

Abstract
Microenvironment molecular cues direct T helper (Th) cell differentiation; however, Th17 fate determination is still imprecisely understood in humans. To assess the role of prostaglandin E2 (PGE2) in Th expansion, we activated peripheral blood mononuclear cells by CD3 cross-linking. In the presence of exogenous PGE2, peripheral blood mononuclear cells produced higher interleukin-17 (IL-17), C-C chemokine ligand 20 (CCL20)/macrophage inflammatory protein 3α (MIP-3α), CXC chemokine ligand 8 (CXCL8)/IL-8, and lower interferon-γ and IL-22 levels than in control cultures. Exogenous PGE2 and IL-23 synergized in inducing IL-17, whereas indomethacin and IL-23 blockade drastically reduced IL-17 but not interferon-γ production. Furthermore, IL-1 but not tumor necrosis factor was absolutely required for IL-17 production. PGE2 doubled the frequency of CD4+ T cells producing IL-17 and within the CD4+ subset enhanced C-C chemokine receptor 6 (CCR6) and CCR4 while decreasing CXC chemokine receptor 3 (CXCR3) expression. Furthermore, in CD4+ T-cell lines, the production of IL-17 segregated with the CCR6+ subset. In the presence of CCR6+ compared with CXCR3+ Th cells, monocytes/macrophages produced much higher levels of matrix metalloproteinase-1, -3, and -9 but similar levels of CXCL10 and IL-1β. These results identify PGE2 and IL-23 as participating in the expansion of CD4+ T cells endowed with high IL-17 production capacity, which in turn favors monocyte production of mediators important for host defense and tissue destruction.
Introduction
T helper (Th) cells may differentiate into distinct functional subsets according to the circumstances in which naive precursors encounter the nominal antigen for which they are specific. In the presence of interleukin-12 (IL-12), Th precursor cells become Th1 and high producers of interferon-γ (IFN-γ), whereas in the presence of IL-4 they become Th2 and high producers of IL-4 and IL-5. These subsets have fundamentally different effector functions, in particular for their capacity to induce monocytes activation.1 Recently, a distinct Th subset has been characterized in mice2–6 and humans7–11 for its high production of IL-17, the use of retinoid-related orphan receptor γt (RORγt) as master transcription factor,12 its role in protection against extracellular bacteria and fungi, and in the pathogenesis of several autoimmune conditions, such as collagen-induced arthritis, experimental autoimmune encephalomyelitis, and an animal model of inflammatory bowel disease.4,13,14 Of interest, the generation of Th17 from naive precursors may follow different requirements in mice and humans. In mice, commitment to the Th17 lineage is dependent on transforming growth factor-β (TGF-β), IL-1β, and IL-6, whereas in humans IL-1β and IL-6 but not TGF-β appear to be required.10,15–18 The role of IL-23 in Th17 differentiation and effector function is still debated. IL-23 is a member of the IL-6 family of cytokines that shares with IL-12 the p40 subunit and has a unique p19 subunit.19 IL-23 promotes Th17 responses in vivo but may be more important for the survival and population expansion of Th17 cells than for Th17 lineage commitment.3,5,16 However, in conjunction with IL-1, IL-23 is sufficient for inducing naive human T cells to produce IL-17A, IL-17F, IL-22, IL-26, IFN-γ, C-C chemokine ligand 20 (CCL20)/macrophage inflammatory protein 3α (MIP-3α), and the transcription factor RORγt.10 Moreover, mice lacking IL-23 are fully resistant to experimental autoimmune encephalomyelitis, collagen-induced arthritis, and inflammatory bowel disease
The pattern of chemokine receptors expressed varies according to the differentiation stage and effector function of T cells.20 The preferential expression of C-C chemokine receptor 6 (CCR6) distinguishes human Th17 from other Th subsets.7,8,21,22 It should be stressed, however, that expression of CXC chemokine receptor 3 (CXCR3) within CCR6+ T cells identifies T cells with preferential IFN-γ production in response to purified protein derivative, whereas IL-17 production in response to Candida albicans hyphae is restricted to CCR6+CCR4+ cells.7
IL-17, when overexpressed in murine knee joint, causes inflammation and bone erosion.23 Likewise, erosion observed in streptococcal cell wall-induced arthritis is highly reduced in IL-17R−/− and is associated with impaired expression of matrix metalloproteinases (MMPs).24 T-cell clones producing IL-17 were generated from the synovium and synovial fluid of rheumatoid arthritis patients,25 and IL-17 was shown to directly favor the production of MMP-9 in human monocytes/macrophages.26 However, the effect of Th17 cells on monocytes has not yet been described.
Prostaglandin E2 (PGE2), a metabolite of arachidonic acid, is the most widely produced prostanoid in the body, particularly in response to inflammatory cytokines.27,28 PGE2 affects the capacity of antigen-presenting cells to produce a variety of cytokines reducing the production of IL-12 p35, IL-6, tumor necrosis factor (TNF), while increasing the production of IL-10, IL-12p40, and IL-23, and may therefore impact on the functional phenotype of T cells during priming.29,30 In the present paper, we explored the capacity of PGE2 to affect IL-17 production by human peripheral blood mononuclear cells. We specifically asked the question whether exogenous and endogenous PGE2 could impact on Th17 expansion and chemokine receptor expression in Th cells and the role of IL-23, IL-1, and TNF in these settings. We further ascertained whether CD4+ T cells cultured in the presence of PGE2 could be enriched in cells particularly potent in inducing monocytes/macrophages to produce mediators involved in inflammation, host defense, and tissue destruction.
Methods
Reagents
Phorbol myristate acetate (PMA), β-mercaptoethanol, brefeldin A, PGE2, and indomethacin were from Sigma-Aldrich (St Louis, MO); ionomycin from Calbiochem (San Diego, CA); RPMI 1640 medium, phosphate-buffered saline, penicillin, streptomycin, L-glutamin, nonessential amino acids, and sodium pyruvate from Invitrogen (Carlsbad, CA); fetal calf serum (FCS) from Amimed (BioConcept, Allschwil, Switzerland); Lymphoprep from Axis-Shild PoCAS (Oslo, Norway); phytohemagglutinin from EY Laboratories (San Mateo, CA); DNase I from Roche Diagnostics (Basel, Switzerland); anti-CD3 CD28 beads from Dynal Biothech ASA (Oslo, Norway); human recombinant (r) IL-2 and IL-1β from Biogen (Cambridge, MA); human rIL-4 from Sandoz (Basel, Switzerland); human rIL-12, rIL-23, and anti–IL-12 monoclonal antibody (mAb) from R&D Systems (Minneapolis, MN); anti–IFN-γ was a kind gift from Dr W. Ferlin (Novimmune, Geneva, Switzerland.); anti–IL-4 25D2.11 mAb and OKT3 from ATCC (Manassas, VA); anti-CD4, CD8, CD14, CD16, CD19, CD45RO, and irrelevant, isotype-matched controls from Dako (Copenhagen, Denmark); streptavidin-phycoerythrin, anti–IFN-γ, anti–IL-4, anti-CCR6, anti-CCR4, and anti-CXCR3 from BD Biosciences PharMingen (San Diego, CA); anti–IL-17A from eBioscience (San Diego, CA); anti-TNF (recombinant-methionyl soluble TNF-type I pegylated receptor [TNF-bp]); IL-1Ra from Amgen (Thousand Oaks, CA); and anti–IL-23p19 (M56) was a kind gift from Dr J. E. Sims (Amgen).
PBMCs and generation of T-cell lines
Peripheral blood was obtained by venipuncture from healthy members of the laboratory. Permission to perform this investigation was granted by the ethical committee of our institution and written informed consent in accordance with the Declaration of Helsinki from all blood donors. PBMCs were purified by Lymphoprep gradient centrifugation and cultured at 1 × 106 cells/mL in 24-well plates in RPMI 1640 medium supplemented with 10% FCS, penicillin (50 U/mL), streptomycin (50 μg/mL), 1% glutamine, 1% nonessential amino acids, 1% sodium pyruvate, and 50 μM 2-β mercaptoethanol (complete medium). Soluble OKT-3 (0.1 μg/mL) was used to activate PBMCs, and 20 U/mL IL-2 was added 48 hours later. When used, PGE2 (50 ng/mL), IL-23 (10 ng/mL), indomethacin (10−5 M), anti–IL-23 mAb (10 μg/mL), IL-1Ra (1 μg/mL), TNF-bp (10−8 M), and IL-1β (500 ng/mL) were added at the beginning of the culture. After 7 days of culture, the supernatants were collected and frozen until cytokine determination, and the cells were harvested and used for fluorescence-activated cell sorter (FACS) analysis.
Generation of polarized T-cell lines
Peripheral blood and naive CD4+ T cells were obtained by negative selection using Dynabeads pan mouse IgG and a cocktail of mAbs to CD8, CD14, CD16, and CD19 followed by sorting in CD4+CD45RO− and CD4+CD45RO+ T cells using FACSAria (BD Biosciences, San Jose, CA). Purified naive (CD4+CD45RO−) and memory (CD4+CD45RO+) T cells were activated by anti-CD3, anti-CD28 coated beads (Dynabeads CD3/CD28; Dynal Biotech, Oslo, Norway) in the presence or absence of PGE2 and expanded for 7 days with IL-2. To obtain Th1, naive CD4+ T cells were activated in the presence of IL-12 and anti–IL-4 and expanded by adding feeder cells and OKT-3 every 15 to 20 days.1,31 To generate Th17 cell lines, CD4+CD45RO+ T cells were cultured in the presence of 1.5 × 106 irradiated allogeneic PBMCs (feeder cells) and OKT-3 (0.1 μg/mL) in medium supplemented with IL-2, IL-23, and PGE2 and expanded by adding feeder cells and OKT-3 every 15 to 20 days always in the presence of IL-2, IL-23, and PGE2. Long-term Th17 cell lines from 2 distinct patients were further enriched in CCR6+ and CXCR3+ cell subsets by sorting using FACSAria.
Cocultures of T cells with monocytes
Fresh PBMCs were obtained by aggregation in the cold as described.32 Briefly, PBMCs were incubated at 50 × 106/mL for 30 minutes at 4°C under rotation leading to monocyte aggregation, followed by 10 minutes of incubation on ice. Pellets of aggregated monocytes were separated from nonaggregated PBMCs by a gradient of FCS. Monocyte-enriched aggregates were further depleted in T and NK cells by rosetting with neuraminidase-treated sheep red blood cells. Polymyxin B (1 μg/mL) was present throughout the whole procedure performed in polypropylene tubes (BD Biosciences). Monocyte purity routinely consisted of more than 85% CD14+ cells, less than 1.5% CD3+ cells, and less than 1% CD19+ cells. Fresh monocytes were seeded at 5 × 104 cells/well in 96-well plates. T cells were added at a T-cell/monocyte ratio of 1 to 1 to 200-μL final volume of RPMI-FCS 10%. After 48 hours of culture in 5% CO2-humidified air at 37°C, supernatants were harvested and stored at −20°C for further MMP or cytokine determination. In all cases, monocytes and T cells alone were used as controls in the presence or absence of 104 anti-CD3 anti-CD28 Dynabeads.
Flow cytometry
Surface expression of CD4, CCR6, CCR4, and CXCR3 was performed by 4-color FACS analysis acquiring more than 50 000 events per sample using FACSCalibur, and data were analyzed by Cellquest software (BD Biosciences). Four-color flow cytometry was used to simultaneously detect surface expression of accessory molecules and intracellular accumulation of cytokines as described.31 Briefly, cells were cultured for 270 minutes at 37°C in RPMI 1640 medium supplemented with 10% FCS, in the presence of PMA (50 ng/mL), ionomycin (1 μM), and brefeldin A (2.5 μg/mL). On fixation and permeabilization with Cytofix/Cytoperm (BD Biosciences PharMingen), cells were labeled with anti–IFN-γ fluorescein isothiocyanate and anti–IL-17 phycoerythrin mAb. In selected experiments, the cells were labeled with biotinylated anti-CD8 mAb (OKT8), washed, and incubated with streptavidin–Cy-Chrome and with anti-CD4 R-phycoerythrin.
Cytokine and MMP assays
IL-17, IFN-γ, IL-4, IL-1β, MMP-1, MMP-3, and MMP-9 were quantified in culture supernatants by a commercially available multiplex beads immunoassay (Fluorokine MAP Multiplex Human Cytokine Panel and Human MMP Panel; R&D Systems, Minneapolis, MN) according to the supplier's instructions, using a Bioplex 200 array reader (Bio-Rad, Hercules, CA), which uses Luminex xMAP Technology (Luminex, Austin, TX). IL-22, CXC chemokine ligand 8 (CXCL8)/IL-8, and CCL20/MIP-3α were assessed by commercial ELISA (R&D Systems).
Statistical analysis
The Student t test for paired samples was used to compare the different experimental conditions to the nil culture condition. P values less than .01 were considered significant.
Results
The role of PGE2 and IL-23 on IL-17 production and Th17 expansion
We first asked whether PGE2 would favor IL-17 production when nonfractionated PBMCs are activated by CD3 cross-linking and IL-2 is added 48 hours later. IL-17 levels almost doubled when PBMCs were cultured in the presence of exogenous PGE2 (Figure 1A). An effect similar in magnitude was observed when adding exogenous IL-23 to PBMCs (Figure 1A). Of major interest, added together, PGE2 and IL-23 had a synergistic effect with a more than 4-fold increase in IL-17 production (P = .009; Figure 1). Distinctly different effects of PGE2 were observed in IFN-γ because in the same culture supernatants IFN-γ decreased in the presence of PGE2 (Figure 1B). Of interest, in the same culture supernatants, the levels of CCL20/MIP-3α, which is also thought to be preferentially produced by Th17, were enhanced in the presence of PGE2 (Figure 1D). However, neither PGE2 nor IL-23 enhanced but rather reduced the production of IL-22 (Figure 1E). Finally, CXCL8/IL-8, mostly produced by monocytes/macrophages under T-cell influence, was greatly enhanced in the presence of PGE2 but not in the presence of IL-23, and no synergistic effect between IL-23 and PGE2 could be observed (Figure 1F). We also assessed the levels of IL-1β as an additional control for the effects of PGE2 on monocytes/macrophages because IL-1β may have a role on Th17 cell differentiation. However, the levels of IL-1β were similar across all the culture conditions (Figure 1C).
Figure 1.

PGE2 and IL-23 specifically enhance the production of IL-17 and some of the related cytokines by human PBMCs. (A) IL-17; (B) IFN-γ; (C) IL-1β; (D) CCL20/MIP-3α; (E) IL-22; (F) CXCL8/IL-8. PBMCs (1 × 106) were activated by CD3 cross-linking and cultured for 7 days before harvesting of supernatants. PGE2 (50 ng/mL) and IL-23 (10 ng/mL) were added at the beginning, and IL-2 (20 U/mL) was added after 48 hours of culture. IL-17, IFN-γ, and IL-1β were assessed by multiplex immunoassay, whereas CCL20/MIP-3α, IL-22, and CXCL8/IL-8 were assessed by ELISA. Box plots represent the 10th, 25th, 50th, 75th, and 90th percentiles of 11 distinct individual donors (except CXCL8/IL-8, n = 7). Circles represent outliers. *Significant differences (P < .01) compared with the nil culture condition by paired Student t test. Note that the IL-17 scale is logarithmic.
We then asked whether endogenous PGE2 and IL-23 could influence the production of IL-17. Consistent with the findings presented in Figure 1, a reduction of IL-17 was observed when endogenous PGE2 and IL-23 were blocked by indomethacin and anti–IL-23 mAb, respectively (Figure 2). The same intervention did not modify the levels of IFN-γ (Figure 2). When added jointly, indomethacin and anti–IL-23 decreased IL-17 production by 41% plus or minus 9% (mean ± SD, n = 5, P = .001), IFN-γ by 15% plus or minus 8.5% (not significant), and IL-1β by 20% plus or minus 20% (not significant). These data indicate that both endogenous and exogenous PGE2 and IL-23 specifically favor the production of IL-17 by PBMCs.
Figure 2.
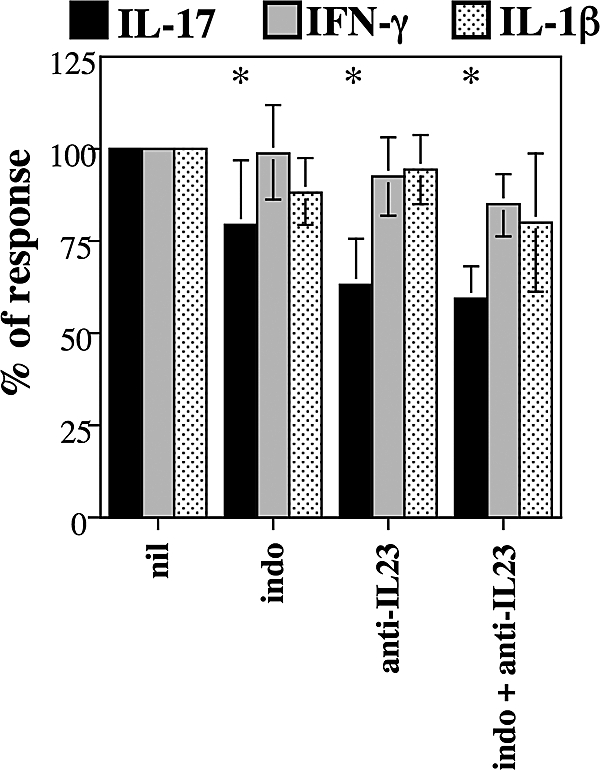
Endogenous PGE2 and IL-23 affect the production of IL-17 by human PBMCs. Culture conditions as in the legend of Figure 1. Indomethacin (10−5 M) and anti–IL-23 mAb (10 μg/mL) were added at the beginning of the culture. Cytokines were assessed by multiplex immunoassay. Bars represent the mean percentage plus or minus SD of the response of the nil culture condition of 9 distinct individual donors. In the nil culture condition, IL-17 was 198.9 (± 182.4) ng/mL, IFN-γ was 1693.9 (± 1365.5) ng/mL, and IL-1β was 617.0 (± 257.2) ng/mL. *Significant differences (P < .01) compared with nil by paired Student t test.
The increased production of IL-17 by PBMCs cultured in the presence of PGE2 and IL-23 could be the result of an increased capacity of the cells to produce this cytokine or to an increased number of cells producing IL-17. We found that PGE2 increased the frequency of IL-17–producing cells as assessed by intracellular staining, an effect enhanced to a marked extent by the addition of exogenous IL-23 (Figure 3A,B). Again, the increase in the frequency of IL-17+ cells was specific because in the same culture conditions the frequency of cells producing IFN-γ was reduced (Figure 3A,B). Of note, PGE2 increased the percentage of single-positive (IL-17+/IFN-γ−) as well as of double-positive (IL-17+/IFN-γ+) cells (Figure 3A). To assess whether the effect of PGE2 on T cells was directly or indirectly resulting from accessory cells, we activated freshly sorted naive and memory CD4+ T cells by CD3 and CD28 cross-linking in the absence of accessory cells. PGE2 profoundly affected the cytokine production also in these culture conditions. In memory CD4+ T cells, PGE2 induced a substantial increase in the proportion of both IL-17+ and IL-17/IFN-γ double-positive cells, whereas in both naive and memory CD4+ cells, PGE2 decreased IFN-γ single-positive cells (Figure 3C). Overall, these results indicate that PGE2 and IL-23 jointly favor the expansion of Th17 cells from the memory subset and that this effect does not require accessory cells.
Figure 3.
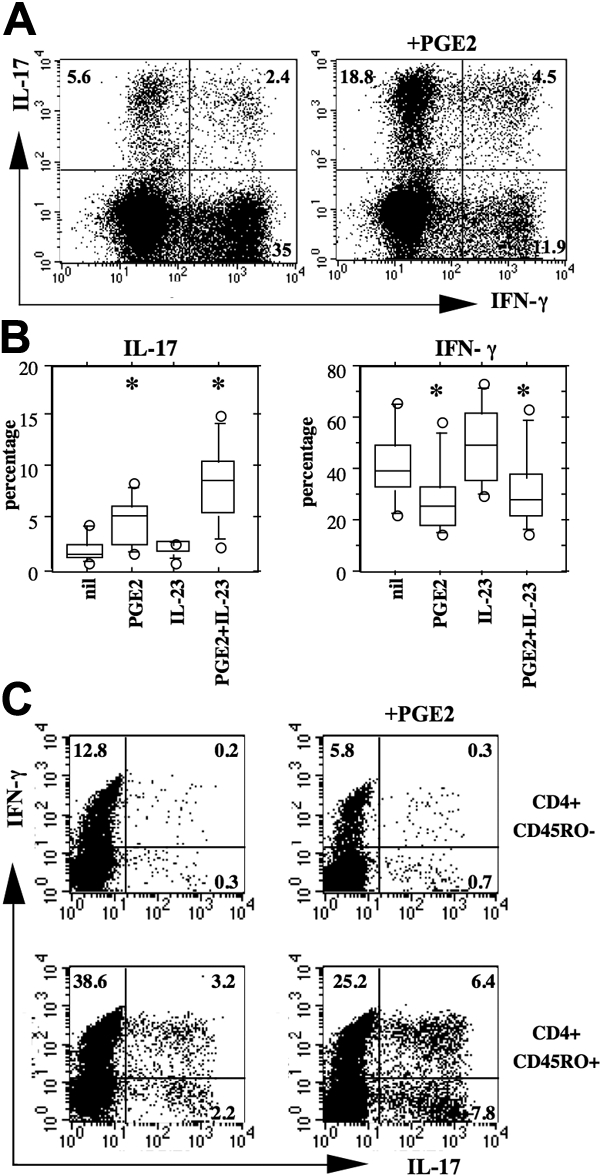
PGE2 and IL-23 favor Th17 expansion. (A,B) Culture conditions as in the legend of Figure 1. Cells were harvested after 7 days of culture and processed for intracellular staining on PMA and ionomycin activation as described in “Flow cytometry.” (A) Representative example of a culture in the presence of IL-23 (10 ng/mL) with or without PGE2 (50 ng/mL). (B) Box plots represent the 10th, 25th, 50th, 75th, and 90th percentiles of 7 distinct individual donors. Circles represent outliers. *Significant differences (P < .01) compared with the nil culture condition by paired Student's t test. Note that IL-17 positivity was restricted to CD4+ T cells. (C) Peripheral blood CD4+ T cells were sorted into CD45RO− (naive) and CD45RO+ (memory) subsets, activated by CD3/CD28-coated beads, and cultured in the presence of IL-2 (20 U/mL), IL-23 (10 ng/mL), with or without PGE2 (50 ng/mL). Experiment representative of similar results with cells from three distinct donors.
IL-1 requirement for the effect of PGE2 and IL-23 on IL-17 production by PBMCs
IL-1 has been identified as being a prerequisite for Th17 differentiation from human naive Th cells.9,10,18 We asked whether IL-1 was required for IL-17 production in PBMCs activated by CD3 cross-linking and cultured in the presence of PGE2 and IL-2. This was dramatically the case because IL-1Ra abated the production of IL-17 (Figure 4A). The IL-1Ra effect was particularly evident on IL-17 production, as far as in the same culture supernatants IFN-γ was decreased by only a fourth and IL-1β production was minimally reduced (Figure 5B,C). Furthermore, in contrast with IL-1 blockade, TNF blockade did not decrease but rather enhanced the production of IL-17. Thus, IL-1 and TNF do not have redundant roles in human Th17 expansion promoted by PGE2. We further explored the relationship between IL-1 and PGE2. When PBMCs were activated in the presence of an excess of IL-1β, the production of IL-17 and IFN-γ was not affected compared with basal conditions. However, PGE2 significantly increased IL-17 and decreased IFN-γ production in the presence of an excess of IL-1β (Figure 4D). Furthermore, the synergistic effect of PGE2 and IL-23 was observed also in the presence of an excess of IL-1β (Figure 4D). On the other hand, indomethacin did not significantly reduce the production of IL-17 observed in the presence of an excess of IL-1β. Thus, IL-1 is required and favors Th17 expansion but does not mediate the effect of PGE2.
Figure 4.

IL-1 but not TNF is required for the production of IL-17 by human PBMCs cultured in the presence of PGE2. Culture conditions as in the legend of Figure 1. IL-1Ra (1 μg/mL), TNF-bp (10−8 M), IL-1β (500 ng/mL), and indomethacin (10−5 M) were added at the beginning of the culture in the absence of exogenous IL-23 except when mentioned. IL-17, IFN-γ, and IL-1β were assessed by multiplex immunoassay in 7-day culture supernatants. Bars represent the mean plus or minus SD of 6 (A-C) or 4 (D) distinct individual donors. *Significant differences (P < .01) compared with the nil culture condition by paired Student t test.
Figure 5.

PGE2 favors the expression of CCR6 and CCR4 in CD4+ T cells. PBMCs were cultured as described in the legend of Figure 1. The cells were harvested after 7 days and submitted to 4-color FACS analysis. (A) Representative histogram of CD4 expression in PBMCs cultured in the absence (thin line) or presence (thick line) of PGE2 (50 ng/mL). (B) Representative histogram of CCR6, CCR4, and CXCR3 expression (thick line) in CD4+ T cells (thin line represents isotype control). (C) Representative dot blot of CCR4 and CXCR3 expression in CD4+CCR6+ T cells. (D) The bars represent the mean percentage plus or minus SD of CD4 and chemokine receptor expression of 11 distinct individual donors. *Significant differences (P < .01) compared with the nil culture condition by paired Student t test.
PGE2 favors the expression of CCR6 and CCR4 on CD4+ T cells
Th17 cells may preferentially express chemokine receptors that distinguish them from other Th subsets.7,8,21,22 We investigated whether PGE2 and IL-23 could affect the expression of chemokine receptors on PBMCs activated by CD3 cross-linking. First, we observed that PGE2 favors the in vitro expansion of CD4+ T cells as the percentage of these cells almost doubled in the presence of PGE2 (Figure 5A). Second, in the presence of PGE2, the percentage of CD4+-expressing CCR6 and CCR4 increased, whereas those expressing CXCR3 decreased (Figure 5B,D). In particular, PGE2 increased in the CD4+CCR6+ subset the CCR4+CXCR3− cells (Figure 5C). In this respect, IL-23 contribution was marginal and not significant (Figure 5D). These data indicate that the expression of chemokine receptors considered characteristic of Th17 cells is particularly enhanced by PGE2.
CCR6+IL-17+CD4+ T cells promote a proinflammatory and prodestructive phenotype in monocytes/macrophages
Taking advantage from the enhanced expansion of Th17 cells in the presence of PGE2 and IL-23, we generated long-term CD4+ T-cell lines enriched in Th17 cells from 2 distinct healthy persons. We further fractionated these cell lines in CCR6+ and CXCR3+ cells by cell sorting. IL-17–producing cells segregated with the CCR6+ subset with almost 70% of the cells producing IL-17 as detected by intracellular staining, more than half of which being IL-17 single positive (Figure 6B). In contrast, the CXCR3+ subset was virtually depleted in IL-17 and enriched in IFN-γ–producing cells (Figure 6C) and very much resembled Th1 cells (Figure 6D).
Figure 6.

IL-17 production in Th17 cell generated under the influence of PGE2 segregates with CCR6 expression. (A-C) CD4+ T cells were activated and expanded thereafter in the presence of irradiated allogeneic PBMCs and OKT-3 (0.1 μg/mL) in medium supplemented with IL-2, IL-23, and PGE2. CCR6+ (B) and CXCR3+ (C) were obtained from the parental Th17 cell line (A) by sorting using FACSAria. (D) Naive CD4+ T cells were activated by CD3 cross-linking in the presence of IL-12 and anti–IL-4 to obtain Th1 and expanded thereafter in the presence of IL-2. (A-D) The cells were harvested 10 days after sorting and processed for chemokine receptor expression and intracellular staining on PMA and ionomycin activation as described in “Flow cytometry.” The cells were all from the same patient and cultured for the same amount of time. The results here shown are representative of results generated with T-cell lines from two healthy persons.
The presence of IL-17 has been associated with several inflammatory conditions and tissue destruction including but not limited to the rheumatoid joint. We were therefore interested in exploring the capacity of cell lines highly enriched in Th17 cells to activate monocytes/macrophages and assess their production of MMPs and other inflammatory mediators. First, we assessed IL-17, IFN-γ, and IL-4 production by T cells cocultured with allogeneic monocytes/macrophages. As expected, substantial levels of IL-17 were produced by the parental Th17 lines, more so by the CCR6+ subsets but virtually not by the CXCR3+ subsets and Th1 cells (Figure 7A). Furthermore, the parental Th17 cell lines and their CCR6+ subsets were extremely potent in inducing the production by monocytes/macrophages of MMP-1, MMP-3, and MMP-9 (Figure 7B). In particular, the induction of all MMPs by the CCR6+ subsets was 2 orders of magnitude greater than that observed in the presence of Th1 cells and of the CXCR3+ subsets (Figure 7B). Of note, the CCL20/MIP-3α levels were much higher in the presence of the parental Th17 and CCR6+ than CXCR3+ or Th1 cells (Figure 7C), whereas the induction of CXCL10 and IL-1β was in the same range for all the Th cell lines tested (Figure 7C). It should be stressed, however, that T cells and not monocytes/macrophages were the cells principally involved in CCL20/MIP-3α production as opposed to CXCL10. Indeed, T cells activated by anti-CD3 CD28-coated beads in the absence of monocytes/macrophages produced as much CCL20/MIP-3α but not CXCL10 as in the presence of allogeneic monocytes/macrophages. Overall, these data point to Th17 cells as very potent in inducing the production by monocytes/macrophages of MMPs with a role in tissue remodeling and destruction.
Figure 7.

Th17 are more potent than Th1 cells to induce MMP production by monocytes/macrophages. (A) IL-17, IFN-γ, IL-4; (B) MMP-1, MMP-3, MMP-9; (C) IL-1β, CCL20/MIP-3α, CXCL10/IP-10. T-cell lines were generated as described in the legend of Figure 6. A total of 50 000 T cells were cocultured with equal numbers of allogeneic monocytes for 48 hours. IL-1β, IL-4, IL-17, IFN-γ, MMP-1, MMP-3, and MMP-9 were assessed by multiplex immunoassay, whereas CCL20/MIP-3α and CXCL10 were assessed by ELISA. Bars represent the mean of duplicate cultures; the SD was within 15% of the mean for all conditions. The results are representative of similar data obtained with T cells generated from 2 healthy donors.
Discussion
PGE2 has function in homeostasis, but its production is enhanced during inflammation in response to TNF and IL-127,28 and is known to dramatically modulate immune responses. However, the effect of PGE2 on Th17 cell differentiation and expansion has not yet been systematically addressed in humans. Thus, the findings we report here are new as far as we demonstrate that in vitro PGE2 specifically increases the frequency of CD4+ T cells producing IL-17 (Th17), simultaneously decreasing that of IFN-γ–producing T cells (Th1). In addition, the Th17 cells expanded in the presence of PGE2 have the interesting characteristic of enhanced production of CCL20/MIP-3α but decreased production of IL-22, thus representing an example of dissociation in the production of cytokines thought to be the product of the same cell type. Because several reports have stressed that PGE2 favors the production of IL-23 while inhibiting that of IL-12 by dendritic cells,29,30,33 it could have been expected that, in the presence of PGE2, PBMCs would preferentially produce IL-17. However, our data clearly show that PGE2 synergizes with IL-23 in expanding Th17 cells and, most importantly, that its effect was evident on purified memory CD4+ T cells activated in the absence of accessory cells, thus indicating a direct effect on T cells. It remains to be investigated whether this depends on enhanced responses to IL-23 or to additional and distinct effects of PGE2 on T cells. In this respect, it should be emphasized that previous reports have shown a preferential inhibitory, rather than stimulatory, activity of PGE2 on T cells.34–37 Furthermore, at variance with findings reported by others, we could not observe preferential expansion of Th2 cells or of FoxP3 gene-expressing cells (not shown) in the presence of PGE2.38,39
Although the simultaneous presence of IL-17 and IL-22 has been proposed to be the “signature” of Th17 cells,40 our results show that PGE2 enhances IL-17 and decreases IL-22 production and are reminiscent of a similar dissociation observed in mouse Th17 cells generated in the continuous presence of TGF-β and IL-6 as opposed to priming in the presence of TGF-β and IL-6 followed by expansion in IL-23.41 IL-22 is considered a proinflammatory cytokine with a distinct role in the generation of psoriatic skin lesions42 and in mediating mucosal host defense against Gram-negative bacteria.43 It is possible that Th17 cells generated in the presence or in the absence of PGE2 may have distinct functional capabilities. It should be stressed, however, that our Th17 cells and in particular the CCR6+ subset were much more potent in inducing MMP production by monocytes/macrophages than Th1 cells.
IL-2 has been shown to constrain Th17 expansion in a STAT5-dependent manner in the mouse.44 However, in agreement with data reported by others,18 exogenous IL-2 favored Th17 cell growth in our culture system, whereas very few cells were recovered in the absence of IL-2 (not shown). Furthermore, we could confirm that IL-1 played a major role in IL-17 production because IL-1Ra abated its production in our system, being much more inhibitory on Th17 than Th1 cells.9,10,18 However, although required to expand Th17 cells, IL-1 did not mediate the effect of PGE2. In addition, because TNF neutralization specifically enhanced IL-17 but not IFN-γ production, our results indicate that TNF may have an inhibitory role on IL-17 production, an effect that, to the best of our knowledge, has not been reported in humans, and it is at variance with findings in the mouse where TNF in conjunction with IL-1 and TGF-β appears to favor IL-17–producing cells.15
The presence of PGE2, but not that of exogenous IL-23, profoundly affected chemokine receptor expression on CD4+ T cells. This effect has not been reported previously and accompanies the enhanced production of IL-17 we observed. The preferential expression of CCR6, CCR4, and the expansion of CCR6+CCR4+CXCR3−CD4+ T cells is consistent and extend previous findings indicating a skewed chemokine receptor repertoire in Th17 cells.7,8,21,22 More important, we were able to enrich IL-17–producing cells by sorting CD4+ T cells on the base of their CCR6 expression and assess their function in cocultures with monocytes/macrophages. Within the CCR6+ subset, there were cells producing also IFN-γ, most of which were IL-17+/IFN-γ+ double-positive cells. However, the amount and pattern of cytokines and of MMPs produced by monocytes/macrophages cultured in the presence of CCR6+ T cells were clearly different from that in the presence of T cells depleted in CCR6 (CXCR3+), which produced almost exclusively IFN-γ, thus proving that CCR6+CD4+ T cells (Th17) generated in the presence of PGE2 have specific functional capabilities that distinguish them from CXCR3+ and Th1 cells. The amount of MMP-1, MMP-3, and MMP-9 produced by monocytes/macrophages stimulated by Th17 compared with Th1 cells is impressive and stresses the pathologic potential of Th17 cells,4 which is most probably preferentially the result of IL-17 itself.26,45 The recent observation, both in humans and mice, that CCR6 expression guides Th17 cells to inflamed joints straightly put our data in a relevant physiopathologic context.46 Of further interest, when assessing the production of chemokines in cocultures with monocytes/macrophages, we observed similar levels of CXCL10, an IFN-induced chemokine binding to CXCR3, in the presence of Th17 and Th1 cells, whereas the levels of CCL20/MIP-3α were much higher in the presence of Th17 cells (Figure 7C). CCL20/MIP-3α is a ligand of CCR6 and is produced by a variety of cell types, including T cells and monocytes.47 We observed that PGE2 increases CCL20/MIP-3α production in PBMCs (Figure 1); and when assessed in cocultures, most, if not all, CCL20/MIP-3α was produced by T cells and in particular by CCR6+ T cells (Th17) and not by Th1 and CXCR3 cells. Thus, this is an interesting example of cells that express both the receptor and, when activated, produce its ligand, which may provide a positive amplification signal allowing the recruitment of additional CCR6+ cells, eventually leading to increased numbers of Th17 cells at sites of inflammation.46
Our experimental system was not designed to test whether PGE2 could contribute to differentiate naive CD4+ T cells into Th17 cells. Nonetheless, recent evidence indicates that dendritic cells recovered in the draining lymph node of inflamed skin are capable of producing PGE2, which then impacts on T-cell priming and differentiation.48 Further, in experimental bowel disease and collagen-induced arthritis, the proinflammatory activity of PGE2 was shown to be mediated by the IL-23/IL-17 axis,49,50 thus stressing the relevance of our in vitro findings.
In conclusion, the data here reported show that PGE2 profoundly affects the cytokine production and chemokine receptor expression of activated helper T cells, enhancing, in an IL-1–dependent manner, the expansion of Th17 cells, which in turn promote the production of high amounts of MMPs by monocytes/macrophages. Thus, PGE2, the production of which is mainly the result of cells of mesenchymal origin under the influence of IL-1 and TNF,27,28 may participate in an amplification loop of immuno-inflammatory events favoring the recruitment at sites of inflammation of Th17 cells, which then may contribute to control infection or aggravate tissue destruction.
Acknowledgments
This work was supported in part by the Swiss National Science Foundation (grant 310000-112180/1).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.C. designed research, analyzed data, and wrote the paper; R.C., M.A., and C.d.R. performed research; P.R.-L. and J.-M.D. designed research and analyzed data; S.F.-L. performed research and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carlo Chizzolini, Immunology and Allergy, University Hospital, 1211 Geneva 14, Switzerland; e-mail: chizzolini@medecine.unige.ch.
References
- 1.Chizzolini C, Chicheportiche R, Burger D, Dayer JM. Human Th1 cells preferentially induce interleukin (IL)-1beta while Th2 cells induce IL-1 receptor antagonist production upon cell/cell contact with monocytes. Eur J Immunol. 1997;27:171–177. doi: 10.1002/eji.1830270125. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 3.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 4.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 6.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 8.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 17.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 18.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 19.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- 21.Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J Immunol. 2008;180:214–221. doi: 10.4049/jimmunol.180.1.214. [DOI] [PubMed] [Google Scholar]
- 22.Lim HW, Lee J, Hillsamer P, Kim CH. Human Th17 cells share major trafficking receptors with both polarized effector T cells and FOXP3+ regulatory T cells. J Immunol. 2008;180:122–129. doi: 10.4049/jimmunol.180.1.122. [DOI] [PubMed] [Google Scholar]
- 23.Koenders MI, Lubberts E, van de Loo FA, et al. Interleukin-17 acts independently of TNF-alpha under arthritic conditions. J Immunol. 2006;176:6262–6269. doi: 10.4049/jimmunol.176.10.6262. [DOI] [PubMed] [Google Scholar]
- 24.Lubberts E, Schwarzenberger P, Huang W, et al. Requirement of IL-17 receptor signaling in radiation-resistant cells in the joint for full progression of destructive synovitis. J Immunol. 2005;175:3360–3368. doi: 10.4049/jimmunol.175.5.3360. [DOI] [PubMed] [Google Scholar]
- 25.Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–1251. [PubMed] [Google Scholar]
- 26.Jovanovic DV, Martel-Pelletier J, Di Battista JA, et al. Stimulation of 92-kd gelatinase (matrix metalloproteinase 9) production by interleukin-17 in human monocyte/macrophages: a possible role in rheumatoid arthritis. Arthritis Rheum. 2000;43:1134–1144. doi: 10.1002/1529-0131(200005)43:5<1134::AID-ANR24>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med. 1985;162:2163–2168. doi: 10.1084/jem.162.6.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dayer JM, de Rochemonteix B, Burrus B, Demczuk S, Dinarello CA. Human recombinant interleukin 1 stimulates collagenase and prostaglandin E2 production by human synovial cells. J Clin Invest. 1986;77:645–648. doi: 10.1172/JCI112350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalinski P, Vieira PL, Schuitemaker JH, de Jong EC, Kapsenberg ML. Prostaglandin E(2) is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood. 2001;97:3466–3469. doi: 10.1182/blood.v97.11.3466. [DOI] [PubMed] [Google Scholar]
- 30.Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 2004;18:1318–1320. doi: 10.1096/fj.03-1367fje. [DOI] [PubMed] [Google Scholar]
- 31.Parel Y, Aurrand-Lions M, Scheja A, Dayer JM, Roosnek E, Chizzolini C. Presence of CD4+CD8+ double-positive T cells with very high interleukin-4 production potential in lesional skin of patients with systemic sclerosis. Arthritis Rheum. 2007;56:3459–3467. doi: 10.1002/art.22927. [DOI] [PubMed] [Google Scholar]
- 32.Ribbens C, Dayer JM, Chizzolini C. CD40-CD40 ligand (CD154) engagement is required but may not be sufficient for human T helper 1 cell induction of interleukin-2- or interleukin-15-driven, contact-dependent, interleukin-1beta production by monocytes. Immunology. 2000;99:279–286. doi: 10.1046/j.1365-2567.2000.00948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnurr M, Toy T, Shin A, Wagner M, Cebon J, Maraskovsky E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: a novel role for the cAMP pathway. Blood. 2005;105:1582–1589. doi: 10.1182/blood-2004-05-1718. [DOI] [PubMed] [Google Scholar]
- 34.He X, Stuart JM. Prostaglandin E2 selectively inhibits human CD4+ T cells secreting low amounts of both IL-2 and IL-4. J Immunol. 1999;163:6173–6179. [PubMed] [Google Scholar]
- 35.Shin HC, Benbernou N, Fekkar H, Esnault S, Guenounou M. Regulation of IL-17, IFN-gamma and IL-10 in human CD8(+) T cells by cyclic AMP-dependent signal transduction pathway. Cytokine. 1998;10:841–850. doi: 10.1006/cyto.1998.0375. [DOI] [PubMed] [Google Scholar]
- 36.Shin HC, Benbernou N, Esnault S, Guenounou M. Expression of IL-17 in human memory CD45RO+ T lymphocytes and its regulation by protein kinase A pathway. Cytokine. 1999;11:257–266. doi: 10.1006/cyto.1998.0433. [DOI] [PubMed] [Google Scholar]
- 37.Nataraj C, Thomas DW, Tilley SL, et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demeure CE, Yang LP, Desjardins C, Raynauld P, Delespesse G. Prostaglandin E2 primes naive T cells for the production of anti-inflammatory cytokines. Eur J Immunol. 1997;27:3526–3531. doi: 10.1002/eji.1830271254. [DOI] [PubMed] [Google Scholar]
- 39.Baratelli F, Lin Y, Zhu L, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175:1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 40.Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 42.Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000;12:1092–1099. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 46.Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 48.Nagamachi M, Sakata D, Kabashima K, et al. Facilitation of Th1-mediated immune response by prostaglandin E receptor EP1. J Exp Med. 2007;204:2865–2874. doi: 10.1084/jem.20070773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheibanie AF, Yen JH, Khayrullina T, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23[rarr]IL-17 axis. J Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 50.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007;56:2608–2619. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]