

Abstract
Damage in transcribed DNA presents a challenge to the cell because it can partially or completely block the progression of an RNA polymerase, interfering with transcription and compromising gene expression. While blockage of RNA polymerase progression is thought to trigger the recruitment of the transcription-coupled DNA repair (TCR), bypass of the lesion can also occur, either error-prone or error-free. Error-prone transcription is often referred to as transcriptional mutagenesis (TM). Elucidating why some lesions pose blocks to transcription elongation while others do not remains a challenging problem. As part of an effort to understand this, we studied transcription past a 5-guanidino-4-nitroimidazole (NI) lesion, using two structurally different RNA polymerases, human RNA polymerase II (hRNAPII) and bacteriophage T7 RNA polymerase (T7RNAP). The NI damage results from the oxidation of guanine in DNA by peroxynitrite, a well known, biologically important oxidant. It is of structural interest because it is a ring-opened and conformationally flexible guanine lesion. Our results show that NI acts as a partial block to T7RNAP while posing a major block to hRNAPII, which has a more constrained active site than T7RNAP. Lesion bypass by T7RNAP induces base misincorporations and deletions opposite the lesion (C > A > –1 deletion > G ≫> U), but hRNAPII exhibits error-free transcription although lesion bypass is a rare event. We employed molecular modeling methods to explain the observed blockage or bypass accompanied by nucleotide incorporation opposite the lesion. The results of the modeling studies indicate that NI’s multiple hydrogen-bonding capabilities and torsional flexibility are important determinants of its effect on transcription in both enzymes. These influence the kinetics of lesion bypass and may well play a role in TM and TCR in cells.
Keywords: 5-guanidino-4, nitroimidazole, Transcription, DNA damage, RNA polymerase, Molecular modeling
1. Introduction
Cells are susceptible to the detrimental effects of reactive oxygen and nitrogen species that are formed during a variety of fundamental cellular processes, especially aerobic respiration, or that result from exposure to exogenous agents, such as ionizing radiation, ultraviolet light, oxidative stress, and smoke [1]. The oxidative products interact with cellular DNA, producing oxidized bases, strand breaks, and abasic sites. Oxidative DNA damage has been implicated in a range of diseases, including different types of cancers, atherosclerosis, neurodegenerative maladies such as Parkinson’s and Alzheimer’s diseases, and aging. The relationships between oxidative DNA damage and disease have been comprehensively reviewed by Evans et al., [2] and Pacher et al., [3].
The powerful oxidant peroxynitrite (ONOO−) is formed via the reaction of nitric oxide (NO·) with superoxide anion (O2·−) [4]. Inflammatory cells, such as macrophages and neutrophils, and the inner membrane of mitochondria, which is the site of the electron transport chain and oxidative phosphorylation, are known to be the sites of overproduction of NO· and O2·−, suggesting that exposure to ONOO is unavoidable [5–7]. Peroxynitrite-related anions, radicals, or neutral peroxynitrous acid can traverse cell membranes by diffusion through anion channels [8,9], ultimately reaching the nucleus where they can react with cellular DNA [3,10]. It has been shown in vitro that peroxynitrite reacts with DNA to form 8-nitroguanine [11], 8-oxoguanine [12] and NI [13] as the major products [14]. The lesion 8-nitroguanine readily depurinates [15], while 8-oxo-guanine, which possesses a lower oxidation potential relative to its parent purine, is prone to further oxidation, giving rise to other lesions [14,16]. However, NI, which is an oxidative DNA lesion formed only through the direct reaction of peroxynitrite with guanine, is very stable, making it a potential candidate as a biomarker for studying peroxynitrite-induced DNA damage and mutagenesis [13,17]. In oligonucleotides, in vitro, the reaction of nitrogen dioxide with the 5- and 8-positions of guanine radicals -- derived from the one-electron oxidation of guanine -- gives rise to NI and 8-nitroguanine, respectively, and in comparable proportions [18]. While 8-nitroguanine has been observed in vivo [19–21], the existence of NI in cellular environments remains to be demonstrated.
The NI lesion is a ring-open purine that lacks the hydrogen-bonding sites of the parent deoxyguanosine but contains novel hydrogen bond donor and acceptor atoms, namely a guanidine group and a nitroimidazole ring (Fig. 1). Furthermore, computational studies have revealed that NI is torsionally flexible, permitting this damaged base to adopt multiple conformations. Specifically, quantum mechanical (QM) studies of the NI lesion were carried out on the base and nucleoside level [22,23], and molecular dynamics (MD) simulations were performed for the NI-damaged DNA duplexes [22]. The QM studies revealed a planar conformation for the damaged base [22], but there were a number of non-planar conformations at the nucleoside level [22,23]. The MD studies showed that NI in the DNA duplex is also non-planar and multi-conformational [22]. The various non-planar conformations of NI are adopted in order to avoid steric crowding between the NI base and adjacent sugar moieties and base neighbors.
Fig. 1.
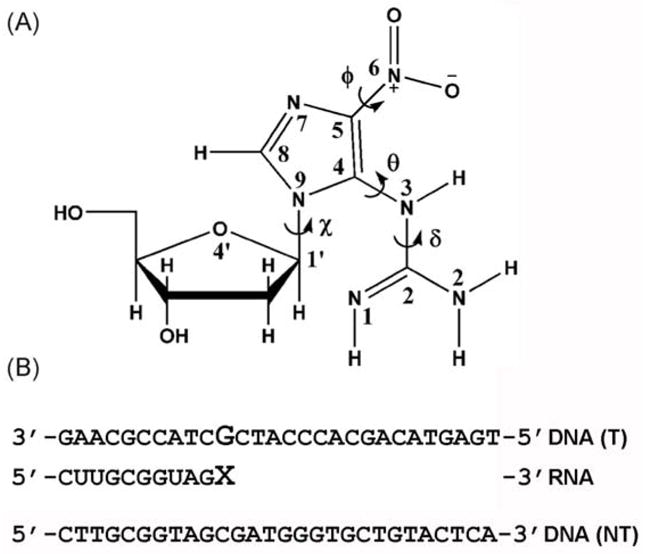
Structure of the NI lesion and sequences of DNA and RNA. (A) NI structure; torsion angles are indicated by the arrows and defined as follows: glycosidic torsion angle χ, O4′-C1′-N9-C4; θ, N9-C4-N3-C2; δ, C4-N3-C2-N2; and ϕ, C4-C5-N6-O6. The two oxygens at the N6 position are equivalent. (B) DNA and RNA sequences; the enlarged G indicates the templating base (guanine or NI) and the enlarged X denotes the incoming ribonucleotide. T: denotes the DNA template strand. NT: denotes the non-template DNA strand.
DNA polymerase (DNAP) primer extension assays past NI showed that the adduct could block replication catalyzed by calf thymus DNAP αand E. coli DNAP I, but not human DNAP β, and that bypass events induced G → T and G → C transversions [13]. Neeley et al. [24] found that in E. coli, replication was significantly blocked while lesion bypass was 50% mutagenic, generating G → A and G → T mutations. The same group recently found that E. coli DNAP V allows error-prone bypass of NI, preferentially incorporating adenine opposite the lesion [25]. Interestingly, NI is only slightly cleaved by formamidopyrimidine-DNA glycosylase (FaPy) and is not removed by endonuclease III [13] in the base excision DNA repair (BER) pathway [26]. Since NI appears refractory to BER, it may be difficult to remove from the genome; hence, this lesion could accumulate over time, unless an as of yet unidentified repair mechanism clears it from the genome. In addition to being mutagenic, this poorly repairable lesion could also be cytotoxic since it can block DNA synthesis [13,24,25].
Lesions in DNA not only affect replication, but they can also exert deleterious effects on transcription. When a gene is expressed, DNA damage located on the transcribed strand can cause RNA polymerase (RNAP) to halt at the modified site, producing a truncated transcript, or the transcription complex can bypass the lesion, producing full-length RNA [27]. Transcription stalling is thought to act as a signal for transcription-coupled DNA repair (TCR) that can clear the damaged site [27,28]. In contrast, lesion bypass can be accompanied by base misincorporation opposite the damage, which changes the RNA sequence in a process referred to as transcriptional mutagenesis (TM) [29]. While transcription past a number of DNA adducts has been studied, and many adducts completely or partially block transcription elongation, there is no simple, direct correlation between the size and type of a DNA lesion and its influence on RNA synthesis.
Here we report the effect of NI on transcription by human RNAP II (hRNAPII) and the bacteriophage T7 RNAP in an effort to understand the impact of this oxidative lesion on RNA synthesis. These two polymerases are structurally different, with the T7 RNAP containing a more open active site region while that of RNAPII is closely packed [30,31]. The resulting transcripts were sequenced, and computer modeling was employed to provide structural rationales for the experimental findings. While NI acts as a partial block to T7 RNAP, it poses a major block to hRNAPII. T7 RNAP bypasses NI in an error-prone manner, inducing base misincorporations and deletions opposite the lesion (C > A > –1 deletion > G ≫>U), but hRNAPII exhibits error-free transcription during its rare lesion bypass events. Molecular modeling reveals hydrogen-bonding patterns that offer explanations for the observed blockage and nucleotide incorporation events opposite the lesion, and indicates that the multiple hydrogen-bonding capabilities of the lesion and its torsional flexibility are important determinants of its effect on transcription.
2. Materials and Methods
Unless stated otherwise, all reagents, enzymes, and biologicals were purchased from Promega, Sigma-Aldrich, New England Biolabs, Ambion, or BioRad. DNA oligomers were obtained from Sigma-Aldrich. Radioactive materials were purchased from Perkin Elmer. All DNA sequencing reported was performed using the GeneAmp PCR System 9700 (BioRad) and 3100 Capillary Sequencer (Applied Biosystems).
2.1 Synthesis of site-specifically modified 2′-deoxyribooligonucleotides containing a single 5-Guanidino-4-nitroimidazole Oligodeoxynucleotide lesion
The detailed protocols for synthesizing 2′-deoxyribooligonucleotides containing a single NI lesion at a defined position have been described earlier [18,32]. Briefly, air-equilibrated phosphate buffer solutions (pH 7.5) containing 0.1 μmol of the 5′-d(CCATCGCTACC) sequence, 300 μmol NaHCO3, 1 μmol NaNO2, and 25 μmol Na2S2O8 in 1 mL were photoexcited with unfocussed 308 nm laser light pulses (~ 15 mJ pulse−1 cm−2, 10 pulse s−1) from a XeCl excimer laser for 10 to 20 s. The nitrated oligodeoxynucleotides, which contained a mixture of NI and 8-nitroguanine adducts, were isolated, purified, and desalted by reverse-phase HPLC techniques as described in more detail in Supporting Information.
2.2. Assembly of DNA templates
DNA templates suitable for transcription by hRNAPII and T7 RNAP were prepared as described [33,34] (Fig. 2). In brief, pCI-neo-G-less-T7 was linearized using the restriction endonuclease BbsI, (Fig. S1) and ligated to a double-strand tail using T4 DNA ligase for 16 h at 16 °C. The tail was assembled by annealing three synthetic, single-strand DNA oligomers, an 11-mer (either unmodified or modified with the NI lesion), and a 96-mer to a complementary 90-mer containing a 3′-biotin tag. DNA oligomers used are shown in Table 1. The product was precipitated using streptavidin-linked beads, gel purified, and tested for nicks as described elsewhere [33,35].
Fig. 2.
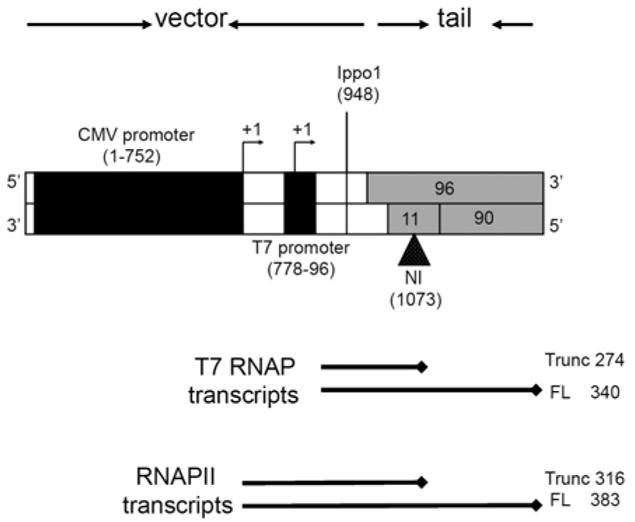
The NI-modified DNA templates prepared for transcriptional assays. The vector (white) is comprised of the cloned T7 promoter and the CMV promoter/enhancer (black boxes). The tail is formed by annealing a 96-mer, a 90-mer, and an 11-mer (grey). The NI position is indicated by a black triangle. The arrowheads designate the +1 transcription start site. The size of the template is 1,140 base pairs. The expected sizes of the transcripts obtained during transcription by T7 RNAP and hRNAPII are indicated below the template.
TABLE 1.
Sequence of the DNA oligomers used in the study
| Oligomer name | DNA Sequence |
|---|---|
| 96-mera | 5′-TTGCGGTAGCGATGGGTGCTGTACTCAGGTGTGGAATCAACCCACAGCTGACAGGGCAGGTCTTGGCCAGTTGGGATATCCAAAACATCTTGTTGA-3′ |
| 90-mera | 5′-TTTTTTTTTTCAACAAGATGTTTTGGATATCCCAACTGGCCAAGACCTGCCCTGTCAGCTGTGGGTTGATTCCACACCTGAGTACAGCAC-3′ |
| 11-mera | 5′-CCATCGCTACC - 3′ |
| 49Lb | 5′-GCTTCCTCATCCTTCTCCATCTCTCTGTGCAGCTGTGGGTTGATTCACG |
| 24Fc | 5′-AACGCAGTCAGTGCTTCTGACACA -3′ |
| 25Rc | 5′-GAGAGATGGAGAAGGATGAGGAAGC -3′ |
| 22R trunc | 5′-TACAGTGCTCGCATACGCTCAA -3′ |
| 24chimera | 5′-UUUUGAGCGTATGCGAGCACTGTAidt -3′ |
The 96-mer, 90-mer and the 11-mer were annealed to make the tail of the DNA templates. The underscored G in the 11-mer represents either the undamaged guanine or the damaged O6-MeG.
49L is the primer used in the reverse transcription of the full-length RNA obtained from transcription of T7 RNA polymerase past the DNA templates.
24F and 25R are the forward and reverse PCR primers. 22R: The reverse transcription primer used in the ligation mediated reverse transcription. It is complimentary
to the chimera 24chimera that was ligated to the RNA prior to reverse transcription.
2.3. Transcription with the bacteriophage T7 RNA polymerase
Transcription using T7 RNAP was performed as described elsewhere [33]. Briefly, the reactions contained 20 units1 of enzyme, 25 ng (0.033 pmoles) of DNA template, 2.5 mM each of ATP, GTP, and UTP, 200 μM [α-32P]CTP (50 Ci/mmol), 100 mM DTT, 10 units RNasin, 40 mM Tris-HCl (pH 7.9 at 25 °C), 10 mM NaCl, 6 mM MgCl2, and 2 mM spermidine in a final volume of 20 μl. The reaction was incubated at 37 °C for 60 min. Unincorporated nucleotides were removed using bio-spin size exclusion columns by Biorad, and the RNA was resolved using an 8% denaturing PAGE.
Time-course transcription reactions were performed to test for lesion bypass efficiency over time as described above but with the addition of a radioactively labeled 103-base DNA oligomer to the reaction mix, which acted as a loading standard during denaturing PAGE. An aliquot of the reaction mixture was removed every 10 min, quenched with loading dye, and heated to 65 °C for 15 min to inactivate the polymerase. ImageQuant (Molecular Dynamics) software was used to visualize and quantify the products.
2.4. Transcription reactions with hRNAPII
HeLa nuclear extracts (Promega Inc.) were utilized to provide the hRNAPII machinery. Multiple rounds of transcription reactions were performed as described in [33,35,36]. Transcription was carried out using 8 units of HeLa nuclear extract2 suitable for one reaction as determined by the manufacturer, Promega Inc., 100 ng (0.15 pmols) DNA template, 10 units RNase inhibitor, 10 mM each of ATP, GTP, and UTP, 0.4 μM [α-32P]CTP (25 Ci/mmol), 20 mM HEPES (pH 7.9 at 25 °C), 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, and 20% glycerol, to a final reaction volume of 25 μl. The reaction was incubated at 30 °C for 60 min and quenched using a stop solution (0.3M Tris-HCl (pH 7.4 at 25°C), 0.3M sodium acetate, 0.5% SDS, 2mM EDTA, 3μg/ml tRNA) as described in the manufacturer’s protocol (Promega).
Transcription was also carried out under conditions in which only a single round of initiation could occur. During these experiments, GTP was omitted from the reaction mix, and the polymerase complex was allowed to transcribe up to the point at which the first GTP was required. After 10 min, heparin was added to bind all free hRNAPII. Finally, the addition of 100 μM GTP allowed transcription to resume, and the reaction was continued for an additional 40 min. RNA from both experiments was purified by phenol:chloroform extraction and resolved by denaturing 8% PAGE.
2.5. Sequencing the full-length transcripts
Transcription was performed in the absence of radioactive nucleotides. All DNA templates were digested with RNase-free DNase at 37 °C for 30 min. The resulting RNA was purified and sequenced using reverse transcriptase and the polymerase chain reaction (RT-PCR). In brief, 20 pmols of RT primer were annealed to the RNA product, and ImProm II (Promega) enzyme was used to perform the reverse transcription by following the manufacturer’s protocol. Residual RNA was then digested with RNase, and the reverse transcriptase was subsequently heat inactivated. Excess primers and small RNA fragments were eliminated by using Microcon Centrifugal Filter Columns. Finally, PCR was performed with the forward and reverse primers 24F and 25R, respectively (Table 1), as described by the manufacturer (Promega). The RT-PCR products were purified in a 1.2% non-denaturing agarose matrix and subsequently inserted into the pDrive cloning vector (Qiagen). Subcloning was performed as described in the Qiagen protocol, and 100 clones were selected for DNA extraction using the QIAprep Spin Miniprep Kit (Qiagen) and sequenced.
2.6. Sequencing the truncated transcripts
To sequence the truncated transcripts, 1 μM of a chimeric RNA-DNA oligomer was ligated to the 3′-ends of the transcripts using T4 RNA ligase and 10% DMSO in a reaction volume of 50 μL [33,37]. The product was concentrated using Microcon Centrifugal Filter Columns (MC-100). RT-PCR was initiated by annealing the reverse transcriptase primer 22RTrunc (Table 1) to the RNA. This approach not only selected the truncated transcripts but all RNA ligated to the chimeric oligomer. The same procedures for subcloning the PCR products was performed as described for the full-length transcripts.
2.7. Molecular modeling of NI in T7 RNAP and yRNAPII
We used a crystal structure containing the substrate insertion complex of T7 RNAP during one cycle of nucleotide addition in the elongation phase of transcription, PDB ID 1S76 [30], as a basis for the lesion modeling in this enzyme. For RNAPII, the X-ray crystal structure of S. cerevisiae RNAPII (yRNAPII), PDB ID 2NVZ [31], was used for modeling NI in the active site of RNAPII. Crystal structures for hRNAPII are not available at this writing, but many crystal structures for yRNAPII are available [38]. The yeast polymerase is a reasonable substitute model for the human enzyme, since the two polymerases share an overall 80% identity [39,40]. Both proteins were crystallized in a ternary complex, each containing a DNA duplex, an RNA chain, and an incoming ribonucleotide. Sequences of the template/non-template DNA and the RNA transcript were remodeled to match the sequence used in the experiments (Fig. 1B).
Because of the flexibility in NI, the observed nucleotide selectivity may be the result of a number of conformational possibilities, which we explored. Our presented models offer feasible structural interpretations of the observed data, without necessarily being entirely unique. Overall, the modeling procedures utilized the following protocol. The initial conformations used here for NI employed our previous QM-computed lowest energy conformations for the NI nucleoside in the syn and anti conformations of the glycosidic bond (Tables S1 and S4)[22]. This structure replaced the templating base in the crystal structure. The incoming NTP in the crystal was replaced by the relevant NTP under investigation. The multidimensional torsional space, involving glycosidic bond torsions χ for the lesion and the NTP, together with the NI flexible torsions ϕ, δ, and θ (Fig. 1), was then manually explored extensively through concerted iterative adjustments, with the goal of locating conformers with the following properties: free of collisions; optimal hydrogen-bonding interactions between the NI and the incoming NTP (where relevant); optimal stacking between the imidazole ring of NI and the previously transcribed DNA/RNA pair; and optimal stabilizing hydrogen bonds between the NI and the polymerase. In the case of T7RNAP, the side chain torsions of Arg632 (Table S1) were finally rotated to prevent collisions with the NI and to subsequently locate hydrogen bonds between this residue and NI. The manual torsional procedures were visually assessed for these properties aided by Insight 2000 (Accelrys, CA) utilities that evaluated collisions (bump check) and reported hydrogen-bonding interactions. While the specific NI rotamers selected for illustration may not be altogether unique, there is a hierarchy of robustness. The glycosidic torsion domains are almost exclusively governed by steric effects between the NI and the rest of the system and are very robust. Furthermore, the torsion θ is governed largely by steric effects involving the nitro group of the NI itself, and also plays a predominant role in determining the hydrogen-bonding possibilities between NI and the incoming NTPs, hence its domain is also quite robust. The torsions ϕ and δ are interdependent. There is a range of ϕ, δ combinations (± 10º) within selected regions that can fulfill the criteria of minimal close contacts and optimal hydrogen bonding between NI and the NTP. In sum, then, our models, while not the only solution to the structural interpretation of the data, do represent within rather narrow ranges the best structural families that are consistent with the observations.
Criteria for hydrogen-bonds in our models are in observed ranges [41–45]: (1) donor to acceptor atom distance: 2.5 Å < X–A< 3.9 Å; (2) hydrogen to acceptor atom distance: 1.5 Å < H–A < 2.9 Å; (3) angle θ defined by the donor-hydrogen-acceptor atoms (X–H–A) > 90°.
All modeling was carried out with the Biopolymer module of Insight 2000 and rendering of the structures and all images were employed using the PyMOL program [46].
3. Results
3.1. Unmodified and NI modified DNA templates suitable for transcription were assembled and confirmed to be free of nicks
The NI lesion was placed on the transcribed strand of the DNA templates downstream from the CMV enhancer/promoter and the T7RNAP promoter (Fig. 2). This design allows for comparison between the two RNAPs when each is transcribing past NI. Unmodified templates were synthesized to act as the control DNA. Following synthesis, the integrity of the templates was checked by digestion with I-PpoI. Fragments were radiolabeled at the 5 ′-ends and analyzed on PAGE. The presence of DNA fragments of length 187, 191, and 953 bases and the absence of fragments of length 73, 77, 110, or 118 bases indicate the complete ligation of the DNA templates and the absence of nicks (Fig. S2).
3.2. T7RNA polymerase transcription is modestly blocked by NI
T7 RNAP transcription was carried out on undamaged and NI-damaged templates at 37 °C for 60 min under the experimental conditions described in detail in Methods. While transcription past the unmodified DNA produced only full-length RNA 340 bases in length, transcription of the NI modified DNA template resulted in a mixture of full-length and truncated RNA transcripts of 340 and 274 bases in length, respectively (Fig. 3A). The percentage of truncated RNA averaged from 8 separate reactions was 13 ± 5% under the reaction conditions specified in the Methods section (Fig. S3). The shorter transcript coincides with the position of the NI on the template strand, indicating that this lesion can block the elongation catalyzed by T7RNAP. Note that, with both undamaged and damaged templates, transcripts were observed between positions 274 and 340. These are caused by inherent pause sites on the DNA and are not lesion dependent. However, additional RNA molecules shorter than 274 bases were observed with the damaged template, but were absent when the undamaged control template was transcribed (Fig. 3A, grey arrows). These may be due to lesion-induced DNA distortions that are propagated upstream. Additionally, elongating RNA polymerases may be backed up behind the blocked ones, which could also cause the release of shorter transcripts [47].
Fig. 3.
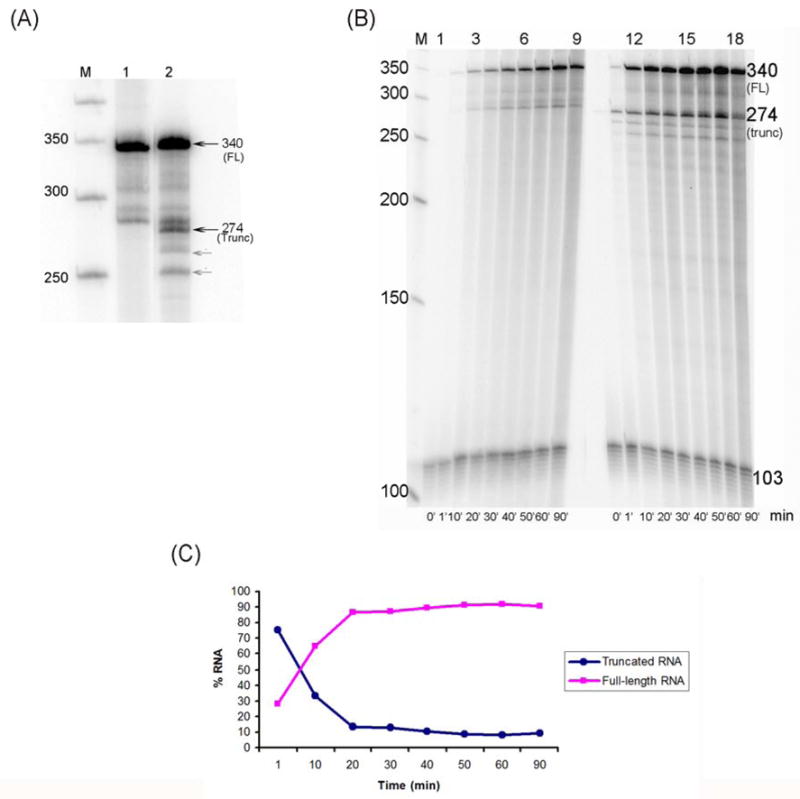
Effect of NI modified-DNA on transcription by T7 RNAP. (A) T7 RNAP transcription was performed as described in Methods for 60 min. The unmodified DNA produced full-length RNA (FL) approximately 340 bases in length (lane 1) while the NI-modified template produced two RNA species, approximately 274 and 340 bases in length (lane 2). A 50 base pair DNA step ladder is labeled M. Shorter RNA fragments are indicated by the grey arrows. These may be due to lesion-induced DNA distortions that are propagated upstream and/or elongating RNA polymerases that are backed up behind the blocked ones, releasing these fragments. (B) Time course experiment to determine lesion bypass efficiency. Transcription was performed as described in Methods and over 90 min with aliquots taken at 0, 1, 10, 20, 30, 40, 50, 60 and 90 min. The undamaged template generates increasing amounts of full-length (FL) transcripts (lanes 1–9) while transcription of the NI-damaged template produces a mixture of truncated and full-length RNA (lanes 10–18). Transcription past NI slows the reaction rate, as indicated by the slow increase in the formation of full-length RNA. The intensity of the bands was compared to a DNA loading control of 103 bases in length. A 50 base pair DNA step ladder is shown (M). Lane numbers are denoted at the top of the gel and time points in minutes at the bottom. (C) Time dependence for formation of full-length and truncated transcripts (displayed as percent of total RNA) over 90 minutes.as determined by band intensity. The values represent the average of three separate transcription trials. As shown, at 1 min the percent of truncated RNA (75% of total RNA) is about 2.5 times greater than that of the full-length transcript (~30% of total RNA); this percentage decreases continuously and after 20 minutes, it stabilizes at ~ 10% of the total RNA produced.
Transcription reactions were also carried out to determine changes in the bypass of the NI lesion over time (Fig. 3B). For each time point, the RNA band intensity was compared to a DNA standard (103 bases in length). The values represent the average of three separate transcription trials. As expected, the undamaged template produced increasing amounts of full-length RNA as the reaction progressed (Fig. 3B, lanes 1–9). However, the NI-damaged templates initially produced a greater amount of truncated than full-length transcripts (Figure 3B and C). At 1 min the percent of truncated RNA (75% of total RNA) is about 2.5 times greater than that of the full-length transcript (~30% of total RNA); this percentage decreases continuously, and after 20 min, it stabilizes at ~ 10% of the total RNA produced. The results suggest that rapid RNA synthesis occurs up to the lesion, which is followed by much slower translesion bypass.
3.3. Human RNA polymerase II transcription is primarily blocked by NI
HeLa nuclear extracts were used to provide the human RNAPII and its associated general transcription factors. To confirm that the effect observed in our experiments was due to the presence of the damage in the templates and to eliminate the possibility of lesion repair, templates were incubated with HeLa nuclear extracts for 1 h followed by exposure to piperidine, an agent that cleaves abasic sites. No template degradation was observed, indicating that NI was not removed in the presence of the HeLa extract.
Both single- and multiple-rounds of transcription were carried out. For single-rounds of transcription, a G-less cassette built into the template was utilized to synchronize the reaction so that each template was transcribed once by one hRNAPII complex. As for multiple-rounds of transcription, each template was transcribed multiple times by different polymerase molecules. As expected, transcription of the unmodified template yielded only full-length transcripts 383 bases in length (Fig. 4A, lanes 1 and 1 ′). On the other hand, a single round of transcription past the NI lesion showed that it posed an absolute barrier to the hRNAPII complex (Fig. 4A, lane 2 of left panel), while multiple rounds of transcription gave rise to a small amount of full-length RNA (here, ~ 14 % of the total RNA, under our experimental conditions) (Fig. 4A, lane 2 ′ of right panel), which is not due to removal of the NI and repair of the template (see below). Note that the amount of RNA obtained from sixteen separate trials, shown in Fig. 4B, indicates that the average percent of lesion bypass by hRNAPII is 9 ± 5% under our experimental conditions (See Methods) and after an incubation time of 60 min.
Fig. 4.
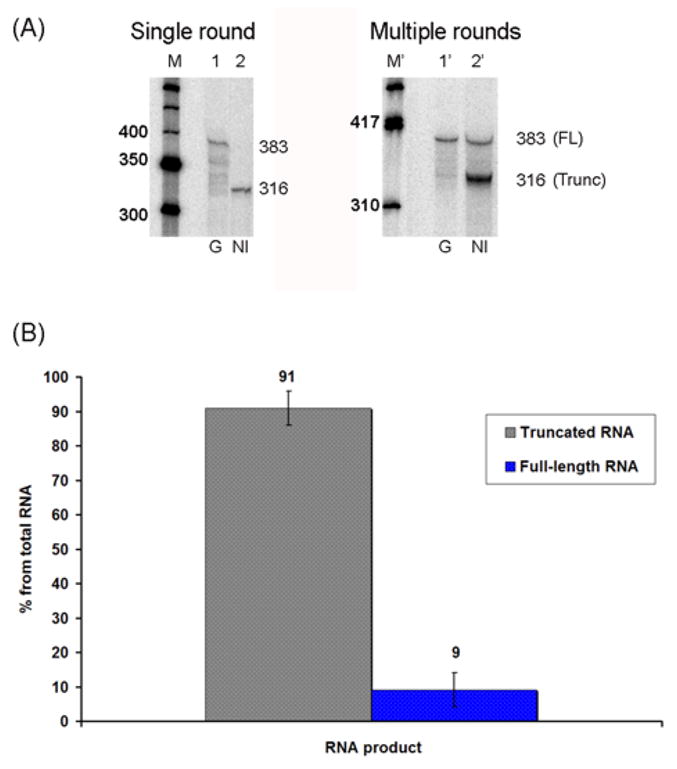
Effect of NI modified-DNA on transcription by hRNAPII as detailed in Methods. (A) Single round (left panel) and multiple rounds (right panel) of transcription using HeLa nuclear extracts. Left Panel: Single rounds of transcription indicate that the undamaged template generates full-length (FL) transcripts of 383 bases in length (lane 1) while the NI damaged templates produces only truncated RNA of 316 bases in length (lane 2). A 50 base pair DNA step ladder is shown (M). Right Panel: Multiple rounds of transcription show the resultant full-length transcripts (~383 bases) generated from the undamaged templates (lane 1′) while the damaged template produced mainly truncated as well as full-length RNA (In this case, ~ 86% and ~ 16 % of total RNA, respectively) (lane 2′). A ϕX174 DNA/Hinf I DNA ladder is shown (M′). (B) Bar graph illustrating the average quantities of RNA obtained in sixteen separate trials of multiple rounds of transcription.
3.4. NI favors the incorporation of cytosine in the full-length RNA
RT-PCR was used to sequence the resulting RNA. The PCR products obtained contained a heterogeneous population of DNA, necessitating subcloning to analyze individual RT-PCR products. The full length RNA generated by both RNA polymerases revealed that the incorporation of the correct nucleotide, cytosine, opposite the NI lesion is favored.
In full-length transcripts resulting from T7 RNAP transcription past the NI templates, the lesion primarily directed the incorporation of nucleotides in the following order: C > A > (–1 deletion) > G ≫> U (Table 2). In contrast, cytosine was exclusively incorporated opposite the NI lesion when the full-length RNA was obtained with hRNAPII (Table 2). As expected, the unmodified templates produced only full-length transcripts having the correct base insertions (data not shown).
TABLE 2.
Bases incorporated opposite NI in the full-length RNA during transcription with T7 RNAP and hRNAPII
Note the incorporation of C opposite the lesion was observed in the very few full-length RNAs obtained during transcription by hRNAPII.
3.5. Inhibition of nucleotide incorporation is a feature of the blocking NI lesion
Truncated transcripts obtained during transcription by T7 RNAP indicate that the polymerase stopped exactly at the site of the lesion, and for the most part, it did not incorporate a base opposite to NI. However, in some cases, the polymerase misincorporated bases in a non-specific pattern. Interestingly, we noticed the addition of one or multiple adenines or a cytosine followed by two non-template instructed adenines in some of the RNA transcripts (Table 3), a feature also observed with other lesions such as O6-methylguanine (O6-meG) [48] and 1,N2-ethenoguanine (1,N2-εG) [33].
TABLE 3.
Bases incorporated opposite NI in the truncated RNA during transcription with T7 RNAP and hRNAPII
| Sequence | RNA length(bases) | % | |
|---|---|---|---|
| Expected RNA Sequence | …UUGCGGUAGCGAUGG | ||
| Sequence obtained with T7 RNAP | …UUGCGGUA | 271 | 8 |
| …UUGCGGUAG | 272 | 28 | |
| …UUGCGGUAGA | 273 | 8 | |
| …UUGCGGUAGAA | 274 | 8 | |
| …UUGCGGUAGAAA | 275 | 8 | |
| …UUGCGGUAGCAA | 276 | 8 | |
| …UUGCGGUAGAGUC | 276 | 16 | |
| …UUGCGG GUUC | 8 | ||
| …UUGCGGUAGGGUC | 8 | ||
| Sequence obtained with hRNAPII | …UUGCGGU | 270 | 6 |
| …UUGCGGUAG | 272 | 67 | |
| …UUGCGGUAGC | 273 | 27 | |
Additionally, truncated transcripts generated by hRNAPII indicated that the enzyme typically stopped at the NI lesion and that nucleotide incorporation opposite it was not observed. In a few cases, however, the enzyme added cytosine opposite the lesion and then ceased to extend further (Table 3).
3.6. The unique hydrogen-bonding opportunities and structural flexibility of NI rationalize the observed lesion processing by T7 RNA polymerase and hRNA polymerase II
3.6.1. The NI-modified DNA in T7 RNAP
Molecular modeling, as described in Methods, was employed to provide a structural explanation for the observed experimental blockage and bypass results. We utilized the NI QM optimized nucleoside structures [22] that were lowest energy in the syn and anti glycosidic domains to initiate the modeling.
To interpret the observed transcription blockage, the lesion was modeled into the polymerase in the lowest energy nucleoside structure which placed the lesion in a syn glycosidic bond conformation. In this orientation, collisions between the NI and the T7 RNAP residues Ser-641 and Thr-636 were engendered (Fig. 5A), which could explain the blockage events exhibited by NI. To explain the observed bypass with purine incorporation opposite NI, we alleviated the collisions by adjusting the NI torsion angles θ, δ, and ϕ as well as the glycosidic torsion angle χ, which however remained in the syn domain. This placed the lesion in an empty pocket in the enzyme on the major groove side of the damaged guanine (Table S1). A collision-free structure was obtained which was stabilized by hydrogen bonds between the syn-NI and the neighboring protein side chains of Arg-632 and Gln-649 (Fig. 6 and Table S2). These hydrogen-bonding possibilities were also observed in models of the guanine lesions 1,N2-εG [33] and O6-meG [48] in T7 RNAP. The NI in this conformation can account for the order of purine NTP incorporation opposite the lesion through the extent of hydrogen bonding between the syn-NI and each purine. For ATP, two hydrogen bonds between N6 of adenine and O61 of NI, and between an N1-protonated adenine, observed in a number of crystals [49–51], and N7 of NI offer an explanation as to why this base could be incorporated. For GTP, a single hydrogen bond between N1 of guanine and N7 of NI is possible, consistent with the lesser incorporation of guanine into the nascent RNA strand (Fig. 7A,B and Table S3A).
Fig. 5.
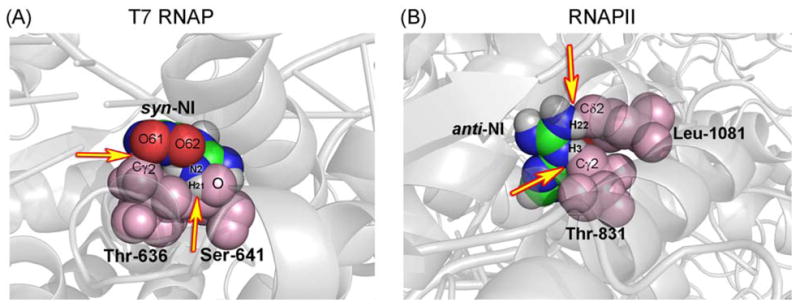
Space filling representation of the blocking models of NI in T7 RNAP and hRNAPII. (A) In T7 RNAP, overlap occurs between the syn-NI atoms, O61 and H of the amino group with the methyl carbon of Thr-636, and the oxygen of Ser-641 respectively. (B) In yRNAPII, the collision involves an overlap between H1 of anti-NI with the carbon of Thr-831 and a close contact between O62 of anti-NI and the carbon of Thr-831. The yellow arrows indicate colliding atoms. Colliding protein residues are shown in light pink.
Fig. 6.
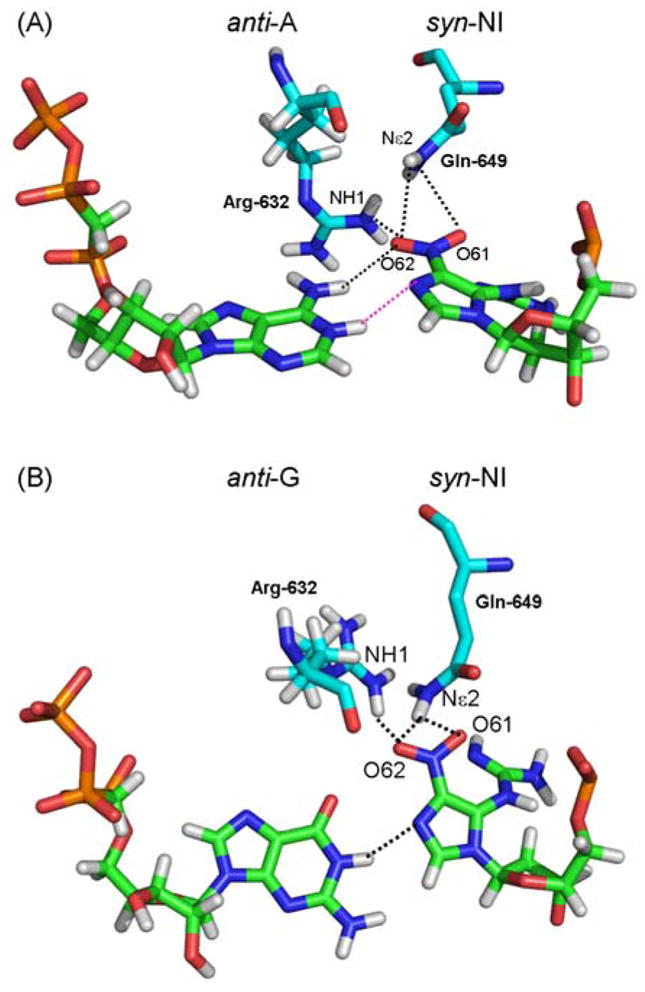
Hydrogen bond network between syn-NI and T7 RNAP residues Arg632 and Gln649. (A) anti-A:syn-NI and (B) anti-G:syn-NI pairs show the NI in a syn conformation, which is stabilized by one hydrogen bond involving the O62 of NI and the NH1 of Arg632, and a bifurcated hydrogen bond between O61 and O62 of NI and Nε2 of Gln649. The ATP and GTP partners are shown to illustrate the difference in torsions adopted by the NI lesion in each case. Hydrogen bonds are shown as dotted black lines. Hydrogen bond involving the protonated adenine is shown in pink.
Fig. 7.
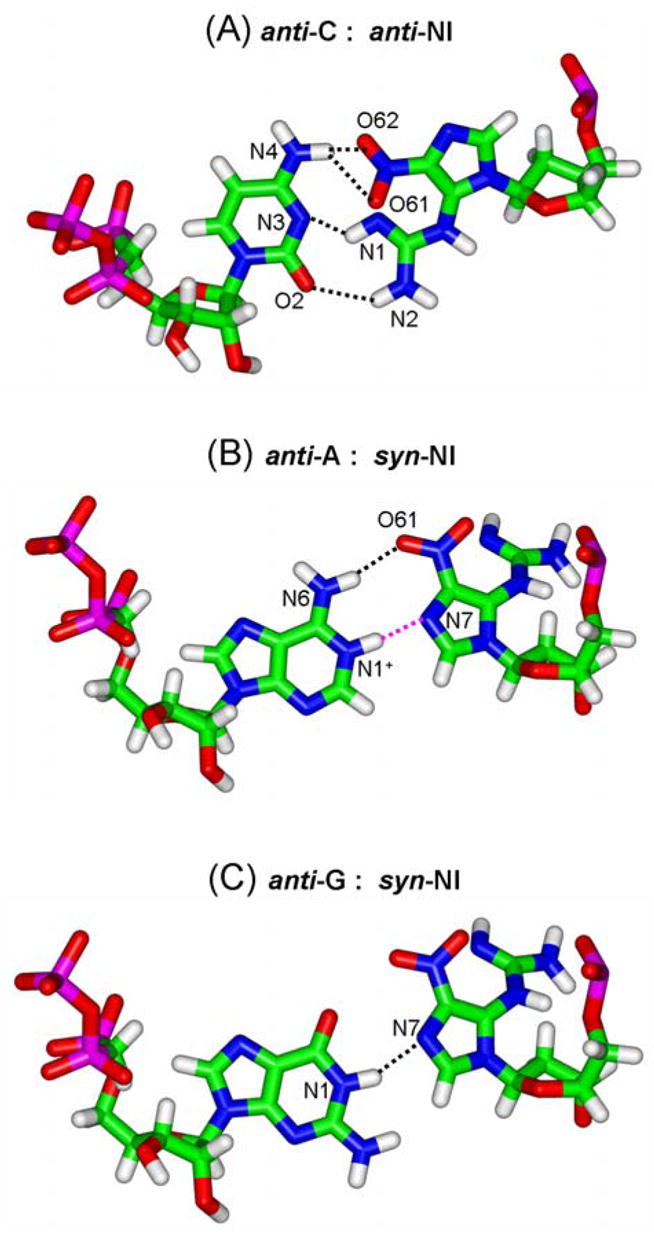
Hydrogen-bonding interactions between NI and the different incoming NTPs in T7 RNAP. (A), anti-NI:anti-C pair; NI(O61):C(N4), NI(O62):C(N4), NI(N1):C(N3) and NI(N2):C(O2). (B) syn-NI:anti-A pair; NI(O61):A(N6) and NI(N7): A(N1+). (C) syn-NI:anti-G pair; NI(N7):G(N1). Table S3 describes the details of the potential hydrogen-bonding partners.
To explain the incorporation of cytosine opposite the damage, it was necessary to model the NI in the anti glycosidic bond conformation, since the syn conformation was unable to provide hydrogen-bonded structures to cytosine. Therefore, we investigated the QM-optimized lowest energy anti conformation (6.6 kcal/mol higher in energy than syn on the nucleoside level [22]). Hydrogen bonding and other favorable interactions between the lesion and the enzyme could readily compensate for this energy difference. Adjustments of the χ, θ, δ and ϕ of NI and χ of the incoming cytosine (C) (Table S1) provided a collision-free structure with favorable stacking interactions with the previous RNA:DNA pair, and three potential hydrogen bonds between the anti-NI and anti-C. Two of these were between the N2 of NI and O2 of C, and N1 of NI and N3 of C, and a third was a bifurcated hydrogen bond between O61 and O62 of NI and N4 of C (Fig. 7C and Table S3B). Purine incomers overlap with NI in the anti conformation.
From the models, we can therefore interpret the observed nucleotide incorporation selectivity C > A > G on the basis of the number of hydrogen bonds between the lesion and the NTPs incorporated; three potential hydrogen bonds in the anti-NI:anti-C pair, two hydrogen bonds in the case of the syn-NI pairing with a protonated adenine and finally only one hydrogen bond in the syn-NI:anti-G pair. Also, the better co-planar alignment of the anti-NI:anti-C pair compared to the syn-NI:anti-(A or G) pairs (Fig. S4) offers an additional reason for the favored incorporation of a cytosine opposite the lesion.
3.6.2 The NI-modified DNA in yRNAPII
Our results showed (1) blockage of hRNAPII elongation with nucleotide incorporation opposite NI was either prevented or permitted but without further extension, and (2) error-free lesion bypass producing full-length RNA.
Blockage can be explained in part on the basis of a syn-NI conformation (Table S4A). All syn structures had collisions except one which, however, contained repulsive interactions between O61 and O62 of NI with oxygen of Ala-828 of the polymerase subunit 1, and a narrow feasible conformational range for all the torsions (<10°) (Fig. S5). Thus, all syn structures are more or less plausible for polymerase blockage. In addition, when the anti conformation was modeled there were again significant collisions which would also correlate with blocking: between H3 of NI and Cγ2 of Thr-831 of the bridge helix, and between O61 with Cδ1, and N2, H22 of NI with Cδ2 of Leu 1081 of subunit 1 (Fig. 5B). These blockage models explain why nucleotide incorporation opposite NI does not occur in the truncated RNA.
The error-free bypass can be explained on the basis of one collision-free anti-NI conformation (Table S4B) which could accommodate an anti-NI:anti-C pair, with three hydrogen bonds identical to those in T7 RNAP (Fig. 7C and Table S5). Thus, this modeled anti conformation constitutes a plausible interpretation for the observed cytosine incorporation. As in T7 RNAP, purine incomers cause collisions in the anti-NI conformation.
Full details for the torsion angles and the resulting hydrogen bonds in the modeled structures are provided in Tables S1–S5.
4. Discussion
In the study described here, we used a site-specific, lesion-modified guanine template to investigate lesion bypass by two different RNAPs, T7 RNAP and hRNAPII. We show that NI on the transcribed strand of DNA affects transcription elongation by modestly blocking the progression of T7 RNAP and largely arresting hRNAPII. RNA sequencing indicates that blockage of both enzymes, albeit to different extents, occurs at the site of NI without any nucleotide incorporation opposite this lesion. However, during lesion bypass, T7 RNAP allows for the misincorporation of various bases into the growing RNA, with the selectivity C > A > (−1 deletion) > G ≫ U, while the rare hRNAPII bypass is error-free. We also provide a structural rationales based on modeling for the observed experimental findings, and conclude that the combination of NI flexibility and inherent structural differences between the enzymes’ active sites can explain the experimental observations. This study further extends our understanding of how different DNA adducts, including 1,N2-εG [33], O6-meG [48] and benzo[a]pyrene- [36,52] and benzo[c]phenanthrene- derived adducts [35,53] affect RNA synthesis. Specifically, our work and that of others has led to an appreciation that the impact of a DNA lesion on transcription is a function of many factors, notably the size, shape, stereochemistry, and hydrogen bonding-properties of the adduct, the local DNA sequence surrounding the damage, and the nature of the enzyme active site [27]. The present NI studies add conformational flexibility as an additional element that impacts transcriptional bypass.
Time course experiments showed that NI slows the rate of transcription elongation (Fig. 3B). This effect is plausibly due to the presence of specific conformations of the NI inside the active site of the enzyme, which do not allow bypass while the predominant conformations do permit bypass. The models show that the lesion in the specific syn conformation that is lowest in energy on the nucleoside level [22] exhibits severe collisions between NI and protein residues Ser-641 and Thr-636 (Fig. 5A). This can explain the observed transcriptional blockage. However, the models show that the flexible NI moiety can also adopt a conformation that produces a collision-free syn-NI. This feasible structure is stabilized by hydrogen bonds between syn-NI and the T7 RNAP side chain of Arg-632 and Gln-749 (Fig. 6 and Table S2). These two residues occupy an empty pocket which can accommodate the NI guanidino group. We propose that the hydrogen bonds hold the lesion in the syn conformation which allows NTP incorporation and favors purines. The observed experimental selectivity for A > G is consistent with the number of hydrogen bonds formed between each purine and the syn-NI: two hydrogen bonds in the syn-NI:anti-A pair and only one hydrogen bond in the syn-NI:anti-G pair (Fig. 7). Interestingly, such syn lesion pairing schemes were also noted in our studies on 1,N2-εG [33] and O6-meG [48] lesions. Thus, more globally, T7 RNAP translesion bypass accompanied by purine incorporation seems to rely on the adoption by a guanine lesion of a syn glycosidic torsion orientation, which facilitates NTP incorporation with a consistent preference for adenine via maximal hydrogen-bonding interactions.
In addition to purine misincorporation in the full-length RNA, sequencing data revealed the presence of RNA species with a deletion opposite NI. We hypothesize that when NI is held in the abnormal syn conformation, the T7 RNAP can also slip past this lesion. The T7 RNAP has been shown to slip on undamaged templates as well [54]. We have previously observed one base deletions with T7 RNAP transcribing the guanine lesion, 1,N2-εG [33]. However, in that case, our models suggested that the deletion could be a result of the damaged base adopting an anti conformation. In the case of NI, only a syn conformer explains deletions, since a collision-free anti conformation accounts for the insertion of cytosine opposite the damage.
To explain the experimentally determined and favored error-free incorporation of cytosine opposite NI by T7 RNAP, our models have identified an anti-NI:anti-C pair with three hydrogen bonds (Fig. 7C). This anti-NI:anti-C pair, with its almost co-planar anti-NI alignment (Fig. S4), can favorably stack with the previously formed DNA:RNA hybrid. In this anti-NI structure, purine incorporation is not feasible due to overlap between the NI and an incoming purine NTP.
In contrast to T7 RNAP, hRNAPII was completely blocked by NI during single rounds of transcription and mostly blocked during multiple rounds of transcription. The percent blockage was rather variable, ranging from 86% to 96% for hRNAPII and 8% to 18% for T7 RNAP under the experimental conditions employed and after an incubation time of 60 min (Fig. 4B). The fact that we detect some bypass by hRNAPII during multiple rounds of transcription suggests that a small fraction of the polymerases can accommodate and bypass the lesion in the active site, but at a much lower frequency than with the bacteriophage enzyme. This likely reflects the intrinsic flexibility of the NI lesion, which can adopt multiple conformations, a few of which can allow the rare bypass observed; multiple rounds of transcription enhance the opportunity to observe such low frequency events.
In this connection, our models showed that most NI conformations, whether syn or anti, exhibited collisions with the enzyme. One syn-NI conformation, characterized by very narrow ranges of the NI and glycosidic torsions, was collision-free but had unfavorable repulsive interactions involving oxygens from NI and the polymerase residue Ala-828, (Fig. S3). The repulsive interactions between syn-NI and RNAPII are in sharp contrast to the stabilizing hydrogen bonding interactions of syn-NI with Arg632 and Gln-649 in T7 RNAP. In addition, one anti-NI conformation was identified which explained cytosine incorporation involving three hydrogen bonds with the lesion (Fig. 7C). Thus, unlike the T7 RNAP case, the models suggest that cytosine incorporation in RNAPII is difficult. Subsequent to C incorporation, further full-length extension in RNAPII is also rare; in this case, we speculate that the difficulty lies in the further translocation, as the non-planar NI could impede the motion of the next templating base into the active site. A translocation barrier has been suggested from crystal structures of thymine dimers and cisplatin lesions in yRNAPII [55,56]. However, again, the flexibility of NI could permit eventual rare translocations to produce full-length RNA transcripts.
T7 RNAP fits more loosely into the active site, while RNAPII is much more crowded (Fig. 8). It is plausible that conformational flexibility of NI inside the active site of T7 RNAP is easily tolerated since there is empty space surrounding the lesion, allowing for conformational adjustments that permit extensive bypass. However, the low bypass efficiency of NI exhibited in the case of hRNAPII can result from the difficulty in inserting the lesion into the active site without collisions which likely occurs only rarely. This effect is analogous to DNAPs where translesion synthesis polymerases with their looser fit and more spacious active sites bypass damaged DNA template bases at the cost of a lowered fidelity [57]. In contrast, the highly accurate and processive DNAPs are characterized by a tight fit around the nascent base pair and have a limited space for specifically accommodating normal Watson-Crick base pairs [58].
Fig. 8.
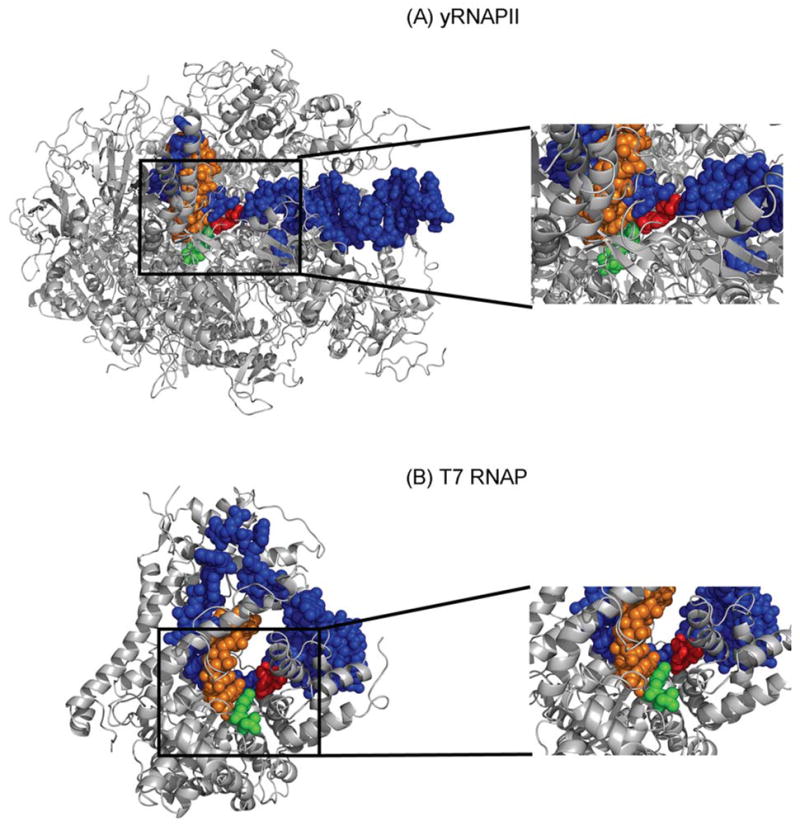
Views of yRNAPII and T7 RNAP with a templating NI opposite a CTP ribonucleotide. The RNAPII active site is much more crowded than the single subunit T7 RNAP. The active site of each enzyme is shown as an inset. The black boxes outline the area shown in the insets. Color code: Protein, grey; DNA: blue; RNA:orange; anti-NI:red and anti-C:green.
The mechanism of stalling in the NI case appears different from the mechanism that was described for yRNAPII stalling associated with two lesions, the cis-syn thymine-thymine dimers (CPD) and the 1,2-d(GpG) interstrand crosslink (GG cisplatin) [55,56]. Stalling of the polymerase at the GG lesion results from a translocation barrier that prevents the damaged nucleotides from entering the active site of the enzyme. Stalling opposite CPD requires uracil misincorporation opposite the 5′ thymine of the dimer, which surprisingly can overcome the translocation barrier to enter the active site. This dimer 5′-thymine:uracil mismatch is thought to inhibit translocation because advancing the distorting T:U pair would destabilize the elongation complex. In the case of NI, this lesion can enter the RNAPII active site, thus overcoming the translocation barrier; however, its presence induces subsequent blockage with or without cytosine incorporation, due to crowding by this conformationally flexible lesion. In the case of the rigid 1,N2-ethenoguanine, the small bulk of this lesion allows for its insertion into the active site, but the additional rigid ring causes a significant disturbance of the normal polymerase complex structure, which cannot easily rearrange itself to accommodate this lesion [33]; the essentially unaltered structure of RNAPII - cisplatin and - thymine dimer complexes was noted in the crystal structures by the Cramer group [55,56]. In the case of O6-methylguanine [48], the lesion is small enough to enter the active site and the 35% blockage (produced under identical experimental conditions to those used here) exhibited by the polymerase is triggered by distortions resulting from the rotational flexibility around the C6-O6 bond of this lesion. It is intriguing to consider the possibility that translocation barriers of various types may be important contributors to polymerase stalling and the subsequent assembly of the TCR machinery. Pull-down assays for TCR components with damaged and undamaged templates will be needed to delineate if TCR is triggered by the NI lesion.
5.Conclusion
Understanding the factors governing transcriptional stalling will aid in elucidating the determinants that invoke TCR and/or induce TM. We conclude that the mechanisms of polymerase blockage or bypass at template lesion sites, and the initiation of TCR versus TM, is a function of individual properties of the DNA lesions such as size, shape, stereochemistry, hydrogen bonding-properties, and as revealed here, lesion flexibility. Intriguing evidence that local base sequence context surrounding lesions also impacts the transcriptional processing is in need of further investigation [36,59]. Hence the factors that regulate polymerase progression are lesion specific and warrant case by case investigation.
Supplementary Material
Acknowledgments
We thank the members of the Scicchitano Laboratory for critical reading and evaluation of the manuscript. The work was supported by NIH ES010581 to DAS, and NIH 2R01 CA75449 and 5R01 CA28038 to SB, as well as NIH 2R01 ES11598 to VS and NEG. AD was supported in part by a graduate fellowship from New York University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations
- TCR
transcription coupled repair
- hRNAPII
human RNA polymerase II
- T7RNAP
T7 RNA polymerase
- NI
5-guanidino-4-nitroimidazole
- ONOO−
peroxynitrite
- NO·
nitric oxide
- O2·−
superoxide anion
- QM
quantum mechanical
- MD
molecular dynamics
- DNAP
DNA polymerase
- BER
base excision repair
- RNAP
RNA polymerase
- yRNAPII
S. cerevisiae RNAPII
- FaPy
formamidopyrimidine-DNA glycosylase
- O6-meG
O6-methylguanine
- 1
N2-εG, 1,N2-ethenoguanine
- RT-PCR
reverse transcriptase-polymerase chain reaction
- DTT
dithiothreitol
- BSA
bovine serum albumin
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- EDTA
diaminoethanetetraacetic acid
- PAGE
polyacrylamide gel electrophoresis
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
Footnotes
One unit is defined as the amount of enzyme required to catalyze the incorporation of 5nmol of CTP into acid-insoluble product in 60 minutes at 37°C in a total volume of 100μl.
One unit is defined by the manufacturer as the amount of extract required for the incorporation of 50 fmol of nucleotides into a 363-nucleotide runoff transcript generated from the CMV immediate early promoter fragment per hour at 30°C
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Rev Physiol Biochem Pharmacol. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 2.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huie RE, Padmaja S. The reaction of NO with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 5.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci U S A. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carreras MC, Pargament GA, Catz SD, Poderoso JJ, Boveris A. Kinetics of nitric oxide and hydrogen peroxide production and formation of peroxynitrite during the respiratory burst of human neutrophils. FEBS Lett. 1994;341:65–68. doi: 10.1016/0014-5793(94)80241-6. [DOI] [PubMed] [Google Scholar]
- 7.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci U S A. 1998;95:3566–3571. doi: 10.1073/pnas.95.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romero N, Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite in the presence of carbon dioxide. Arch Biochem Biophys. 1999;368:23–30. doi: 10.1006/abbi.1999.1272. [DOI] [PubMed] [Google Scholar]
- 10.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424:37–49. doi: 10.1016/s0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 11.Yermilov V, Rubio J, Becchi M, Friesen MD, Pignatelli B, Ohshima H. Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis. 1995;16:2045–2050. doi: 10.1093/carcin/16.9.2045. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy LJ, Moore K, Jr, Caulfield JL, Tannenbaum SR, Dedon PC. Quantitation of 8-oxoguanine and strand breaks produced by four oxidizing agents. Chem Res Toxicol. 1997;10:386–392. doi: 10.1021/tx960102w. [DOI] [PubMed] [Google Scholar]
- 13.Gu F, Stillwell WG, Wishnok JS, Shallop AJ, Jones RA, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligo 2′-deoxynucleotides and DNA containing guanine: formation and stability of a 5-guanidino-4-nitroimidazole lesion. Biochemistry. 2002;41:7508–7518. doi: 10.1021/bi020148q. [DOI] [PubMed] [Google Scholar]
- 14.Burney S, Niles JC, Dedon PC, Tannenbaum SR. DNA damage in deoxynucleosides and oligonucleotides treated with peroxynitrite. Chem Res Toxicol. 1999;12:513–520. doi: 10.1021/tx980254m. [DOI] [PubMed] [Google Scholar]
- 15.Yermilov V, Rubio J, Ohshima H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995;376:207–210. doi: 10.1016/0014-5793(95)01281-6. [DOI] [PubMed] [Google Scholar]
- 16.Tretyakova NY, Niles JC, Burney S, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligonucleotides containing 8-oxoguanine. Chem Res Toxicol. 1999;12:459–466. doi: 10.1021/tx980235c. [DOI] [PubMed] [Google Scholar]
- 17.Niles JC, Wishnok JS, Tannenbaum SR. A novel nitroimidazole compound formed during the reaction of peroxynitrite with 2′,3′,5′-tri-O-acetyl-guanosine. J Am Chem Soc. 2001;123:12147–12151. doi: 10.1021/ja004296k. [DOI] [PubMed] [Google Scholar]
- 18.Joffe A, Mock S, Yun BH, Kolbanovskiy A, Geacintov NE, Shafirovich V. Oxidative generation of guanine radicals by carbonate radicals and their reactions with nitrogen dioxide to form site specific 5-guanidino-4-nitroimidazole lesions in oligodeoxynucleotides. Chem Res Toxicol. 2003;16:966–973. doi: 10.1021/tx025578w. [DOI] [PubMed] [Google Scholar]
- 19.Akaike T, Okamoto S, Sawa T, Yoshitake J, Tamura F, Ichimori K, Miyazaki K, Sasamoto K, Maeda H. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc Natl Acad Sci U S A. 2003;100:685–690. doi: 10.1073/pnas.0235623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma N, Adachi Y, Hiraku Y, Horiki N, Horiike S, Imoto I, Pinlaor S, Murata M, Semba R, Kawanishi S. Accumulation of 8-nitroguanine in human gastric epithelium induced by Helicobacter pylori infection. Biochem Biophys Res Commun. 2004;319:506–510. doi: 10.1016/j.bbrc.2004.04.193. [DOI] [PubMed] [Google Scholar]
- 21.Ding X, Hiraku Y, Ma N, Kato T, Saito K, Nagahama M, Semba R, Kuribayashi K, Kawanishi S. Inducible nitric oxide synthase-dependent DNA damage in mouse model of inflammatory bowel disease. Cancer Sci. 2005;96:157–163. doi: 10.1111/j.1349-7006.2005.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Flexible 5-guanidino-4-nitroimidazole DNA lesions: structures and thermodynamics. Biochemistry. 2006;45:6644–6655. doi: 10.1021/bi0601757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neeley WL, Henderson PT, Essigmann JM. Efficient synthesis of DNA containing the guanine oxidation-nitration product 5-guanidino-4-nitroimidazole: generation by a postsynthetic substitution reaction. Org Lett. 2004;6:245–248. doi: 10.1021/ol036188j. [DOI] [PubMed] [Google Scholar]
- 24.Neeley WL, Delaney JC, Henderson PT, Essigmann JM. In vivo bypass efficiencies and mutational signatures of the guanine oxidation products 2-aminoimidazolone and 5-guanidino-4-nitroimidazole. J Biol Chem. 2004;279:43568–43573. doi: 10.1074/jbc.M407117200. [DOI] [PubMed] [Google Scholar]
- 25.Neeley WL, Delaney S, Alekseyev YO, Jarosz DF, Delaney JC, Walker GC, Essigmann JM. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J Biol Chem. 2007;282:12741–12748. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 26.Fromme JC, Verdine GL. Base excision repair. Adv Protein Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 27.Scicchitano DA, Olesnicky EC, Dimitri A. Transcription and DNA adducts: what happens when the message gets cut off? DNA Repair (Amst) 2004;3:1537–1548. doi: 10.1016/j.dnarep.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Svejstrup JQ. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem Sci. 2007;32:165–171. doi: 10.1016/j.tibs.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Doetsch PW. Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat Res. 2002;510:131–140. doi: 10.1016/s0027-5107(02)00258-0. [DOI] [PubMed] [Google Scholar]
- 30.Yin YW, Steitz TA. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 31.Wang D, Bushnell DA, Westover KD, Kaplan CD, Kornberg RD. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shafirovich V, Mock S, Kolbanovskiy A, Geacintov NE. Photochemically catalyzed generation of site-specific 8-nitroguanine adducts in DNA by the reaction of long-lived neutral guanine radicals with nitrogen dioxide. Chem Res Toxicol. 2002;15:591–597. doi: 10.1021/tx015593l. [DOI] [PubMed] [Google Scholar]
- 33.Dimitri A, Goodenough AK, Guengerich FP, Broyde S, Scicchitano DA. Transcription processing at 1,N2-ethenoguanine by human RNA polymerase II and bacteriophage T7 RNA polymerase. J Mol Biol. 2008;375:353–366. doi: 10.1016/j.jmb.2007.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perlow RA, Schinecker TM, Kim SJ, Geacintov NE, Scicchitano DA. Construction and purification of site-specifically modified DNA templates for transcription assays. Nucleic Acids Res. 2003;31:e40. doi: 10.1093/nar/gng040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schinecker TM, Perlow RA, Broyde S, Geacintov NE, Scicchitano DA. Human RNA polymerase II is partially blocked by DNA adducts derived from tumorigenic benzo[c]phenanthrene diol epoxides: relating biological consequences to conformational preferences. Nucleic Acids Res. 2003;31:6004–6015. doi: 10.1093/nar/gkg771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perlow RA, Kolbanovskii A, Hingerty BE, Geacintov NE, Broyde S, Scicchitano DA. DNA adducts from a tumorigenic metabolite of benzo[a]pyrene block human RNA polymerase II elongation in a sequence- and stereochemistry-dependent manner. J Mol Biol. 2002;321:29–47. doi: 10.1016/s0022-2836(02)00593-4. [DOI] [PubMed] [Google Scholar]
- 37.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 40.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 41.Torshin IY, Weber IT, Harrison RW. Geometric criteria of hydrogen bonds in proteins and identification of “bifurcated” hydrogen bonds. Protein Eng. 2002;15:359–363. doi: 10.1093/protein/15.5.359. [DOI] [PubMed] [Google Scholar]
- 42.Fabiola F, Bertram R, Korostelev A, Chapman MS. An improved hydrogen bond potential: impact on medium resolution protein structures. Protein Sci. 2002;11:1415–1423. doi: 10.1110/ps.4890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 44.Tabernero L, Bella J, Aleman C. Hydrogen bond geometry in DNA-minor groove binding drug complexes. Nucleic Acids Res. 1996;24:3458–3466. doi: 10.1093/nar/24.17.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panigrahi SK, Desiraju GR. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007 doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 46.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 47.Zhou Y, Martin CT. Observed instability of T7 RNA polymerase elongation complexes can be dominated by collision-induced “bumping”. J Biol Chem. 2006;281:24441–24448. doi: 10.1074/jbc.M604369200. [DOI] [PubMed] [Google Scholar]
- 48.Dimitri A, Broyde S, Scicchitano DA. Transcription bypass past O6-methylguanine by human RNA polymerase II and bacteriophage T7 RNA polymerase. Biochemistry. doi: 10.1093/nar/gkn657. (to be submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown T, Leonard GA, Booth ED, Chambers J. Crystal structure and stability of a DNA duplex containing A(anti).G(syn) base-pairs. J Mol Biol. 1989;207:455–457. doi: 10.1016/0022-2836(89)90268-4. [DOI] [PubMed] [Google Scholar]
- 50.Williams LD, Shaw BR. Protonated base pairs explain the ambiguous pairing properties of O6-methylguanine. Proc Natl Acad Sci U S A. 1987;84:1779–1783. doi: 10.1073/pnas.84.7.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan B, Mitra SN, Sundaralingam M. Crystal structure of an RNA 16-mer duplex R(GCAGAGUUAAAUCUGC)2 with nonadjacent G(syn).A+(anti) mispairs. Biochemistry. 1999;38:2826–2831. doi: 10.1021/bi982122y. [DOI] [PubMed] [Google Scholar]
- 52.Choi DJ, Marino-Alessandri DJ, Geacintov NE, Scicchitano DA. Site-specific benzo[a]pyrene diol epoxide-DNA adducts inhibit transcription elongation by bacteriophage T7 RNA polymerase. Biochemistry. 1994;33:780–787. doi: 10.1021/bi00169a020. [DOI] [PubMed] [Google Scholar]
- 53.Roth RB, Amin S, Geacintov NE, Scicchitano DA. Bacteriophage T7 RNA polymerase transcription elongation is inhibited by site-specific, stereospecific benzo[c]phenanthrene diol epoxide DNA lesions. Biochemistry. 2001;40:5200–5207. doi: 10.1021/bi0024355. [DOI] [PubMed] [Google Scholar]
- 54.Macdonald LE, Zhou Y, McAllister WT. Termination and slippage by bacteriophage T7 RNA polymerase. J Mol Biol. 1993;232:1030–1047. doi: 10.1006/jmbi.1993.1458. [DOI] [PubMed] [Google Scholar]
- 55.Brueckner F, Hennecke U, Carell T, Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- 56.Damsma GE, Alt A, Brueckner F, Carell T, Cramer P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1314. [DOI] [PubMed] [Google Scholar]
- 57.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci U S A. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 59.Tornaletti S, Donahue BA, Reines D, Hanawalt PC. Nucleotide sequence context effect of a cyclobutane pyrimidine dimer upon RNA polymerase II transcription. J Biol Chem. 1997;272:31719–31724. doi: 10.1074/jbc.272.50.31719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.