaka D.; Matsumura S. Tetrahedron Lett. 2001, 42, 8873–8876. [Google Scholar]; m Handa M.; Scheidt K. A.; Bossart M.; Zheng N.; Roush W. R. J. Org. Chem. 2008, 73, 1031–1035. [DOI] [PubMed] [Google Scholar]; n Handa M.; Smith I. I. I.; W J.; Roush W. R. J. Org. Chem. 2008, 73, 1036–1039. [DOI] [PubMed] [Google Scholar]
- a Nicolaou K. C.; Fylaktakidou K. C.; Monenschein H.; Li Y. W.; Weyershausen B.; Mitchell H. J.; Wei H. X.; Guntupalli P.; Hepworth D.; Sugita K. J. Am. Chem. Soc. 2003, 125, 15433–15442. [DOI] [PubMed] [Google Scholar]; b Nicolaou K. C.; Li Y.; Fylaktakidou K. C.; Mitchell H. J.; Wei H. X.; Weyershausen B. Angew. Chem., Int. Ed. 2001, 40, 3849–3854. [DOI] [PubMed] [Google Scholar]; c Nicolaou K. C.; Li Y. W.; Fylaktakidou K. C.; Mitchell H. J.; Sugita K. Angew. Chem., Int. Ed. 2001, 40, 3854–3857. [DOI] [PubMed] [Google Scholar]; d Nicolaou K. C.; Li Y. W.; Sugita K.; Monenschein H.; Guntupalli P.; Mitchell H. J.; Fylaktakidou K. C.; Vourloumis D.; Giannakakou P.; O’Brate A. J. Am. Chem. Soc. 2003, 125, 15443–15454. [DOI] [PubMed] [Google Scholar]; e Wehlan H.; Dauber M.; Fernaud M. T. M.; Schuppan J.; Keiper S.; Mahrwald R.; Garcia M. E. J.; Koert U. Chem. Eur. J. 2006, 12, 7378–7397. [DOI] [PubMed] [Google Scholar]; f Wehlan H.; Dauber M.; Fernaud M. T. M.; Schuppan J.; Mahrwald R.; Ziemer B.; Garcia M. E. J.; Koert U. Angew. Chem., Int. Ed. 2004, 43, 4597–4601. [DOI] [PubMed] [Google Scholar]
- a Crimmins M. T.; Christie H. S.; Chaudhary K.; Long A. J. Am. Chem. Soc. 2005, 127, 13810–13812. [DOI] [PubMed] [Google Scholar]; b Schuppan J.; Wehlan H.; Keiper S.; Koert U. Angew. Chem., Int. Ed. 2001, 40, 2063–2066. [PubMed] [Google Scholar]; c Schuppan J.; Wehlan H.; Keiper S.; Koert U. Chem. Eur. J. 2006, 12, 7364–7377. [DOI] [PubMed] [Google Scholar]; d Wu B.; Liu Q. S.; Sulikowski G. A. Angew. Chem., Int. Ed. 2004, 43, 6673–6675. [DOI] [PubMed] [Google Scholar]
- Isolation:; a Hatfield G. M.; Woodard R. W.; Son J. K. J. Nat. Prod 1992, 55, 753–759. [DOI] [PubMed] [Google Scholar]; b Meyer M.; Kellerschierlein W.; Drautz H.; Blank W.; Zahner H. Helv. Chim. Acta 1985, 68, 83–94. [Google Scholar]; Total synthesis:; c Scheidt K. A.; Bannister T. D.; Tasaka A.; Wendt M. D.; Savall B. M.; Fegley G. J.; Roush W. R. J. Am. Chem. Soc. 2002, 124, 6981–6990. [DOI] [PubMed] [Google Scholar]
- a Crimmins M. T.; King B. W.; Tabet E. A.; Chaudhary K. J. Org. Chem. 2001, 66, 894–902. [DOI] [PubMed] [Google Scholar]; b Crimmins M. T.; King B. W.; Tabet E. A. J. Am. Chem. Soc. 1997, 119, 7883–7884. [Google Scholar]
- Cho B. T.; Chun Y. S. J. Org. Chem. 1998, 63, 5280–5282. [Google Scholar]
- Paquette L. A.; O’Neil S. V.; Guillo N.; Zeng Q. B.; Young D. G. Synlett 1999, 1857–1866. [Google Scholar]
- The ratio of isomers 12 and 13 was determined by GC analysis. The double bond geometry of silylenol ethers 12 and 13 was assigned by analysis of the corresponding NOESY spectra
- Meyer C.; Marek I.; Normant J. F. Synlett 1993, 386–388. [Google Scholar]
- Jehanno E.; Vaultier M. Tetrahedron Lett. 1995, 36, 4439–4442. [Google Scholar]
- a Roush W. R.; Halterman R. L. J. Am. Chem. Soc. 1986, 108, 294–296. [Google Scholar]; b Roush W. R.; Ando K.; Powers D. B.; Palkowitz A. D.; Halterman R. L. J. Am. Chem. Soc. 1990, 112, 6339–6348. [Google Scholar]
- Brown H. C.; Bhat K. S. J. Am. Chem. Soc. 1986, 108, 5919–5923. [DOI] [PubMed] [Google Scholar]
- Saito S.; Hasegawa T.; Inaba M.; Nishida R.; Fujii T.; Nomizu S.; Moriwake T. Chem. Lett. 1984, 1389–1392. [Google Scholar]
- Masse C. E.; Yang M.; Solomon J.; Panek J. S. J. Am. Chem. Soc. 1998, 120, 4123–4134. [Google Scholar]
- a Fargeas V.; LeMenez P.; Berque I.; Ardisson J.; Pancrazi A. Tetrahedron 1996, 52, 6613–6634. [Google Scholar]; b Kocienski P.; Wadman S.; Cooper K. J. Am. Chem. Soc. 1989, 111, 2363–2365. [Google Scholar]
- Fetizon M.; Jurion M. J. Chem. Soc., Chem. Commun. 1972, 382–384. [Google Scholar]
- Miyaura N.; Yamada K.; Suginome H.; Suzuki A. J. Am. Chem. Soc. 1985, 107, 972–980. [Google Scholar]
- Frank S. A.; Chen H.; Kunz R. K.; Schnaderbeck M. J.; Roush W. R. Org. Lett. 2000, 2, 2691–2694. [DOI] [PubMed] [Google Scholar]
- No aldol products derived from silyl enol ether 13 were observed
- Evans D. A.; Yang M. G.; Dart M. J.; Duffy J. L.; Kim A. S. J. Am. Chem. Soc. 1995, 117, 9598–9599. [Google Scholar]
- a Evans D. A.; Dart M. J.; Duffy J. L.; Yang M. G. J. Am. Chem. Soc. 1996, 118, 4322–4343. [Google Scholar]; b Evans D. A.; Duffy J. L.; Dart M. J. Tetrahedron Lett. 1994, 35, 8537–8540. [Google Scholar]
- For the preparation of carboxylic acid 32, see ref (7g)
- Inanaga J.; Hirata K.; Saeki H.; Katsuki T.; Yamaguchi M. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- a Evans D. A.; Dart M. J.; Duffy J. L.; Rieger D. L. J. Am. Chem. Soc. 1995, 117, 9073–9074. [Google Scholar]; b Evans D. A.; Yang M. G.; Dart M. J.; Duffy J. L. Tetrahedron Lett. 1996, 37, 1957–1960. [Google Scholar]
- The minor geometric isomer could not be completely separated from other by-products and was not fully characterized.
- a Chatterjee A. K.; Choi T. L.; Sanders D. P.; Grubbs R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. [DOI] [PubMed] [Google Scholar]; b Chatterjee A. K.; Grubbs R. H. Angew. Chem., Int. Ed. 2002, 41, 3171–3173. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Dart M. J.; Dugy J. L.; Yang M. G.; Livingston A. B. J. Am. Chem. Soc. 1995, 117, 6619–6620. [Google Scholar]
- Ruck R. T.; Jacobsen E. N. Angew. Chem., Int. Ed. 2003, 42, 4771–4774. [DOI] [PubMed] [Google Scholar]
- A NOE between H-22 and H-24 is not observed because of overlapping signals in the NMR spectrum
- A direct comparison to earlier reported cytotoxicity data is difficult because of the significant dependence on cell culture media and exposure time. A publication providing a detailed description of these effects is in preparation
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.