

Abstract
We have previously observed that soluble urokinase plasminogn activator receptor (suPAR) prevents impairment of cerebrovasodilation induced by hypercapnia and hypotension after hypoxia/ischemia (H/I) in the newborn pig. In this study, we investigated the role of low-density lipoprotein related protein (LRP) receptor and the ERK isoform of mitogen activated protein kinase (MAPK) in uPA-mediated impairment of vasodilation after H/I in piglets equipped with a closed cranial window. CSF uPA increased from 9 ± 2 to 52 ± 8 and 140 ± 21 ng/ml at 1 and 4h after H/I, respectively. The LRP antagonist receptor associated protein (RAP) and anti-LRP antibody blunted the increase in CSF uPA at 1h (17 ± 2 ng/ml) but not 4h post insult. uPA detectable in sham-treated cortex by immunhistochemistry was markedly elevated 4h after H/I. Phosphorylation (activation) of CSF ERK MAPK was detected at 1 and 4h post H/I and blocked by RAP. Exogenous uPA administered at 4h post H/I further stimulated ERK MAPK phosphorylation, which was blocked by RAP. Pre-treatment of piglets with RAP, anti-LRP, and suPAR completely prevented, and the ERK MAPK antagonist U 0126 partially prevented, impaired responses to hypotension and hypercapnia post H/I, but none of these antagonists affected the response to isoproterenol. These data indicate that uPA is upregulated after H/I through an LRP-dependent process and that the released uPA impairs hypercapnic and hypotensive dilation through an LRP- and ERK MAPK dependent pathway. These data suggest that modulation of uPA upregulation and/or uPA-mediated signal transduction may preserve cerebrohemodynamic control after hypoxia/ischemia.
Keywords: cerebral circulation, newborn, plasminogen activators, signal transduction, ischemia
1. Introduction
Perinatal cerebral hypoxia/ischemia has many causes, unclear pathophysiology, no specific mechanism-related treatment, and poor outcome. Neonatal stroke may occur in as many as 1 in 4000 births (27). In newborns with stroke, complications such as hypoxic/ischemic events are common (11). Maternal and perinatal coagulopathy predispose to perinatal stroke (12,22), with 30% of neonatal strokes being due to thrombosis (10). A better understanding of the pathophysiologic responses that occur in children after cerebral hypoxia/ischemia is needed to develop mechanism based approaches to therapy.
One contributor to neurological damage after hypoxia/ischemia is thought to be cerebrovascular dysfunction. For example, hypotension leads to loss of cerebrovascular regulation promoting tissue ischemia, while cerebrovasoconstriction associated with hypocapnia contributes to periventricular leukomalacia in the perinate (30). Using a piglet model, we have shown that pial artery dilation in response to hypotension and hypercapnia is blunted after cerebral hypoxia/ischemia (20,24,25). However, the mechanism underlying loss of compensatory vasodilation and therapeutic avenues to ameliorate its deleterious effects on CNS ischemia remain uncertain.
Urokinase (uPA) and tissue plasminogen activator (tPA) are serine proteases that convert plasminogen to the active protease plasmin (5,9). Recombinant tPA is the only FDA approved for stroke (21). However, tPA exhibits deleterious as well as beneficial effects that profoundly constrain its clinical utility. In addition to its salutary role in reperfusion, tPA contributes to excitotoxic neuronal cell death (28) and increases stroke infarct volume in mice (31).
We have observed that exogenous tPA or uPA applied topically to the piglet cerebral cortex potentiates the impairment of pial artery dilation caused by hypercapnia and hypotension in the setting of hypoxia/ischemia (3). In other studies, we have shown that the endogenous plasminogen activator inhibitor-1 derived peptide, EEIIMD, inhibits tPA and uPA-mediated vascular activity mediated through the low-density lipoprotein-related receptor (LRP) without inhibiting their fibrinolytic activity (4,8,26). Pretreatment with EEIIMD partially prevented, whereas soluble urokinase plasminogen activator receptor (suPAR), which blocks uPA binding to LRP (13), completely prevented impairment of vasodilation caused by hypercapnia and hypotension in the setting of hypoxia/ischemia (3). These data suggest that endogenous uPA is the predominate cause of vascular derangement induced by this form of cerebral injury. However, the intracellular mechanisms involved in this impairment are unknown.
Mitogen activated protein kinase (MAPK), a family of at least 3 kinases, extracellular signal-related kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) is upregulated and may contribute to injury after stroke (1,14,23). For example, activation of ERK MAPK contributes to impaired hypercapnia-induced pial artery dilation seen after hypoxia/ischemia in the piglet (20). However, others have observed neuroprotection with ERK MAPK stimulation after cerebral ischemia (19). We hypothesize that uPA is upregulated after cerebral hypoxia/ischemia and activates ERK MAPK in an LRP dependent manner with the effect of inhibiting adaptive vascular responses to hypercapnia and hypotension post insult.
2. Results
Cerebral hypoxia/ischemia elevates the amount of uPA in cerebral cortex and CSF
Figure 1 shows immunocytochemical and corresponding histopathologic data derived from the same animals and areas of brain parenchyma, obtained from piglets 4h after being placed in either sham control or hypoxia/ischemia conditions. Abundant uPA antigen (3-4 on a 5 point scale) is observed in the parietal cortex of animals subjected to cerebral hypoxia/ischemia, primarily in association with the vessels and parenchyma (Figure 1, Panel A) and diffusely within areas of subarachnoid hemorrhage (Figure 1, Panel B). Yellow arrows show uPA-positive thrombi in three arterioles. uPA can be seen within microthrombi and endothelial fibrin deposition of pial arterioles (Figure 1, Panel C). Close-up views of the hypoxic/ischemic cortex reveals uPA within damaged neurons and glial cells, as well as scattered accumulations within the extracellular space (Figure 1, Panel D). In contrast, no staining is seen with non-immune IgG in the hypoxic/ischemic animal (Figure 1, Panel E), excluding non-specific IgG binding. Minimal uPA (0-1 on a 5 point scale) is detected in a sham control section of piglet parietal cortex (Figure 1, Panel F). H + E staining of the parietal cortex section from an hypoxic/ischemic animal shows subarachnoid hemorrhage and thrombi in arterioles (Figure 1, Panel G), neuronal necrosis and scattered parenchymal hemorrhage (Figure 1, Panel H), whereas the comparable section from a sham control animal shows that the cortical surface is unremarkable (Figure 1, Panel I). To estimate the uPA detected in brain tissue, the concentration of uPA in cortical periarachnoid CSF was determined by ELISA. uPA in CSF increased within 1h of cerebral hypoxia/ischemia and increased further by 4h post insult (Figure 2).
Figure 1.
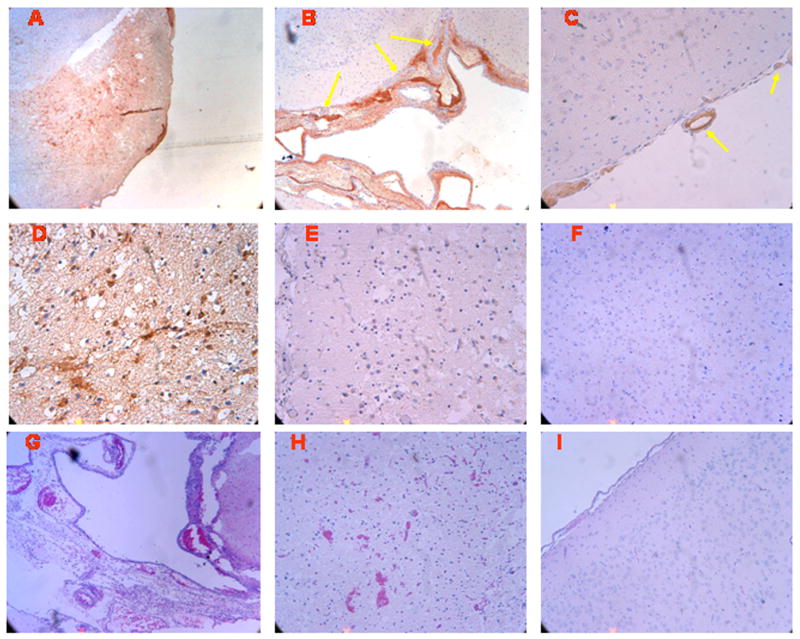
Immunohistochemistry and histopathology four hours after cerebral hypoxia/ischemia. Sections of the parietal cortex from piglet brains after ischemic injury (A-E) and from uninjured control animal (I and F), were subjected to antigen retrieval in citrate buffer and stained with anti-uPA monoclonal antibody (5 μg/ml) (Panels, A-D and F) or with non-immune mouse IgG1 as a negative control (Panel E), secondary biotinylated anti-mouse IgG (1:200), followed by incubation with HRP-conjugated streptavidin. Magnification shown is 100x for Panels A, B, G, and I, 200X for panels, C, E, F, and H, 400x for Panel D. Adjacent sections from the same brains exposed to ischemic injury (Panels G-H) and from uninjured controls (Panel I), were stained by H&E for histological inspection. These data reflect an n of 2 per experimental group.
Figure 2.
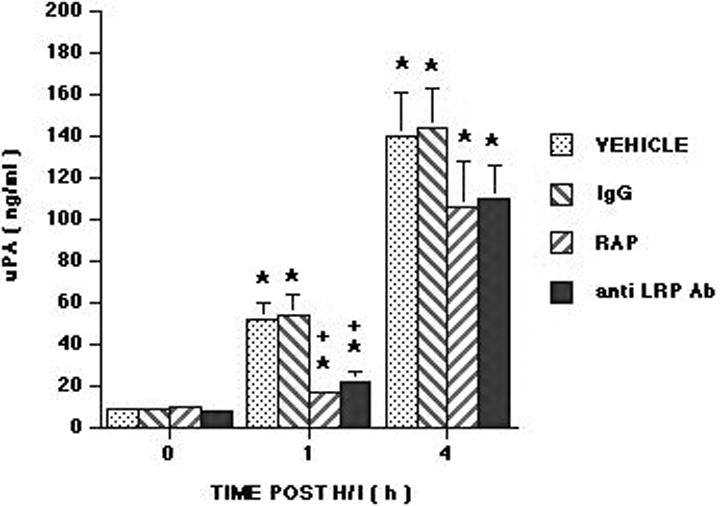
Influence of hypoxia/ischemia (H/I) on CSF uPA (ng/ml) as a function of time post insult (hours) in vehicle (saline), IgG (10 μg/ml), RAP (10-7 M), and anti-LRP antibody (anti LRP Ab) (10 μg/ml) pretreated pigs, n=6 for all except n=3 for IgG. IgG, RAP and anti LRP Ab were administered after the 0 time sample, but 30 min prior to H/I. *P<0.05 versus corresponding 0 time value +P<0.05 versus corresponding vehicle value.
We next studied the effect of RAP, an LRP antagonist (8,26) on CSF uPA concentration. Administration of RAP (10-7 M) blunted the increase in CSF uPA concentration at 1 but not at 4h post cerebral hypoxia/ischemia (Figure 2). RAP was administered after the baseline CSF sample had been collected, 30 min prior to induction of cerebral hypoxia/ischemia, to make certain that the baseline plasminogen activator status of the 2 groups of animals was similar. To corroborate these findings and exclude an effect of residual endotoxin in the RAP preparation, we used an anti-LRP antibody as an independent approach. Anti-LRP (10 μg/ml) blunted the elevation of CSF uPA at 1h but not 4h post insult, the same pattern observed with RAP. In contrast, mouse IgG at the same concentration had no effect on CSF uPA concentration (Figure 2).
Impairment of hypercapnic and hypotensive pial artery dilation after cerebral hypoxia/ischemia occurs through an LRP- dependent process
Two levels of hypercapnia, hypotension, isoproterenol, and uPA elicited reproducible graded pial small artery (120 to 160 μm) and arteriole (50 to 70 μm) dilation in sham control animals (data not shown). Pial small artery dilation in response to hypercapnia and hypotension was blunted 1h after hypoxia/ischemia, whereas responses to isoproterenol were unchanged (Figure 3)(3). Similar reductions in responses were seen in arterioles. Pretreatment with topical exogenous RAP (10-7 M) or anti-LRP (10 μg/ml), administered 30 min prior to hypoxia/ischemia followed by continuous exposure of the cerebral cortical surface to these agents post insult fully protected the responses to hypercapnia and hypotension 1h after injury (Figure 3). In contrast, mouse IgG had no effect on impairment of pial artery dilation 1h after injury (Figure 3). Similar findings were seen 4h post insult. For example, pial small artery dilation in response to moderate and severe hypotension was 14 ± 1 and 29 ± 2% 4h after hypoxia/ischemia in animals pretreated with RAP (10-7 M), which was no different statistically than that observed in animals pretreated with RAP at 1h post insult (16 ± 2 and 26 ± 3%). Pretreatment with uPA blunted hypercapnic and hypotensive pial artery dilation to vasoconstriction after hypoxia/ischemia, while pretreatment with suPAR, an antagonist of the vascular action of uPA, had been previously observed to produce full protection of pial artery dilation induced by hypercapnia and hypotension 1h post hypoxia/ischemia (3). Topical application of uPA (10-7 M) for 1h in the absence of hypoxia/ischemia had no effect on pial artery dilation to hypercapnia, hypotension, or isoproterenol (15 ± 1 and 27 ± 2 versus 14 ± 2 and 26 ± 3% dilation for moderate and severe hypotension before and after uPA, respectively).
Figure 3.

Influence of hypercapnia (lo, hi; pCO2 of 50-55 and 70-75 mm Hg), hypotension (mod, sev; 25 and 45% reductions in mean arterial blood pressure), and isoproterenol (10-8, 10-6 M) (panels A-C) on pial artery diameter. Conditions are before (control), 1h after hypoxia/ischemia (pO2 of 35 mm Hg for 10 min followed by global cerebral ischemia for 20 min) (H/I), 1h after H/I pretreated with IgG (10 μg/ml) 30 min prior to H/I, 1h after H/I pretreated with RAP (10-7 M), 1h after H/I pretreated with anti LRP antibody (anti LRP Ab) (10 μg/ml), and 1h after H/I pretreated with U 0126 (10-6 M), n=6 for all except n=3 for IgG. Baseline small artery diameter for control, H/I, H/I + IgG, H/I + RAP, H/I + anti LRP Ab, and H/I + U 0126 were 144 ± 11, 119 ± 9, 118 ± 8, 138 ± 6, 136 ± 7, and 132 ± 7 μm, respectively. *P<0.05 versus corresponding control value +P<0.05 versus corresponding non pretreated H/I value.
Hypoxia/ischemia produces phosphorylation of ERK MAPK in an LRP dependent process
ERK MAPK contributes to impairment of hypercapnic and hypotensive pial artery dilation after cerebral hypoxia/ischemia
Total and phosphyorylated ERK MAPK in cortical periarachnoid CSF was measured by ELISA under baseline (prior to hypoxia/ischemia) and injury (1,4h) conditions. The activation (phosphorylation) state of the ERK MAPK isoform was determined by expressing the data as a percent of control (total) to normalize these values. The vehicle was dimethyl sulfoxide (DMSO) (100 μl) diluted in 9.9 ml of 0.9% saline. The vehicle or antagonist was administered 30 min prior to hypoxia/ischemia. Hypoxia/ischemia induced marked phosphorylation (activation) of ERK MAPK within 1h post injury (Figure 4). Exogenous adminstration of uPA to the cerebral cortex, in a concentration observed in CSF after hypoxia/ischemia (10-7M, Figure 2), further stimulated phosphorylation of ERK MAPK at 4h post insult (Figure 4). Pretreatment with the LRP antagonist, RAP (10-7 M), anti-LRP (10 μgml), and the ERK MAPK inhibitor U 0126 blocked phosphorylation of ERK MAPK induced by hypoxia/ischemia alone and hypoxia/ischemia accompanied by exogenous administration of uPA (Figure 4). In contrast, CSF ERK MAPK was unchanged with uPA administration under sham control conditions (Figure 5). uPA induced pial artery dilation was unchanged by U 0126 but was blocked by suPAR under sham control conditions. U 0126 and suPAR had no effect on CSF ERK MAPK concentration in the absence or presence of uPA under sham control conditions (Figure 5).
Figure 4.
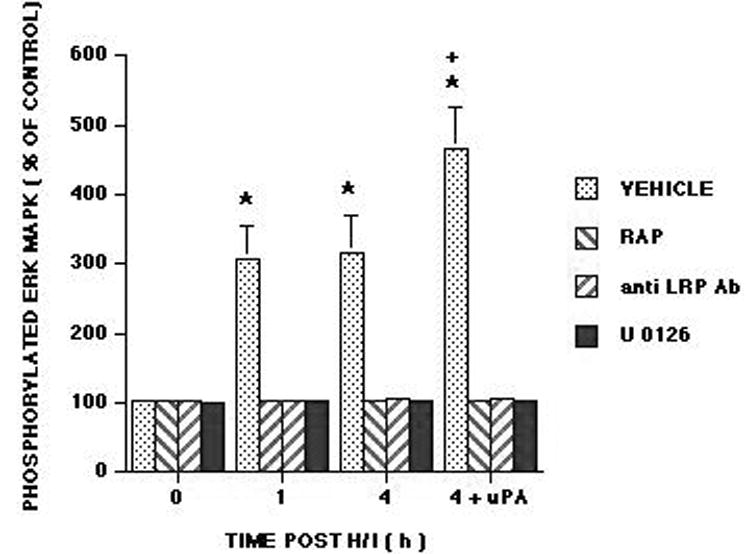
Phosphorylation of ERK MAPK in cortical periarachnoid CSF was determined prior to hypoxia/ischemia (H/I) (0 min), as a function of time (hours, h) after H/I, and 4h after H/I and additional exogenous administration of uPA (10-7 M) in vehicle, RAP (10-7 M), anti LRP Ab (10 μg/ml) and U 0126 (10-6 M) pretreatment animals, n=6. Data expressed as percent of control by ELISA determination of phospho ERK MAPK and total ERK MAPK isoforms and subsequent normalization to total form. *P<0.05 compared with corresponding 0 time value +P<0.05 compared with corresponding 4h non uPA treated value
Figure 5.
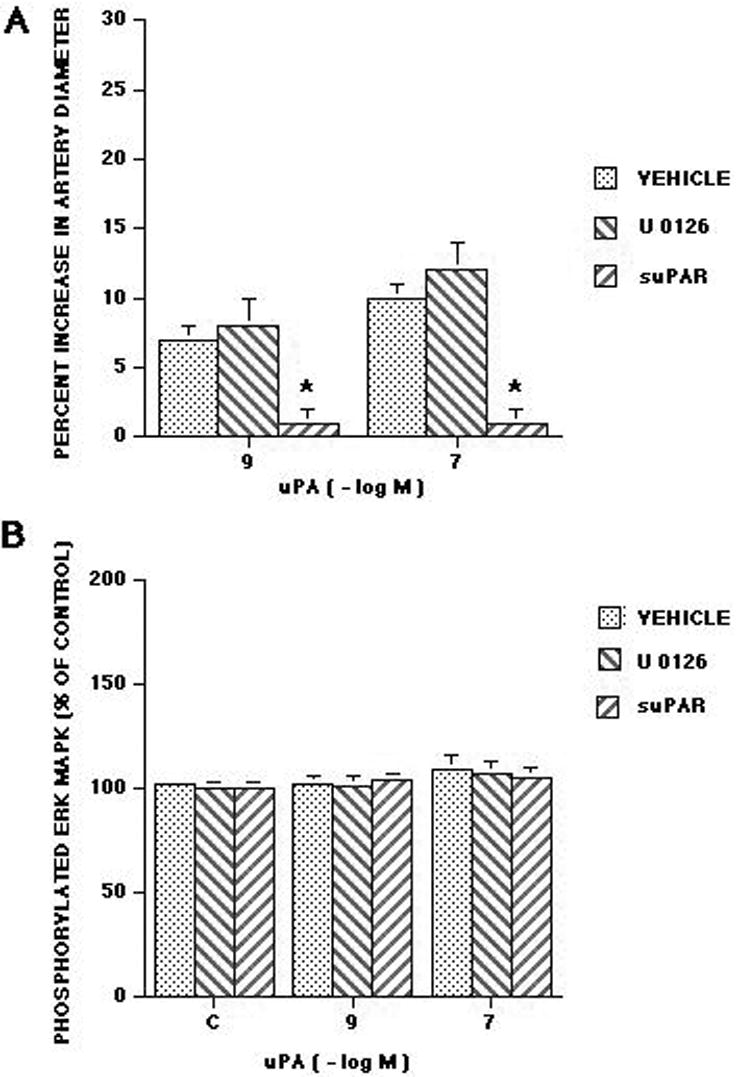
Influence of uPA (10-9, 10-7 M) on A: pial artery diameter and B: CSF phosphorylated ERK MAPK (percent of control) in the absence (vehicle) and presence of U 1026 (10-6 M) and suPAR (10-7 M), n=6. *P<0.05 compared with corresponding vehicle value.
Figure 6 shows immunohistochemical analysis of phospho ERK MAPK expression in piglets 4h after either sham control conditions or cerebral hypoxia/ischemia. Abundant phospho ERK MAPK antigen (3-4 on a 5 point scale) was observed in the parietal cortex of animals subjected to cerebral hypoxia/ischemia (Figure 6, Panels A,B). Closeup of the hypoxic/ischemic cortex reveals phospho ERK MAPK within damaged neurons and glial cells (Figure 6, Panel B). In contrast, minimal phospho ERK MAPK (0-1 on a 5 point scale) was detected in sham control sections of piglet parietal cortex (Figure 6, Panels C, D). No non-immune IgG (0 on a 5 point scale) is seen in an hypoxic/ischemic animal (Figure 6, Panel E), excluding non specific IgG binding. In Figure 7, Western analysis of CSF for phospho p44/p42 ERK MAPK shows elevation at 4h post hypoxia/ischemia which is markedly reduced by suPAR and modestly reduced by U 0126. At 1h post insult, little elevation of phospho p44/p42 MAPK is observed in the absence or presence of suPAR or U 0126. Pretreatment with the ERK MAPK antagonist U 0126 partially prevented the impairment of pial artery dilation induced by hypercapnia and hypotension in the setting of hypoxia/ischemia (Figure 3). U 0126 had no effect on isoproterenol induced dilation (Figure 3).
Figure 6.
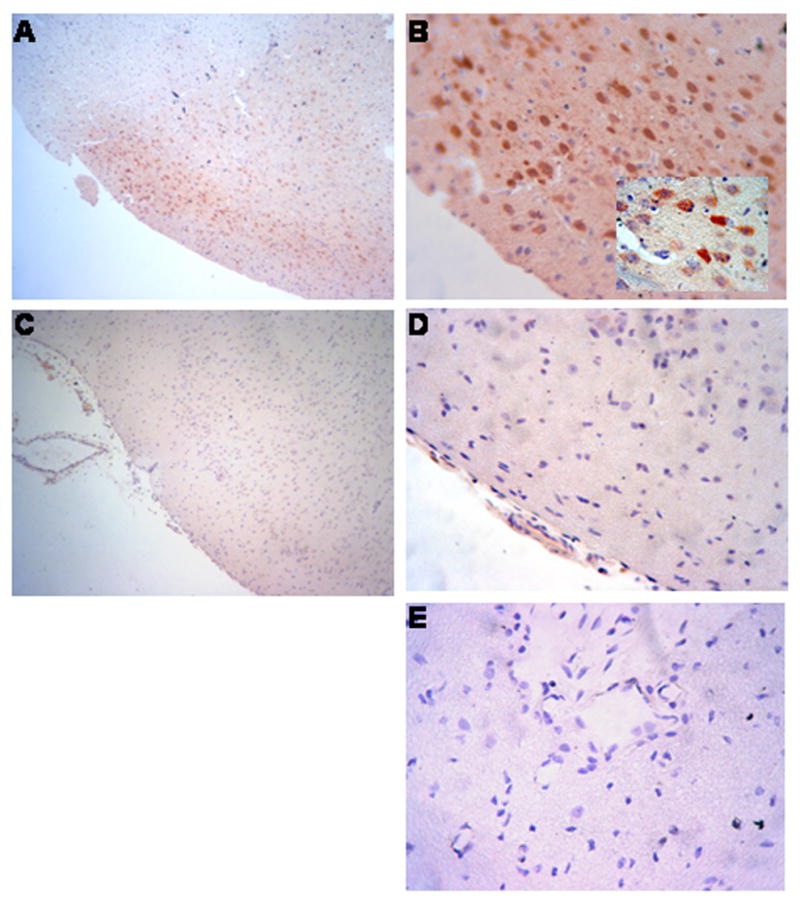
Immunhistochemistry for phosphorylation of ERK MAPK four hours after cerebral hypoxia/ischemia. Sections of the parietal cortex from piglet brains after ischemic injury (A,B, and E) and from uninjured control animal (C and D) were stained with phospho-p44/42 MAPK rabbit monoclonal antibody (Panels A-D) or with rabbit IgG as a negative control (Panel E). Magnification shown is 100X for Panels A and C, 400X for Panels D,B, and E, X1000 for insert in Panel B. These data reflect an n of 2 per experimental group.
Figure 7.
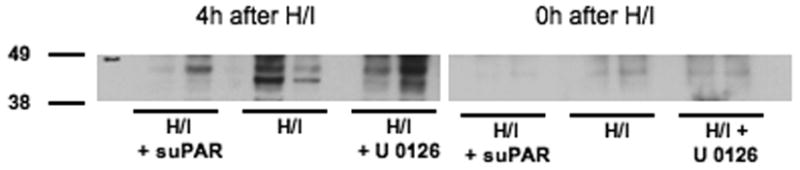
Western analysis of CSF for phospho–p44/42 ERK MAPK at 4h and 0h of hypoxia/ischemia in suPAR, vehicle, and U 0126 pretreated animals, n=2 for each experimental group.
RAP, anti-LRP, and U 0126 have no effect on hypercapnic and hypotensive pial artery dilation in sham control piglets
Administration of RAP, anti-LRP, and U 0126 to the cerebral cortical surface had no effect on pial artery diameter. Similarly, these agents had no effect on hypercapnic and hypotensive pial artery dilation in sham control piglets (17 ± 1 and 28 ± 2 versus 18 ± 1 and 29 ± 2% subjected to high and low hypercapnic conditions in the absence and presence of RAP).
Blood chemistry
Blood chemistry values were collected before and after all experiments. There were no statistically significant differences between sham control, hypoxia/ischemia, and hypoxia/ischemia antagonist treated animals. Hypoxia decreased pO2 to 34 ± 4 mm Hg. Low levels of hypercapnia raised pCO2 to 57 ± 7 and high levels of hypercapnia raised pCO2 to 77 ± 9 mm Hg. Carbon dioxide levels were kept constant during periods of hypoxia and oxygen levels were kept constant during periods of hypercapnia.
3. Discussion
Several principal new findings emerged from this study. First, the presence of uPA and phosphorylated (activated) ERK MAPK in piglet brain was demonstrated by immunohistochemical staining. Second, although it has been previously shown that uPA is increased in rat brain and baboon basal ganglia after focal cerebral ischemia (18,29), our studies are the first to demonstrate to our knowledge that uPA expression increases after global cerebral hypoxia/ischemia. Considerable uPA and ERK MAPK was detected in neurons/glia. These data cannot distinguish between the possibility that neurons/glia serve as the cellular site of origin for uPA and ERK MAPK and/or that uPA synthesized elsewhere post insult binds to these cells (eg to uPAR). Third, in the same animals where robust staining for uPA and ERK MAPK was seen, there was also marked histopathology evident on H + E staining. These observations support the concept that plasminogen activators act at the level of the neurovascular unit, specifically on pial artery reactivity to vasoactive stimuli after cerebral hypoxia/ischemia.
tPA is thought to be transported across the blood brain barrier (BBB) in a process that is mediated by LRP (6). Oxygen glucose deprivation, however, has been observed to switch this transport from an LRP dependent to a progressively LRP independent process within 4h (7). Our data are the first to show that cerebral hypoxia/ischemia also upregulates uPA in an LRP dependent manner. They support the aforementioned studies in that RAP and anti-LRP antibody blunted the elevation of CSF uPA at 1, but not at 4h post cerebral hypoxia/ischemia. These data are consistent with the possibility that some of the uPA detected in CSF post ischemia derives from extracerebral sources due to a functionally disrupted BBB, while the balance originates from within the brain parenchyma. A limitation of the closed cranial window technique to quantify CSF plasminogen activator concentration is that the cellular site of origin cannot be determined. Potential sources include neurons, glia, vascular smooth muscle, and endothelial sources. Importantly, though, our findings demonstrate that mechanisms independent of plasminogen activator catalytic activity are involved early in the process when it may be most amenable to modulation.
Control of cerebrovascular tone after hypoxic/ischemic injury plays a critical role in mediating CNS ischemia and neurologic damage in affected newborns. We found that hypoxia/ischemia impairs dilation of pial arteries in response to hypercapnia and hypotension, while responses to isoproterenol were unchanged, as reported previously (3,20,24,25). Pretreatment with exogenous tPA or uPA potentiated the effect of hypoxia/ischemia, reversing pial artery dilation in response to hypercapnia and hypotension to vasoconstriction (3). This observation is particularly intriguing in that exogenous tPA and uPA by themselves produced modest dilation in control animals (3). Thus, application of a vasodilator would have been predicted to have an additive or synergistic effect in the presence of a second dilator, rather than causing vasoconstriction. The fact that responses to isoproterenol after hypoxia/ischemia remained unchanged in the presence of the plasminogen activators indicates that this effect is specific for hypercapnia and hypotension and likely depends on the signal transduction cascades that they activate. The mechanism which promotes interaction with these alternative signaling systems (eg ERK MAPK, as discussed below) under hypoxic/ischemic conditions is uncertain, but may involve a different binding site for the uPA-suPAR complex, competition with uPA for binding to LRP, or a dose-dependent parabolic effect that is shifted by suPAR. Importantly, new data in the present study show that topical administration of uPA for 1h without hypoxia/ischemia does not have the same effect, eg responses to hypercapnia and hypotension were unchanged. The prior studies using topical exogenous administration of plasminogen activators were designed to simulate the conditions likely to occur within minutes of injury wherein tPA and/or uPA are released from damaged brain parenchymal cells rather than the therapeutic setting of stroke where tPA is given intravenously. Because the study design employed topical pretreatment, caution is urged regarding clinical interpretation as to whether intravenous delivery of tPA or uPA post injury affects vascular contractility similarly and is detrimental or beneficial.
The PAI-1 derived peptide EEIIMD inhibits vasoactivity of tPA and uPA without inhibiting its fibrinolytic activity (4,26), whereas soluble uPA receptor (suPAR) exerts a similar effect on uPA alone (13). Pretreatment with EEIIMD partially prevented and suPAR completely prevented impairment of pial artery dilation induced by hypercapnia and hypotension in the setting of hypoxia/ischemia (3). These data suggest that while both endogenous tPA and uPA may contribute to outcome, uPA is the predominant cause of vascular derangement under these experimental conditions (3). The fact that EEIIMD binds to the docking site of tPA and uPA but does not inhibit their plasminogen activator activity demonstrates that this effect is not mediated through their plasminogen activator activities. However, the protective effect of EEIIMD or suPAR did not result from a general nonspecific potentiation/inhibition of vascular responsiveness, as neither affected dilation induced by isoproterenol (3).
Results of the present study extend these initial observations and indicate that uPA released after cerebral hypoxia/ischemia impairs pial artery dilation caused by hypercapnia and hypotension through an LRP and ERK MAPK dependent process. Similar observations made with the LRP antagonist RAP and anti-LRP antibody strengthened this conclusion. Hypoxia/ischemia itself resulted in enhanced phosphorylation (activation) of CSF ERK MAPK, reflective of what occurs in the brain parenchyma. Exogenous administration of uPA to the cerebral cortex in a concentration observed in CSF post insult further enhanced the phosphorylation of ERK MAPK, indicating that this isoform of MAPK is not maximally activated by hypoxia/ischemia. Therefore, the additional endogenous release (or exogenous administration) of uPA after the initial insult can exacerbate the outcome. Analysis of CSF by Western blot corroborated the results obtained by ELISA, providing additional support for the immunohistochemical data showing that hypoxia/ischemia upregulated ERK MAPK. Results from both ELISA and Western analysis show that U 0126 blocked ERK MAPK phosphorylation. Western analysis showing suPAR blocks ERK MAPK upregulation after hypoxia/ischemia strengthens the connection of uPA in this process. uPA, however, did not upregulate ERK MAPK under sham injury conditions. Potential reasons for the lack of upregulation by uPA are unknown but could relate to lack of upstream substrate availability for release of MAPK under non pathologic conditions.
It appears that coupling of uPA to LRP regulates both the amount of uPA in the CSF and uPA-mediated downstream signal transduction events. By regulating cell surface uPAR, LRP can control the activity of cell signaling factors downstream of uPAR, including ERK MAPK (32). Pretreatment with the ERK MAPK antagonist U 0126 partially prevented the impairment of pial artery dilation caused by hypercapnia and hypotension observed after cerebral hypoxia/ischemia. U 0126 had no effect on isoproterenol induced dilation. These data suggest that the phosphorylation (activation) of ERK MAPK observed after cerebral hypoxia/ischemia contributes to impaired cerebrovascular control post insult. Because exogenous uPA further activates ERK MAPK, these data together suggest that the release of uPA after hypoxic/ischemia activates ERK MAPK, which impairs pial artery dilation induced by hypercapnia and hypotension. Because U 0126 did not completely prevent impairment of dilation to these two stimuli, other undefined mechanisms, which may include the opioid nociceptin/orphanin FQ and/or activation of the N-methyl-D-aspartate receptor (2,20), also contribute to cerebral hypoxic/ischemic vascular derangement induced by hypoxia/ischemia.
In conclusion, our studies indicate that uPA is released in response to cerebral hypoxia/ischemia through an LRP-initiated process and that the released uPA impairs pial artery dilation induced by hypercapnia and hypotension through an LRP- and ERK MAPK dependent pathway (Figure 8). At sites where robust uPA and ERK MAPK staining was present, marked histopathology was evident. These observations support the concept that uPA acts at the level of the neurovascular unit (Figure 8), specifically on pial artery reactivity to vasoactive stimuli after cerebral hypoxia/ischemia. These data suggest that modulation of uPA expression and/or action may preserve cerebrohemodynamic control after hypoxia/ischemia (Figure 8) and thereby improve outcome.
Figure 8.
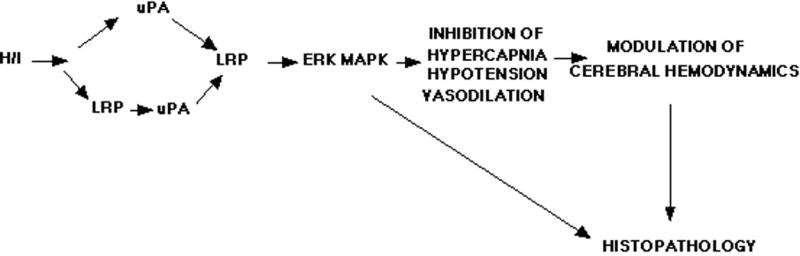
Proposed mechanism relating uPA, LRP, and ERK MAPK to cerebral hemodynamic outcome after hypoxia/ischemia.
4. Experimental Procedure
Materials
The LRP antagonist, recombinant receptor associated protein (RAP) (8,26) was generated in E. coli (DH5α/pGEX-2T plasmid) expressing GST-RAP (kind gift of Dr. T. Willnow, University of Potsdam, Germany) after induction with IPTG. RAP was purified on glutathione-sepharose (Amersham) and was eluted using 10mM glutathione in PBS and buffer exchange to PBS using an Amicon Unit centrifugal device (10,000 MWCO). The preparation of RAP was passed twice through Acticlean Etox (Sterogene) column to remove potential contamination with endotoxin. RAP migrated as a single band on SDS-PAGE and inhibited the internalization of 125I uPA in murine embryonic fibroblasts (MEF-1) cells, which is known to be LRP dependent. Recombinant single chain urokinase (uPA) was expressed in S2 cells, purified by antibody affinity chromatography and HLPC and characterized for affinity to the urokinase receptor, for plasminogen activator activity and for fibrinolytic activity in the presence and absence of suPAR, as described previously (15-17). uPA migrates as a homogeneous -50 kDa protein on SDS-PAGE under reducing and non-reducing conditions.
Closed cranial window technique and cerebral hypoxia/ischemia
Newborn pigs (1-5 days, 1.2-1.6 Kg) of either sex were studied. All protocols were approved by the Institutional Animal Care and Use Committee. Animals were sedated with isoflurane (1-2 MAC). Anesthesia was maintained with a-chloralose (30-50 mg/ kg. supplemented with 5 mg / kg/h i.v.). A catheter was inserted into a femoral artery to monitor blood pressure and to sample for blood gas tensions and pH. Drugs to maintain anesthesia were administered through a second catheter placed in a femoral vein. The trachea was cannulated, and the animals were ventilated with room air. A heating pad was used to maintain the animals at 37° - 39° C, monitored rectally.
A cranial window was placed in the parietal skull of these anesthetized animals. This window consisted of three parts: a stainless steel ring, a circular glass coverslip, and three ports consisting of 17-gauge hypodermic needles attached to three precut holes in the stainless steel ring. For placement, the dura was cut and retracted over the cut bone edge. The cranial window was placed in the opening and cemented in place with dental acrylic. The volume under the window was filled with a solution, similar to CSF, of the following composition (in mM): 3.0 KCl, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3. This artificial CSF was warmed to 37° C and had the following chemistry: pH 7.33, pCO2 46 mm Hg, and pO2 43 mm Hg, which was similar to that of endogenous CSF. Pial arterial vessel diameter was measured with a microscope, a camera, a video output screen and a video microscaler.
Total cerebral ischemia was accomplished by infusing artificial CSF into a hollow bolt in the cranium to maintain an intracranial pressure 15 mm Hg greater than the numerical mean of systolic and diastolic arterial blood pressure (24,25). Intracranial pressure was monitored via a sidearm of the cranial window. To prevent the arterial pressure from rising inordinately (Cushing response), venous blood was withdrawn as necessary to maintain mean arterial blood pressure no greater than 100 mm Hg. As the cerebral ischemic response subsided, the shed blood was returned to the animal. Cerebral ischemia was maintained for 20 min. Hypoxia (PO2 of approximately 35 mm Hg) was produced for 10 min before ischemia by decreasing the inspired O2 via inhalation of N2, which was followed immediately by the total cerebral ischemia. Hypotension was induced by the rapid withdrawal of either 5-8 or 10-15 ml blood/Kg to induce moderate or severe hypotension (decreases in mean arterial blood pressure of 25 and 45%, respectively) (2). Such drops in blood pressure were maintained constant for 10 min by titration of additional blood withdrawal or blood reinfusion (2). Two levels of hypercapnia (low and high) were induced via inhalation of graded levels of a 10% CO2-21% O2-balance N2 gas mixture for 10 min to produce levels of pCO2 of 50-60 mm Hg for the low exposure and 70-80 mm Hg for the high exposure (20).
Protocol
Two types of pial vessels, small arteries (resting diameter, 120-160 μm) and arterioles (resting diameter, 50-70 μm) were examined to determine whether segmental differences in the effects of hypoxia/ischemia could be identified. Typically, 2-3 ml of artificial CSF were flushed through the window over a 30s period, and excess CSF was allowed to run off through one of the needle ports. For sample collection, 300 μl of the total cranial window volume of 500 μl was collected by slowly infusing artificial CSF into one side of the window and allowing the CSF to drip freely into a collection tube on the opposite side.
Nine experimental groups were studied (all n=6): (1) sham control, vehicle treated, (2) sham control, treated with uPA (10-7 M), (3) sham control, treated with the ERK MAPK inhibitor U 0126 (10-6 M topical, Sigma), (4) sham control, treated with suPAR (10-7 M), (5) hypoxia/ischemia, vehicle treated, (6) hypoxia/ischemia treated with mouse IgG (10 μg/ml), (7) hypoxia/ischemia treated with the LRP antagonist RAP (10-7 M), (8) hypoxia/ischemia treated with an anti LRP antibody (Ab) (10 μg/ml topical; American Diagnostica #3402, directed at the alpha chain of α2M-R/LRP), and (9) hypoxia/ischemia treated with U 0126. The vehicle for all agents was 0.9% saline, except for the MAPK inhibitor, which used dimethyl sulfoxide (100 μl) diluted with 9.9 ml 0.9% saline. In sham control animals, responses to hypercapnia, hypotension, isoproterenol (10-8, 10-6 M), and uPA (10-9, 10-7 M) were obtained initially and then again 1 and 4h later in the presence of the agent vehicle. In drug treated sham animals, responses to these stimuli were obtained before and after either uPA, U 0126 or suPAR. Drugs were administered topically for 1h and then co-administered with stimuli. In hypoxia/ischemia animals, responses to vasoactive stimuli were obtained initially and then again 1 and 4h post insult in the presence of the agent vehicle. In drug treated hypoxia/ischemia animals, drugs were administered 30 min before hypoxia/ischemia and the insult protocol followed as described above. CSF samples for ERK MAPK determination were also collected in animals given topical uPA (10-7 M) 4h post hypoxia/ischemia to determine if exogenous plasminogen activator would further augment ERK MAPK in the CSF under insult conditions.
ELISA
Commercially available ELISA Kits were used to quantity CSF uPA (Diapharma) and ERK MAPK (Assay Designs) concentration. The amounts of phosphorylated ERK MAPK enzyme were normalized to total ERK MAPK protein and then expressed as percent of the control condition.
Immunohistochemistry
Four hours after cerebral hypoxia/ischemia, the animal was sacrificed and 2 thin slices of parietal cortex were cut parallel to the brain surface. These slices were placed in 4% paraformaldehyde for 24h at 4° C and then subjected to Paraffin sectioning. Paraffin sections of the parietal cortex from piglet brains after hypoxia/ischemia and from uninjured sham control animals were unwaxed, were incubated in 10mM sodium citrate buffer pH 6.0 inside a food steamer (sub boiling temperature) for 10 minutes to unmask the antigen, endogenous peroxidase was blocked with 0.3 % H2O2, and stained with anti human uPA monoclonal Antibody (5 μg/ml) (#3689 American Diagnostica), anti-Phospho-p44/42 MAPK rabbit monoclonal antibody which recognize the phosphorylated forms of both p42 and p44 kinases (ERK1 and ERK2) (1 μg/ml, Cell Signaling #4376), or with mouse IgG1 as a negative control, secondary biotinylated anti-mouse IgG (1:200), followed by incubation with HRP-conjugated streptavidin. Peroxidase was detected using the avidin-biotin complex ABC Kit (Vector Lab) counterstained with hematoxylin. Positive staining is visualized by the brown colored [3,3-diaminobenzidine (DAB) reaction product. The cross reactivity of #3689 antibodies with porcine uPA was tested and confirmed first by performing a staining using kidney, an organ that is known to express uPA, from the same piglets (data not shown). A 5 point scale (0-1 minimal, 2-3 moderate, 4-5 maximal) was used to denote qualitative changes in immunocytochemical stain detection. Sections were also stained by H&E for histological inspection by investigators blinded to treatment.
Western analysis
Western blotting was performed on CSF samples collected before the injury and 4 hrs after the injury. Protein concentration was measured in each sample by BCA protein assay (Pierce) and 20 ug of total protein was loaded per well on 4-12% Nu-PAGE SDS gel (Invitrogen). Gels were transferred onto nitrocellulose membrane at 25 V for 2 hrs. in TRIS-Glycine Transfer Buffer(12 mM Tris Base, 96 mM Glycine, pH 8.3). After transfer, the membrane was blocked in 5% dried milk in TBS Buffer with 0.05% Tween20 for 1 hr and incubated overnight at 4 C in either p44/42 MAP Kinase Antibody or Phospho-p44/42 MAPK Rabbit mAb (both from Cell Signaling Technology) diluted 1:1000 in 2% BSA in TBS-0.05% Tween20. The membrane was washed in TBS-0.05% Tween20 and incubated for 1 hr. with the Hosreradish Peroxidase (HRP) labeled secondary antibodies diluted 1:10000 in 5% dried milk in TBS-0.05% Tween20. Detection was performed using the ECL Plus Blotting Detection Kit (Amersham Biosciences), per the manufacturer's instructions.
Statistical analysis
uPA and ERK MAPK staining were semi quantitatively determined through the use of a five point scale (0, no stain; 5, maximal stain) by an investigator blinded to treatment. Pial artery diameter, CSF uPA, and ERK MAPK values were analyzed using ANOVA for repeated measures. If the value was significant, the data were then analyzed by Fishers protected least significant difference test. An α level of p<0.05 was considered significant in all statistical tests. Values are represented as mean ± SEM of the absolute value or as percentage changes from control value.
Acknowledgments
This research was funded by grants from the National Institutes of Health, NS53410 and HD57355 (WMA), HL76406, CA83121, HL76206, HL07971, and HL81864 (DBC), HL77760 and HL82545 (AARH), the University of Pennsylvania Research Foundation (WMA), the University of Pennsylvania Institute for Translational Medicine and Therapeutics (DBC), and the Israeli Science Foundation (AARH). The authors thank Amy Christine for her help in the performance of these experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Nat Acad Sci. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstead WM. NOC/oFQ and NMDA contribute to piglet hypoxic ischemic hypotensive cerebrovasodilation impairment. Pediatr Res. 2002;51:586–591. doi: 10.1203/00006450-200205000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Armstead WM, Cines DB, Higazi AA. Plasminogen activators contribute to impairment of hypercapnic and hypotensive cerebrovasodilation after cerebral hypoxia/ischemia in the newborn pig. Stroke. 2005;36:2265–2269. doi: 10.1161/01.STR.0000181078.74698.b0. [DOI] [PubMed] [Google Scholar]
- 4.Armstead WM, Cines DB, Higazi AA. Plasminogen activators contribute to age dependent impairment of NMDA cerebrovasodilation after brain injury. Dev Brain Res. 2005;156:139–146. doi: 10.1016/j.devbrainres.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Bdeir K, Murciano JC, Tomaszewski J, Koniaris L, Martinez J, Cines DB, Muzykantov VR, Higazi AA. Uorkinase mediates fibrinolysis in the pulmonary microvasculature. Blood. 2000;96:1820–1826. [PubMed] [Google Scholar]
- 6.Benchenane K, Berezowski V, Carine A, Fernandez-Monreal M, Lopez-Atalaya JP, Brillault J, Chuquet J, Nouvelot A, MacKenzie ET, Bu G, Cecchelli R, Touzani O, Vivien D. Tissue-type plasminogen activator crosses the intact blood brain barrier by low-density lipoprotein receptor-related protein mediated transcytosis. Circ. 2005;111:2241–2249. doi: 10.1161/01.CIR.0000163542.48611.A2. [DOI] [PubMed] [Google Scholar]
- 7.Benchenane K, Berezowski V, Fernandez-Monreal M, Brillault J, Valable S, Dehouck MP, Cecchelli R, Vivien D, Touzani O, Ali C. Oxygen glucose deprivation switches the transport of tPA across the blood brain barrier from an LRP-dependent to an increased LRP-independent process. Stroke. 2005;36:1059–1064. doi: 10.1161/01.STR.0000163050.39122.4f. [DOI] [PubMed] [Google Scholar]
- 8.Bu G, Williams S, Strickland DR, Schwartz AL. Low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor is an hepatic receptor for tissue-type plasminogen activator. Proc Nat Acad Sci. 1992;89:7427–7431. doi: 10.1073/pnas.89.16.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collen D, Lijnen HR. Basic and clinical aspects of fibrinolysis and thrombolysis. Blood. 1991;78:3114–3124. [PubMed] [Google Scholar]
- 10.DeVeber G, Andrew M. Cerebral sinovenous thrombosis in children. N Engl J Med. 2001;345:417–423. doi: 10.1056/NEJM200108093450604. [DOI] [PubMed] [Google Scholar]
- 11.Ferriero DM. Neonatal brain injury. New Engl J Med. 2004;351:1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 12.Gunther G, Junker R, Strater R, Schobess R, Kurnik K, Kosch A, Nowak-Gottl U. Symptomatic ischemic stroke in full-term neonates. Stroke. 2000;31:2437–2441. doi: 10.1161/01.str.31.10.2437. [DOI] [PubMed] [Google Scholar]
- 13.Haj-Yehia A, Nassar T, Sachais BS, Kuo A, Bdeir K, Al Mehdi AB, Mazar A, Cines DB, Higazi AA. Urokinase-derived peptides regulate vascular smooth muscle contraction in vitro and in vivo. FASEB J. 2000;14:1411–1422. doi: 10.1096/fj.14.10.1411. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi T, Sakai K, Sasaki C, Zhang WR, Warita H, Abe K. c-JUN N-terminal kinase (JNK) and JNK interacting protein response in rat brain after transient middle cerebral artery occlusion. Neuroscience Lett. 2000;284:195–199. doi: 10.1016/s0304-3940(00)01024-7. [DOI] [PubMed] [Google Scholar]
- 15.Higazi AA, Bdeir K, Hiss E, Arad S, Kuo A, Barghouti I, Cines DB. Lysis of plasma clots by urokinase- soluble urokinase receptor complexes. Blood. 1998;92:2075–2083. [PubMed] [Google Scholar]
- 16.Higazi AA, Mazar A, Wang J, Reilly R, Henkin J, Kniss D, Cines D. Single-chain urokinase-type plasminogen activator bound to its receptor is relatively resistant to plasminogen activator inhibitor type 1. Blood. 1996;87:3545–3549. [PubMed] [Google Scholar]
- 17.Higazi AA, Upson RH, Cohen RL, Manuppello J, Bognacki J, Henkin J, McCrae KR, Kounnas MZ, Strickland DK, Preissner KT, Lawler J, Cines DB. Interaction of single-chain urokinase with its receptor induces the appearance and disappearance of binding epitopes within the resultant complex for other cell surface proteins. Blood. 1996;88:542–551. [PubMed] [Google Scholar]
- 18.Hosomi N, Jucero J, Heo JH, Koziol JA, Copeland BR, del Zoppo GJ. Rapid differential endogenous plasminogen activator expression after acute middle cerebral artery occlusion. Stroke. 2001;32:1341–1348. doi: 10.1161/01.str.32.6.1341. [DOI] [PubMed] [Google Scholar]
- 19.Hu BR, Liu CL, Park DJ. Alteration of MAP kinase pathways after transient forebrain ischemia. J Cereb Blood Flow Metab. 2000;20:1089–1095. doi: 10.1097/00004647-200007000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Jagolino AL, Armstead WM. PTK MAPK, and NOC/oFQ impair hypercapnic cerebrovasodilation after hypoxia/ischemia. Am J Physiol. 2003;284:H101–H107. doi: 10.1152/ajpheart.00457.2002. [DOI] [PubMed] [Google Scholar]
- 21.Kim YH, Park JH, Hong SH, Koh JY. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999;284:647–50. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- 22.Kraus FT, Acheen VI. Fetal thrombotic vasculopathy in the placenta: cerebral thrombi and infarcts, coagulopathies, and cerebral palsy. Hum Pathol. 1999;30:759–769. doi: 10.1016/s0046-8177(99)90136-3. [DOI] [PubMed] [Google Scholar]
- 23.Laher I, Zhang JH. Protein kinase C and cerebral vasospasm. J Cerebral Blood Flow & Metabolism. 2001;21:887–906. doi: 10.1097/00004647-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Leffler CW, Busija DW, Armstead WM, Mirro R, Beasley DG. Ischemia alters cerebral vascular responses to hypercapnia and acetylcholine in piglets. Pediatr Res. 1989;25:180–183. doi: 10.1203/00006450-198902000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Leffler CW, Busija DW, Beasley DG, Armstead WM, Mirro R. Postischemic microvascular cerebral responses to norepinephrine and hypotension in newborn pigs. Stroke. 1989;20:541–546. doi: 10.1161/01.str.20.4.541. [DOI] [PubMed] [Google Scholar]
- 26.Nassar T, Haj-Yehia A, Akkawi S, Kuo A, Bdeir K, Mazar A, Cines DB, Higazi AA. Binding of urokinase to low density lipoprotein-related receptor (LRP) regulates vascular smooth muscle cell contraction. J Biol Chem. 2002;277:40499–40504. doi: 10.1074/jbc.M207172200. [DOI] [PubMed] [Google Scholar]
- 27.Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–158. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- 28.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMD receptor-mediated signaling. Nature Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 29.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Volpe JJ. Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol. 1998;5:135–151. doi: 10.1016/s1071-9091(98)80030-2. [DOI] [PubMed] [Google Scholar]
- 31.Wang YF, Tsirka SE, Strickland S, Stiege PE, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 32.Webb DJ, Nguyen DH, Gonias SL. Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and matrigel invasion. J Cell Sci. 2000;113:123–134. doi: 10.1242/jcs.113.1.123. [DOI] [PubMed] [Google Scholar]