

Abstract
Cytochrome P450 enzymes mediate important oxidative processes in biological systems including regio- and stereo-specific hydroxylation and epoxidation reactions. The inherent requirement of these biomolecules for separate redox partner(s) significantly limits their application in biotechnology. To address this challenge, naturally occurring and/or bio-engineered self-sufficient P450 systems with covalently fused redox partners have been utilized to harness their catalytic power. In this study, we describe the first in vitro characterization of a bacterial biosynthetic cytochrome P450 PikC fused to a heterologous reductase domain RhFRED that demonstrates single-component self-sufficiency. This novel fusion system not only produces a more active and effective biocatalyst, but also suggests a general design for a universal reductase to generate diverse self-sufficient fusions for functional identification or industrial applications of biosynthetic P450s.
Cytochrome P450 enzymes (P450s) are highly attractive biocatalysts due to their ability to catalyze a variety of regio- and stereo-specific oxidation reactions of complex organic compounds. These reactions occur under mild conditions by taking advantage of the two-electron activated dioxygen that is often challenging in organic synthesis.1 To activate molecular oxygen, redox partners are required to sequentially transfer two reducing equivalents from NAD(P)H to P450.2 Classically, there are two major redox partner systems, including an FAD containing reductase with a small iron-sulfur (Fe2S2) redoxin for most bacterial and mitochondrial P450s (Class I), and a single FAD/FMN containing flavoprotein for eukaryotic microsomal P450s (Class II).3 The inherent requirement of cytochome P450s for separate protein partner(s) significantly limits their application in biotechnology.
The discovery of the first self-sufficient P450BM3, which is naturally fused to a eukaryotic-like reductase represents an effective solution to this limitation.4 The fusion nature of this enzyme dramatically improves electron transfer efficiency and coupling with the oxidative process, enabling it to be the most efficient P450 enzyme characterized to date.5 Based upon the self-sufficiency of this naturally fused enzyme, a number of engineered proteins of diverse eukaryotic P450s bearing a reductase domain from P450BM3 have been generated with in vitro activities.6 This provides ready access to the great catalytic versatility of the membrane-bound eukaryotic P450s. In contrast, the biosynthetic P450s (Class I) lack such a universal reductase that can be used to engineer diverse self-sufficient P450s for either functional identification or potential industrial application.
Recently, a new class of self-sufficient cytochrome P450s exemplified by P450RhF from Rhodococcus sp. NCIMB 9784 was discovered to be naturally fused to a novel FMN/Fe2S2 containing reductase partner.7 Although the physiological function of P450RhF remains unknown, its reductase domain (RhFRED), which is similar to the phthalate family of dioxygenase reductases, is capable of transferring electrons from NADPH to the heme domain of the monooxygenase, supporting 7-ethoxycoumarin dealkylation activity.8 Moreover, recent reports from Misawa et al. demonstrated that this reductase domain could be used to reconstitute the catalytic activities of various Class I P450s in vivo through expression of corresponding genes fused to RhFRED in Escherichia coli cells.9 This suggests that RhFRED might be developed into a generally effective redox partner for biosynthetic bacterial P450s. However, the lack of corresponding in vitro data could not unambiguously exclude in trans involvement of additional cellular redox partners.
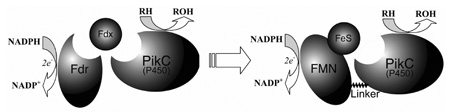
Herein, we describe the first in vitro characterization of a single component bacterial biosynthetic cytochrome P450 fused to RhFRED that demonstrates high catalytic efficiency. The PikC cytochrome P450 in this study is involved in the pikromycin biosynthetic pathway of Streptomyces venezuelae.10 PikC catalyzes the final hydroxylation step toward both the 12-membered ring macrolactone YC-17 (1) and the 14-membered ring macrolactone narbomycin (4) to produce methymycin/neomethymycin (2/3) and pikromycin (5) as major products (Scheme 1).11 Recently, we elucidated the structural basis for the remarkable substrate flexibility by analyzing ligand-free and substrate-bound structures of PikC.12 However, since the native redox partner of PikC remains unknown, its in vitro activity has depended on expensive spinach ferredoxin reductase (Fdr) and ferredoxin (Fdx) (Scheme 2A), as are many other biosynthetic P450s.13 To investigate an alternative electron transfer pathway mimicking the fusion organization in P450RhF (Scheme 2B), the pikC gene was linked to the RhFRED gene including the native 16 amino acid linker sequence. The hybrid gene was cloned into pET28b(+), and overexpressed in E. coli BL21 (DE3) to generate N-terminal His6-tagged PikC-RhFRED. After Ni-NTA chromatography, the purified red-colored recombinant P450 displayed (upon reduction) the signature peak at 450 nm in the CO-difference spectrum. Interestingly, gel filtration chromatography indicated that PikC-RhFRED predominantly dimerizes in storage buffer solution containing 0.2 mM dithioerythritol (DTE). In contrast, wild type (wt) PikC was shown to be monomeric under the same conditions. It was thus unclear whether the inter-monomer electron transfer could occur in the dimeric PikC-RhFRED as in P450BM3.14
Scheme 1.

Major physiological reactions catalyzed by PikC
Scheme 2.

Two redox partner systems (electron transfer pathways) used in this study for PikC. A, three components system; B, one component RhFRED system.
We next tested the ability of PikC-RhFRED to hydroxylate 1 and 4 in vitro when provided electron donor NADPH. We were gratified to observe that this chimeric protein showed significantly improved catalytic activity compared to wt PikC in the presence of exogenous redox partners (spinach Fdr and Fdx), producing higher yields of 2/3 and 5 under identical reaction conditions (Figure 1). This result unambiguously confirms that PikC-RhFRED is a self-sufficient P450 enzyme. Interestingly, we also constructed the pET21b(+)-pikC-RhFRED and obtained the purified C-terminal His6-tagged PikC-RhFRED. This protein showed a similar CO-difference spectrum as its N-terminal His6-tagged counterpart (Data not shown). However, it lacks catalytic activity, which is consistent with a similar C-terminal His6-tagged form of original P450RhF.8 This provides additional evidence for the importance of the C terminus of RhFRED for electron transfer.
Figure 1.

HPLC analysis of reactions (1 h) catalyzed by wt PikC and fusion enzyme PikC-RhFRED. a) Negative control of 1 in absence of P450. b) 1 with wt PikC in presence of Fdr, Fdx, and NADPH. c) 1 with PikC-RhFRED in presence of only NADPH. d) Negative control of 4 in absence of P450. e) 4 with wt PikC in presence of Fdr, Fdx, and NADPH. f) 4 with PikC-RhFRED in presence of only NADPH.
As mentioned above, one benefit of the fusion arrangement is that the covalent linkage presumably stabilizes the interaction between the P450 and redox partner, thus enhancing electron transfer efficiency. As such, one would expect this to improve the catalytic activity in terms of kcat, whereas the substrate specificity would not be changed significantly.15 To test whether this also applies to PikC-RhFRED, we first determined the substrate binding affinity of 1 and 4 toward both PikC and PikC-RhFRED. As expected, 1 and 4 binds to PikC-RhFRED with Kd values of 92.6 ± 0.5 µM and 215.0 ± 4.2 µM, respectively, which are similar to 112.9 ± 1.9 µM (1) and 288.3 ± 7.1 (4) toward wt PikC. This indicates that attachment of the heterologous reductase domain has no significant impact on substrate binding to PikC. Subsequently, we compared the kinetic parameters of PikC-RhFRED with those of the PikC-Fdr-Fdx three component system. As previously reported,11,16 substrate inhibition was observed in all cases when substrate concentration was greater than 250 µM. Moreover, the solubility limitation (less than 500 µM) of macrolides in aqueous solution prevented us from deducing the Ki value. Therefore, we determined the apparent specificity constants (kcat/Km) by fitting the low-concentration data to the linear region of the Michaelis-Menten curve. By directly monitoring the substrate consumption by HPLC, the kcat/Km values of PikC-RhFRED were determined to be 0.96 and 1.20 µM−1·min−1 for 1 and 4, respectively. In contrast, the specificity constants of wt PikC partnered by Fdr and Fdx were 0.24 µM−1·min−1 for 1 and 0.31 µM−1·min−1 for 4. It is evident that the fusion enhanced the catalytic activity approximately 4 fold for both 1 and 4. Notably, the kinetic parameters of wt PikC differ significantly from those previously determined indirectly, using a NADPH depletion assay,11,16 suggesting the stoichiometric ratio between NADPH and substrate hydroxylation could not be 1:1. The presumed de-coupling between electron transfer and hydroxylation might account for this difference.
Finally, when RhFRED was fused to another prototype biosynthetic P450 EryF,13 a more active self-sufficient biocatalyst was obtained once again (See supporting information). Together with previous in vivo work,9 our studies demonstrate that further development of RhFRED as the basis for an efficient cost-effective redox partner for bacterial biosynthetic P450s is warranted. Further efforts to understand this unique reductase, especially the electron transfer process involving heterologous fusion systems are now in progress.
Supplementary Material
Plasmid maps for constructs, SDS-PAGE analysis, UV-visible absorption spectrum for PikC-RhFRED, Gel filtration chromatography results, Substrate binding affinity measurements, Steady-state kinetics data, Mass spectrometry data for EryF-RhFRED reaction analysis, Experimental procedures.
ACKNOWLEDGMENT
We thank Dr. Norihiko Misawa for generously providing the construct pRED containing reductase domain gene from P450RhF, and Dr. Gary W. Ashley for the gift of 6-deoxyerythronolide. The authors thank Dr. Jeffrey D. Kittendorf and Liangcai Gu for helpful discussions. We are grateful for support from NIH RO1 grant GM078553 (to D.H.S. and L.M.P.).
References
- 1.(a) Ortiz de Montellano PR. In: Cytochrome P450: Structure, Mechanism and Biochemistry. 2nd ed. Ortiz de Montellano PR, editor. New York: Plenum Press; 1995. p. 473. [Google Scholar]; (b) Guengerich FP. Chem. Res. Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 2.Hannemann FBA, Ewen KM, Bernhardt R. Biochim. Biophys. Acta. 2007;1770:330. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lewis DFV, Hlavica P. Biochim. Biophys. Acta. 2000;1460:353. doi: 10.1016/s0005-2728(00)00202-4. [DOI] [PubMed] [Google Scholar]; (b) Munro AW, Girvan HM, McLean KJ. Nat. Prod. Rep. 2007;24:585. doi: 10.1039/b604190f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ruettinger RT, Fulco AJ. J. Biol. Chem. 1981;256:5728. [PubMed] [Google Scholar]; (b) Otey CR, Bandara G, Lalonde J, Takahashi K, Arnold FH. Biotechnol. Bioeng. 2005;93:494. doi: 10.1002/bit.20744. [DOI] [PubMed] [Google Scholar]
- 5.Munro AW, Leys DG, McLean KJ, Marshall KR, Ost TWB, Daff S, Miles CS, Chapman SK, Lysek DA, Moser CC, Page CC, Dutton PL. Trends Biochem. Sci. 2002;27:250. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 6.(a) Fairhead M, Giannini S, Gillam EMJ, Gilardi G. J. Biol. Inorg. Chem. 2005;10:842. doi: 10.1007/s00775-005-0033-1. [DOI] [PubMed] [Google Scholar]; (b) Dodhia VR, Fantuzzi A, Gilardi G. J. Biol. Inorg. Chem. 2006;11:903. doi: 10.1007/s00775-006-0144-3. [DOI] [PubMed] [Google Scholar]
- 7.(a) De Mot R, Parret AHA. Trends Microbiol. 2002:502. doi: 10.1016/s0966-842x(02)02458-7. [DOI] [PubMed] [Google Scholar]; (b) Roberts GA, Grogan G, Greter A, Flitsch SL, Turner NJ. J. Bacteriol. 2002;184:3898. doi: 10.1128/JB.184.14.3898-3908.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Roberts GA, Çelik A, Hunter DJB, Ost TWB, White JH, Chapman SK, Turner NJ, Flitsch SL. J. Biol. Chem. 2003:48914. doi: 10.1074/jbc.M309630200. [DOI] [PubMed] [Google Scholar]; (b) Hunter DJB, Roberts GA, Ost TWB, White JH, Müller S, Turner NJ, Flitsch SL, Chapman SK. FEBS Lett. 2005;579:2215. doi: 10.1016/j.febslet.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kubota M, Nodate M, Yasumoto-Hirose M, Uchiyama T, Kagami O, Shizuri Y, Misawa N. Biosci. Biotechnol. Biochem. 2005;69:2421. doi: 10.1271/bbb.69.2421. [DOI] [PubMed] [Google Scholar]; (b) Nodate M, Kubota M, Misawa N. Appl. Microbiol. Biotechnol. 2006;71:455. doi: 10.1007/s00253-005-0147-y. [DOI] [PubMed] [Google Scholar]
- 10.Xue Y, Zhao L, Liu H-w, Sherman DH. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12111. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Xue Y, Wilson D, Zhao L, Liu H-w, Sherman DH. Chem. Biol. 1998;5:661. doi: 10.1016/s1074-5521(98)90293-9. [DOI] [PubMed] [Google Scholar]; (b) Lee SK, Park JW, Kim JW, Jung WS, Park SR, Choi CY, Kim ES, Ahn JS, Sherman DH, Yoon YJ. J. Nat. Prod. 2006;69:847. doi: 10.1021/np060026p. [DOI] [PubMed] [Google Scholar]
- 12.Sherman DH, Li S, Yermalitskaya LV, Kim Y, Smith JA, Waterman MR, Podust LM. J. Biol. Chem. 2006;281:26289. doi: 10.1074/jbc.M605478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Lambalot RH, Cane DE. Biochemistry. 1995;34:1858. doi: 10.1021/bi00006a006. [DOI] [PubMed] [Google Scholar]; (b) Andersen JF, Hutchinson RC. J. Bacteriol. 1992;174:725. doi: 10.1128/jb.174.3.725-735.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ogura H, Nishida CR, Hoch UR, Perera R, Dawson JH, Ortiz de Montellano PR. Biochemistry. 2004;43:14712. doi: 10.1021/bi048980d. [DOI] [PubMed] [Google Scholar]
- 14.Neeli R, Girvan HM, Lawrence A, Warren MJ, Leys D, Scrutton NS, Munro AW. FEBS Lett. 2005;579:5582. doi: 10.1016/j.febslet.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 15.(a) Munro AW, Girvan HM, McLean KJ. Biochim. Biophys. Acta. 2007;1770:345. doi: 10.1016/j.bbagen.2006.08.018. [DOI] [PubMed] [Google Scholar]; (b) Yabusaki Y. Biochimie. 1995;77:594. doi: 10.1016/0300-9084(96)88175-2. [DOI] [PubMed] [Google Scholar]
- 16.Graziani EI, Cane DE, Betlach MC, Kealey JT, McDaniel R. Bioorg. Med. Chem. Lett. 1998:3117. doi: 10.1016/s0960-894x(98)00553-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plasmid maps for constructs, SDS-PAGE analysis, UV-visible absorption spectrum for PikC-RhFRED, Gel filtration chromatography results, Substrate binding affinity measurements, Steady-state kinetics data, Mass spectrometry data for EryF-RhFRED reaction analysis, Experimental procedures.