

Abstract
Objectives
The ability of minocycline to be transported into cardiac cells, concentrate in normal and ischemic myocardium and act as in vivo cardioprotector was examined. We also determined minocycline's capacity to act as a reducer of myocardial oxidative stress and matrix metalloproteinase (MMP) activity.
Background
The identification of compounds with the potential to reduce myocardial ischemic injury is of great interest. Tetracyclines (TTCs) are antibiotics with pleiotropic cytoprotective properties that accumulate in normal and diseased tissues. Minocycline is highly lipophilic and has shown promise as a possible cardioprotector. However, minocycline's potential as an in vivo cardioprotector as well as the means by which this action is attained are not well understood.
Methods
Rats were subjected to 45 min of ischemia and 48 h of reperfusion. Animals were treated 48 h before and 48 h after thoracotomy with either vehicle or 50 mg/kg/day minocycline. Tissue samples were used for biochemical assays and cultured cardiac cells for minocycline uptake experiments.
Results
Minocycline significantly reduced infarct size (∼33%), tissue MMP-9 activity and oxidative stress. Minocycline was concentrated ∼24-fold in normal (0.5 mM) and ∼50-fold in ischemic regions (1.1 mM) vs. blood. Neonatal rat cardiac fibroblasts, myocytes and adult fibroblasts demonstrate a time- and temperature-dependent uptake of minocycline to levels that approximate those of normal myocardium.
Conclusions
Given the high intracellular levels observed and results from the assessment of in vitro antioxidant and MMP inhibitor capacities, it is likely that minocycline acts to limit myocardial ischemic injury via mass action effects.
Keywords: myocardial infarction, cardioprotection, pleiotropic action
Introduction
Tetracyclines (TTCs) are broad-spectrum antibiotics and exert antimicrobial effects by inhibiting protein synthesis. TTCs are classified as natural, semi-synthetic and chemically modified (1). Two of the more common semi-synthetic TTCs used clinically as antibiotics are doxycycline and minocycline. TTCs are known to chelate calcium and as a consequence incorporate into teeth (which during dentition stains them), cartilage and bone (2). TTCs are also found at high levels in gingival tissue/salival fluid and their accumulation has been linked with an active cellular uptake process (3). In the 1970's it was reported that radio-labeled TTCs accumulate in infarcted myocardium in proportion to the degree of tissue damage (4,5). However, the extent and means by which TTCs may accumulate in normal and ischemic myocardium has not been examined.
Doxycycline and minocycline possess cytoprotective properties (1,6-8). Effects are likely secondary to their capacity to act as anti-inflammatory, anti-apoptotic, reactive oxygen species scavenger and matrix metalloproteinase (MMP) inhibitors (1,6-8). However, there is controversy in this area given that for example, doxycycline is not a particularly potent MMP inhibitor since IC50 values are in the micromolar range (9). Thus, the importance of determining compound tissue concentrations since they would ultimately define how effective TTCs may be in exerting cytoprotective actions. Nonetheless, the promise of these class of drugs in preventing and/or limiting organ injury is currently being explored in 30+ ongoing clinical trials (www.clinicaltrials.gov) several of which cover cardiovascular pathologies such as stroke, vascular aneurisms, coronary bypass surgery, post-infarct ventricular remodeling and diseases such as Parkinson's.
Of the TTCs, minocycline is unique in that it not only has excellent bioavailability, absorption and long half life, but is also lipophilic (7). Thus, this agent may reach very high tissue levels (secondary to active transport systems and lipophillicity) and as a consequence have the ability to protect organs from ischemic injury. Indeed, reports from studies using ex vivo heart systems and cultured cells indicate that minocycline can protect myocardium from ischemic injury (10). However, its potential for in vivo cardioprotective actions has not been determined. Thus, the purpose of our study was to determine the extent to which minocycline renders significant cardioprotection from ischemic injury in vivo, to examine myocardial levels attained and, if a cellular uptake process is involved. For this purpose we utilized a rat model of ischemia-reperfusion (I/R) injury. We also wished to establish how myocardial minocycline levels may determine the effectiveness of the compound in exerting cytoprotective actions secondary to its antioxidant and MMP inhibitor properties.
Methods
Minocycline treatment
Male Sprague-Dawley rats (Harlan) of 250-300 g were used. Two groups of rats were generated: control and minocycline-treated rats. Subgroups were utilized for the assessment of I/R injury (n=12 control, 10 minocycline), hemodynamics (n=5/each), and biochemical determinations (n=4 control, 5 minocycline). A separate subgroup was generated for measurements of tissue minocycline (n=5). Minocycline hydrochloride was administered via intraperitoneal (IP) injection at 25 mg/kg every 12 h, a dose within a range known to attain effective cardioprotection ex vivo under regional ischemia (10). In controls water was used. Treatment began 48 h before thoracotomy and continued until 48 h of reperfusion. Procedures were performed according to guidelines by the American Association for Accreditation of Laboratory Animal Care, protocols were approved by the UCSD Institutional Animal Care and Use Committee and conform to published NIH guidelines for animal research.
Surgery
Animals were anesthetized by IP injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), intubated, and positive-pressure ventilated. Hearts were exposed through a left thoracotomy, the left anterior descending (LAD) coronary artery was occluded and released after 45 min and the suture left in place.
Hemodynamics
In subgroups of anesthetized animals prior to sacrifice a micromanometer was inserted into the right carotid artery to measure heart rate, carotid and left ventricular (LV) pressures.
Tissue sampling and staining
At 48 h reperfusion, heparinized/anesthetized animals were euthanized and underwent cardiectomy. To delineate the area at risk the LAD suture was re-tied, hearts perfused with 0.4% trypan blue using a syringe and sliced into 2 mm-thick sections. A single ecuatorial slice was photographed and the extent of the area at risk and infarct areas were quantified by computer planimetry. The remaining slices were used for biochemical assays. The LV freewall was separated and divided into ischemic region and border zone. Right ventricle was used as non-ischemic tissue. To distinguish between viable and infarcted myocardium the equatorial slice was stained in 1% w/v 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate buffer pH 7.4 for 20 min at 37°C. Infarct area (IA) was quantified by digital planimetry and expressed as percentage of area at risk (AAR).
Gelatin zymography
Tissue (50 mg) was homogenized in 10 mM HEPES, pH 7.5,150 mM NaCl, 0.2 mM EDTA, 25% glycerol, 100 μg/ml phenylmethylsulfonylfluoride, and 0.2 kallikrein inhibitory units/ml aprotinin. Samples (10 μg) were analyzed by SDS-PAGE as described (11). An internal control (human MMP-2/MMP-9) was loaded to normalize gels. Gelatinolytic activity was digitally quantified.
Glutathione (GSSG/GSH) Assay
Tissue was homogenized in buffer (154 mM KCl, 5 mM diethylenetriaminepentaacetic acid (DPTA), and 0.1 M potassium phosphate, pH 6.8). After centrifugation, an aliquot was removed for protein determination. One volume of cold acid buffer (40 mM HCl, 10 mM DPTA, 20 mM ascorbic acid, and 10% trichloroacetic acid) was added to one volume of homogenate. The suspension was centrifuged and the supernatant solution filtered. Reduced and oxidized glutathione, GSH and GSSG respectively, levels were determined by using the fluorophore o-phthalaldehyde (12).
Cell culture
Neonatal and adult rat ventricular fibroblasts (NRVF, ARVF) and myocytes (NRVM) were prepared as previously described using a collagenase based method and gradient separations (13). NRVF and ARVF were seeded into 12-well cell culture plates (200,000 cells/well) and allowed to proliferate to confluence (∼350,000 cells/well). NRVM were seeded into 6-well cell culture plates (500,000 cells/well). Cells were cultured in standard serum containing media until used (13). Cells were serum deprived for 24 h before use.
Minocycline transport assay in cardiac cells
Minocycline transport in cardiac cells was measured fluorometrically in each seeded well as described (3,14) with modifications. Briefly, culture plates containing cell monolayers were washed 4× with Hank's Balance Salt Solution (HBSS), overlaid with HBSS and warmed to 37°C prior to assay. Warm HBSS (0.2 ml) containing twice the desired final minocycline concentration (40 μM) was simultaneously added to each well with multichannel pipettes. After incubation at 37°C and 4°C for the indicated times, the minocycline solution was quickly removed. Each well was then rapidly washed 4× according to incubation temperature with either warm (37°C) or cold (4°C) HBSS to eliminate extracellular minocycline. Cell monolayers were lysed with 1 ml of ddH2O and then sonicated on ice 3× for 5 s bursts (resting 10 s in between to prevent overheating). The lysate was centrifuged at 13,000 × g for 10 min. The supernatant was recovered and mixed with 1 ml ethylene glycol containing 200 mM citric acid and 200 mM magnesium acetate prior to fluorescence measurement. Calibration plots were constructed to relate fluorescence to well minocycline content. To estimate the intracellular concentration of minocycline, NRVF and ARVF cell volume was measured using a cell and particle counter (Z2 Coulter Counter; Beckman Coulter) yielding an average diameter of 12 μm. NRVM diameter was obtained from previous published reports (19.5 μm) (15).
Minocycline determination in heart tissue and plasma
Fluorometric assay of minocycline in heart homogenates was done as previously described (16) with modifications. Briefly, 150 mg of heart tissue was homogenized using a Polytron, in 2 ml of 20 mM HEPES and 10 mM MgCl2 solution pH 7.4. Samples were centrifuged at 13,000 × g for 10 min. Supernatants were recovered and deproteinized with 0.6 ml 1.5 M trichloracetic acid. Samples were then vortexed and allowed to sit at room temperature (RT) for 10 min. Samples were centrifuged at 13,000 × g for 10 min. Supernatants were recovered and transferred to 15 ml conical tubes. 0.5 ml of 0.1 M HCl containing 1 mg/ml thiopropionic acid was added along with 1.0 ml of Sorense's buffer (pH 6.0). After mixing, 0.5 ml of 0.75 M aluminum chloride was added and the tubes were shaken vigorously. After 15 min incubation at RT, fluorescence was measured (λex = 384, λem = 450 nm). Similar procedure was used for minocycline determinations in plasma. Calibration plots were constructed to relate fluorescence to tissue minocycline content.
IC50 determinations
Inhibition of human recombinant MMP-7 and MMP-9 (Calbiochem) activity by minocycline was measured using a fluorescent assay kit from BIOMOL International. Assays contained 0.6 μM MMP-7 or MMP-9 in buffer [50 mM N-(2-hydroxyethyl)piperazine-N'-ethanesulfonic acid, 10 mM CaCl2, 0.05% Brij-35, pH 7.5] in the presence of increasing concentrations of minocycline (0, 0.01, 0.1, 0.25, 0.5, 1, 5, 50, 100, 250, 500, 1000, 1500, 2000 μM). Protein was incubated with drug for 1 hr at 37°C in the dark. Cleavage reactions were started by addition of the fluorescent substrate. Fluorescence (λex = 335, λem = 405 nm) was measured at 30 s intervals for 1 h. Cleavage rates were determined from the linear regions of kinetic curves. IC50 was determined from graphs of MMP activity versus log [minocycline].
MMP activity in heart homogenates
Frozen tissue samples were homogenized using a hand-held homogenizer on ice-cold lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM CaCl2, 0.2mM NaN3, 0.1% Triton X-100). Homogenates were centrifuged and supernatants isolated. 100 μg total protein and 20 μM OmniMMP fluorogenic substrate (BIOMOL International) were mixed in buffer. Given that the initial tissue concentration was diluted during homogenization, heart homogenates (in 100 μ l of buffer) derived from minocycline-treated rats were supplemented with minocycline to reach a final concentration of 0.5 mM (non-ischemic regions) and 1.1 mM (ischemic regions), as determined by results from the minocycline tissue uptake assays described in this study. Kinetic fluorescence measurements were performed (λex = 335, λem = 405 nm) and substrate cleavage rates were determined from the linear regions of kinetic curves.
Determination of antioxidant activity
Antioxidant activity for minocycline and Trolox (internal control) was measured in triplicates using an in vitro Antioxidant Assay Kit purchased from Cayman Chemical.
Statistical Analysis
Results are expressed as mean±S.E.M. Comparisons between means were analyzed, as appropriate, by student's t-test or one-way ANOVA followed by Bonferroni t-test. A value of p<0.05 was considered statistically significant.
Results
Minocycline reduced infarct size
Body and heart weights (data not shown) were similar for both groups after I/R. In the control group the mean area at risk (47±3.7% of LV) was not significantly different from that in the minocycline-treated group (41±2.7% of LV) (Fig. 1A). The infarct area in the minocycline-treated group (30±3.5% of the area at risk) was significantly reduced compared to that of the control group (45±4.2% of the area at risk), representing an overall infarct reduction of ∼33% by minocycline (p=0.01) (Fig. 1B). The assessment of hemodynamics yielded non significant differences between control vs. minocycline treated rats in heart rate (385±25 bpm vs. 363±35), LV end-diastolic pressure (6.5±4 mmHg vs. 4.1±3.3), LV systolic pressure (101±5 mmHg vs. 104±7) or mean arterial pressure (86±7 mmHg vs. 86±5). Thus, changes in hemodynamics fail to explain the observed differences in infarct size.
Figure 1. Reduction of myocardial infarct size by minocycline.
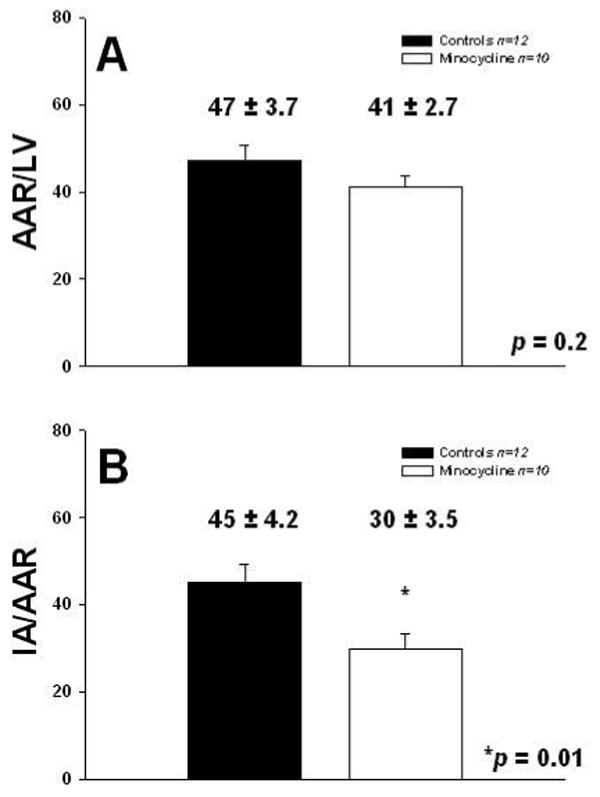
A. Morphometric analysis of area at risk (AAR) as a function of left ventricular area (AAR/LV), and B. Infarct area (IA) as a function of AAR (IA/AAR) in I/R control (n=12) and minocycline-treated (n=10) hearts. Values are mean ± S.E.M.
Minocycline attenuated MMP-9 upregulation in the ischemic region
Gelatin zymography of tissue homogenates from the ischemic and non-ischemic regions reveal visible bands corresponding to 92 kDa MMP-9 (Fig. 2A). Densitometric analysis indicated that MMP-9 levels were reduced by ∼50% in the ischemic region of the minocycline-treated group vs. controls (p<0.01, Bonferroni, 6 comparisons) (Fig. 2B). MMP-9 levels in the non-ischemic regions remained similar between groups. No differences in MMP-2 levels were observed in any group (data not shown). The 86 kDa MMP-9 band was not quantified as it was not visible.
Figure 2. Reduction of MMP-9 levels in the ischemic region with minocycline treatment.
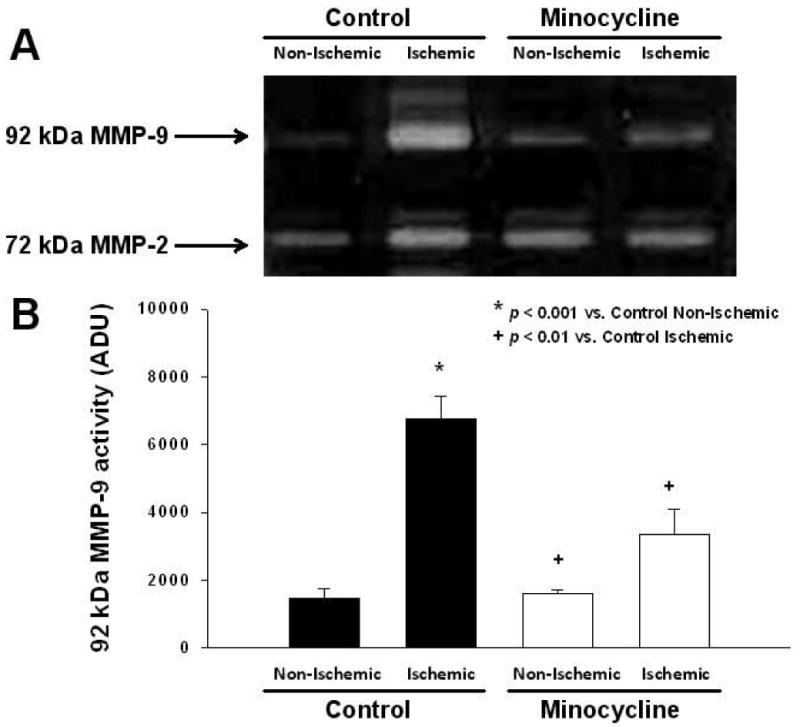
A. Representative gelatin zymography of myocardial MMP-2 and MMP-9 levels in I/R control (n=4) and minocycline-treated (n=5) hearts. B. Bar graph derived from densitometric analysis of 92 kDa MMP-9 zymographic activity in control (non-ischemic and ischemic) and minocycline-treated (non-ischemic and ischemic) hearts regions. Values are mean ± S.E.M.
Minocycline as an antioxidant
We explored whether minocycline ameliorated oxidative stress in our in vivo model of cardiac I/R. GSSG/GSH was used as a measure of tissue oxidative stress. Ischemic tissue from minocycline-treated animals displayed a significant decrease (p<0.05, Bonferroni, 6 comparisons) of GSSG/GSH by 97% vs. control ischemic tissue (Fig. 3). In vitro assays indicate that minocycline bears similar antioxidant potential as the vitamin E analog Trolox when compared at the same concentrations (44, 88, 135, 180, 225 and 330 μM) (Fig. 4).
Figure 3. Reduction of tissue oxidative stress with minocycline treatment.
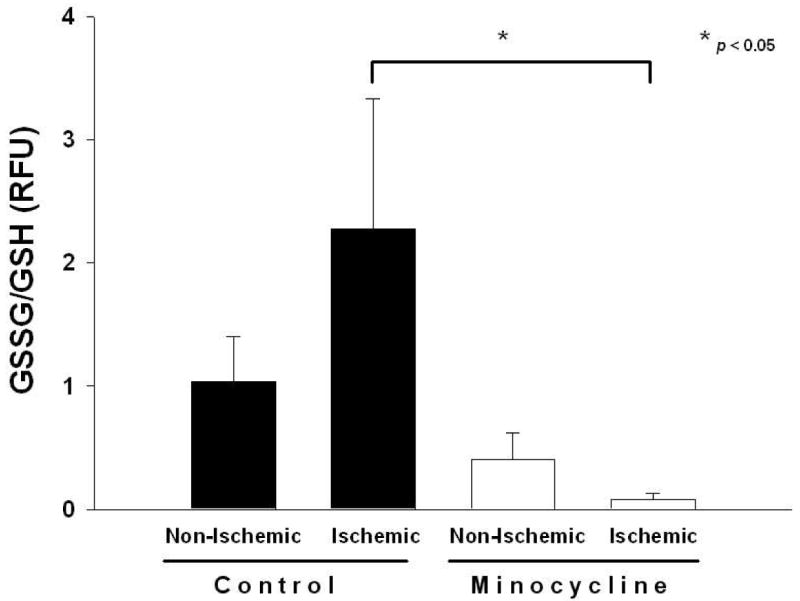
Tissue oxidative stress levels in control (n=4) and minocycline (n=5) treated animals. Decreased ratio of oxidized to reduced glutathione content (GSSG/GSH) was observed in ischemic heart tissue homogenates of minocycline treated animals. Values are mean ± S.E.M.
Figure 4. In vitro minocycline antioxidant potential.
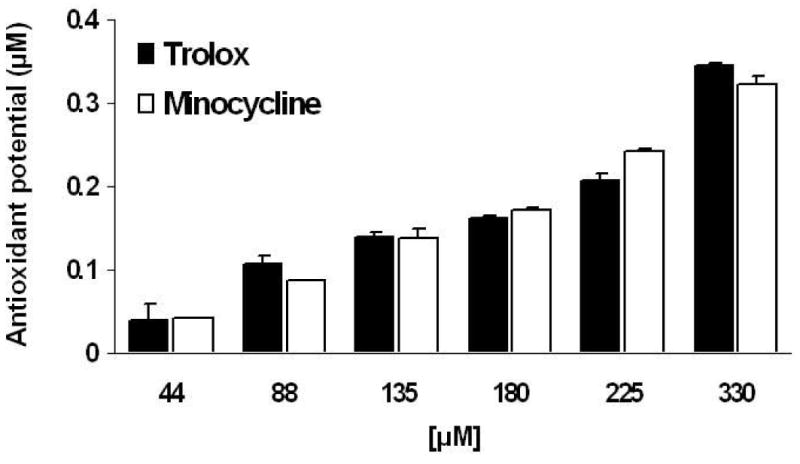
Using an in vitro assay kit, minocycline displays antioxidant activity similar to that of the vitamin E analog Trolox. Values are mean ± S.E.M. of 3 experiments.
Accumulation of minocycline in the heart
Fluorometric analysis indicated that animals treated with minocycline displayed significant (p<0.05, Bonferroni, 4 comparisons) accumulation of the antibiotic in the heart by revealing ∼24-fold increase relative to plasma levels. Moreover, minocycline accumulation was enhanced upon induction of I/R injury, where antibiotic concentrations reached levels ∼2-fold higher vs. those of non-ischemic regions (∼50-fold vs. plasma) (Fig. 5). Previous studies have shown that human gingival fibroblasts display an active transport mechanism for minocycline uptake (3,14). In cultured ARVF (Fig. 6A), as well as in NRVF (Fig. 6B), minocycline accumulation also takes place in a time- and temperature-dependent manner. At 37°C minocycline uptake in cardiac fibroblasts follows a kinetic pattern of active transport reaching a saturation stage at ∼6 min, whereas at 4°C no significant accumulation occurred over time. Myocytes display a similar minocycline uptake process as in fibroblasts yielding a 6-fold concentration of the compound vs. extracellular levels (Fig. 7).
Figure 5. Accumulation of minocycline in plasma, non-ischemic and ischemic cardiac tissue.
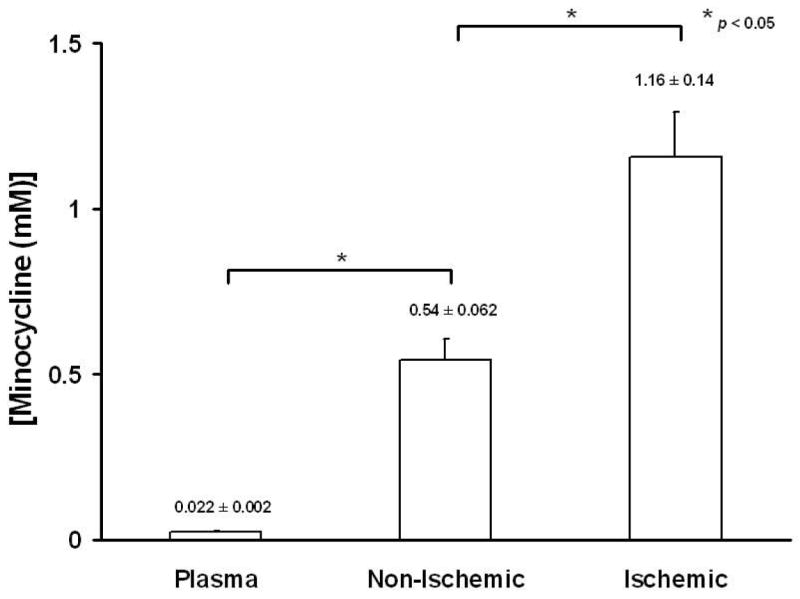
Normal cardiac tissue displays a 24-fold minocycline accumulation vs. plasma, which is further enhanced by 2-fold in response to ischemia. Values are mean ± S.E.M (n=5).
Figure 6. Minocycline accumulation in neonatal (A) and adult (B) cardiac fibroblasts.
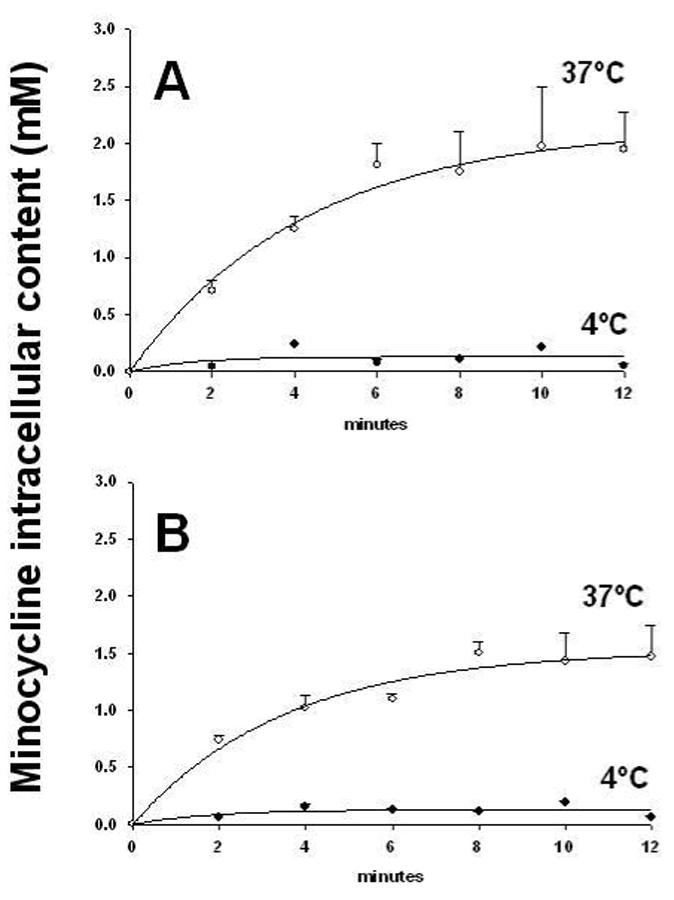
Cells were incubated in a monolayer of 40 μM minocycline in HBSS at 4°C (•) and 37°C (❍) and uptake was monitored over the indicated time intervals. Values represent mean ± SEM of 3 independent experiments.
Figure 7. Minocycline accumulation by cardiac myocytes.
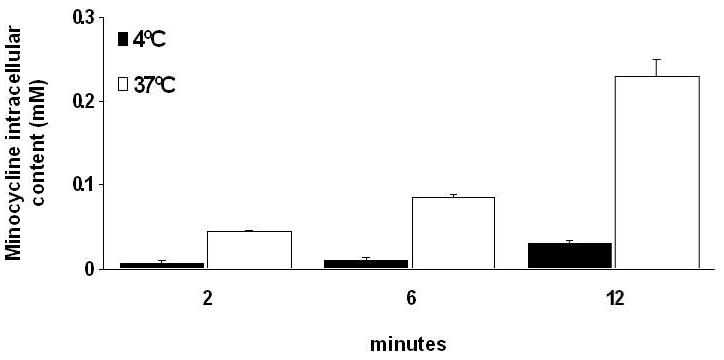
Cells were incubated in a monolayer of 40 μM minocycline in HBSS at 4°C and 37°C and uptake was monitored over the indicated time intervals. Values represent mean ± SEM of 3 independent experiments.
Minocycline as MMP inhibitor
Based on the IC50 values determined, minocycline revealed an IC50 for MMP-7 (125 μM) and MMP-9 (180 μM) (Fig. 8). On the basis of the results obtained regarding minocycline uptake in heart tissue, MMP activity assays were supplemented with 0.5 mM for non-ischemic region and 1.1 mM for ischemic region homogenates to approximate in vivo concentrations. Minocycline at 0.5 mM in the non-ischemic tissue reduced global MMP activity by 74% vs. controls (ischemic and non-ischemic). Minocycline at 1.1 mM in the ischemic-tissue reduced global MMP activity by 91% vs. controls (Fig. 9). Decreases were significant (p<0.0001, Bonferroni, 6 comparisons). In addition, ischemic tissue treated with minocycline displayed a further reduction of 66% in global MMP activity when compared to that of non-ischemic tissue treated with minocycline.
Figure 8. IC50 values of minocycline for human recombinant MMP-7 and MMP-9.
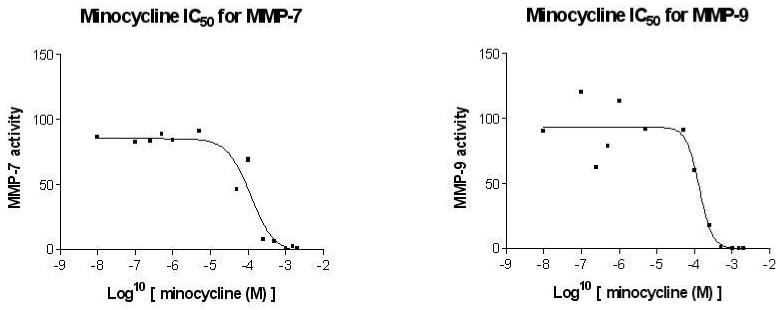
In vitro assays using active MMPs show inhibition at IC50 = 125 μM for MMP-7 and IC50 = 180 μM for MMP-9.
Figure 9. Global heart tissue MMP activity.
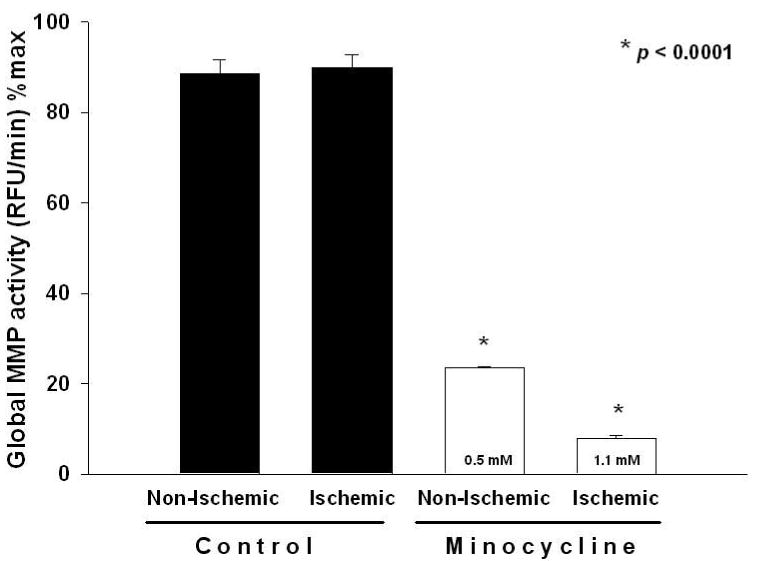
Samples used were from control (n=4) and minocycline (n=5) treated animals. Minocycline significantly inhibited global MMP activity in hearts from non-ischemic and ischemic regions vs. corresponding controls. Values are mean ± S.E.M.
Discussion
Results demonstrate that pre- and post-treatment of rats subjected to I/R yields a significant ∼33% reduction in infarct size. This is the first report for in vivo cardioprotection by minocycline. Scarabelli, et al.(10) reported minocycline cardioprotection in cultured cardiac myocytes and ex vivo rat hearts. The cardioprotective effects were attributed to minocycline actions on apoptotic cell pathways. We have also previously reported on the cardioprotective actions of doxycycline (17,18). The ability of minocycline to limit tissue damage in the setting of ischemia has been documented in kidney and lung (19,20). Minocycline also demonstrates neuroprotection in vitro and in vivo (7,21,22). Neuroprotective mechanisms include reduction of microglial activation, downregulation of pro-inflammatory/cell death mediators and control of mitochondrial permeability (21-23). Minocycline, by potentially acting as an MMP inhibitor, can also prevent blood-brain barrier disruption (7,22,23). Given this evidence, clinical studies in stroke patients have been performed where minocycline was administered orally for 5 days (200mg) with a therapeutic window of time of 6-24 h after onset of stroke (24). Patients had significantly better outcome with minocycline vs. placebo. Currently, clinical trials are being pursued to examine for doxycycline cardioprotection in coronary artery bypass patients and for amelioration of adverse post-infarction remodeling (www.clinicaltrials.gov).
Investigators using radiolabeled TTC noted its capacity to accumulate in damaged myocardium and serve to diagnose infarcts (5,25). Results demonstrated a correlation between infarct size as determined by radiolabeled TTC and serum creatine kinase. The ability of TTCs to concentrate in other tissues is well know (26). Dentists take advantage of the high concentration of doxycycline in saliva as a means to treat periodontal (gum) disease which compromises dental ligaments via MMPs. Periostat (a form of doxycycline) is commercialized for this purpose (6). To investigate TTCs accumulation in gingival tissue, Yang et al. (3) examined the capacity of gingival fibroblasts to uptake the compounds. Gingival fibroblasts transport minocycline in a concentration and temperature-dependent manner. At steady state, the cellular/extracellular concentration ratio was >60 for minocycline. It was concluded that gingival fibroblasts possess active transporters for TTCs that may explain high salival levels of these agents. TTCs uptake has also been observed in neutrophils and may partly explain high levels observed in injured tissues (27).
We explored the capacity of minocycline to accumulate in myocardial tissue and cells. Minocycline accumulates in myocardium severalfold above plasma levels. Accumulation was more pronounced in ischemic vs. normal myocardium or plasma. To identify the mechanism that may account for myocardial accumulation we performed experiments using neonatal and adult rat cardiac fibroblasts, and neonatal rat cardiac myocytes. Cardiac fibroblasts possess a comparable uptake system to that reported for gingival cells (3). Uptake was saturable, time- and temperature-dependent. Cardiac myocytes also demonstrated uptake but with reduced magnitude. The estimation of intracellular minocycline levels indicate a capacity for isolated cardiac cells to accumulate the compound in a magnitude compatible with our in vivo normal tissue results. This observation allows us to conclude that most of the TTCs accumulated in myocardium are found in physical association with cellular structures. However, as observed in gingiva high intracellular concentrations likely lead to high interstitial levels. We speculate that minocycline accumulation in ischemic tissues may also arise from its chelation properties binding to divalent cations such as calcium in cells whose permebility is compromised (28).
We previously reported a significant downregulation of MMP-9 activity in ischemic myocardium with doxycycline treatment 48 h after I/R (18). Minocycline treatment also led to a significant decrease in 92 kDa MMP-9 levels in the ischemic region. No changes were observed in the 86 kDa form of MMP-9 or in MMP-2 levels. We also previously published the IC50 of doxycycline for MMP-7 (28 μM) (9). We performed similar in vitro determinations using minocycline as a means to determine the capacity of this agent to act as an MMP inhibitor. Results yielded an IC50 for MMP-7 of 125 μM and for MMP-9 of 180 μM. Thus, minocycline is a weaker MMP inhibitor vs. doxycycline in vitro, confirming previous reports (29,30). However, since minocycline can attain millimolar concentrations, mass action effects may be possible on intracellular or immediate extracellular MMP targets (such as MMP-2 or MMP-9) which may activate with I/R (31). To investigate this possibility, we performed global MMP activity assays. Samples were homogenized in assay buffer. Since homogenization requires the dilution of the tissue sample, we supplemented minocycline to the assay buffer to generate comparable concentrations to those derived from in vivo measurements. At the minocycyline concentrations tested (0.5 mM for non-ischemic and 1.1 mM for ischemic) a significant and dose dependent reduction in MMP activity was observed. A recent report shows a significant correlation between the degree of MMP inhibition and brain levels of TTCs (32). Thus, it is possible that part of the cardioprotective effects of minocycline may be derived from high tissue levels of the compound.
TTCs are also known as anti-oxidants. Reductions in ROS levels can prevent pro-MMP activation by the cysteine switch mechanism as seen in stroke (33). Studies have demonstrated that the structural features of phenolic compounds and their hydroxyl radicals (as those of TTCs) confer ROS scavenger potential (28). Minocycline treatment led to a significant blunting of ischemic tissue oxidative stress. These data are compatible with previous reports where TTCs have shown to reduce tissue oxidative stress (34,35). Leon, et al. (35) recently reported the capacity of doxycycline to protect cardiac myocytes from peroxynitrite-induced contractile failure independent of MMP inhibitor actions. We compared the anti-oxidant potential of minocycline vs. a known ROS scavenger, Trolox. Results indicate that minocycline has comparable anti-oxidant potential to Trolox. On the basis of the anti-oxidant results observed and the concentrations measured in samples of myocardium, we can assume that minocycline acts as an effective ROS scavenger. Again, this effect would result not only as a consequence of its unique anti-oxidant structural feature, but also because of mass action effects. Minocycline has also been shown to be cytoprotective by attenuating apoptotic events/pathways (7). It is thus, possible that high concentrations of minocycline found in normal and ischemic tissue also allows for notable effects on these other targets.
It is important to note the limitations associated with our study. We utilized a pre-treatment scheme to examine the cardioprotective effects of minocycline. As such the effects apply to ischemic injury and not necessarily to reperfusion (since we did not only give the drug either just before or after it). Thus, the extrapolation of these results to the clinical scenario awaits further studies. Furthermore, the use of an animal model of I/R injury has inherent limitations when compared to patients with coronary heart disease. In conclusion, results from our study indicate that minocycline can act in the in vivo setting to reduce infarct size following ischemic injury. Minocycline cardioprotection may not only be secondary to the capacity of the compound to modulate specific signaling pathways but also from mass action effects on endpoints such as antioxidant and MMP inhibitor actions. The levels of minocycline observed in tissue samples and cells justify a reevaluation by investigators to consider how this class of drugs act to limit tissue injury. Given their unusual capacity to accumulate in tissues and areas of injury one would even be tempted to qualify TTCs as “smart” drugs.
Acknowledgments
We wish to acknowledge the support of a CONACYT-UC MEXUS doctoral fellowship to D. Romero-Perez and a post-doctoral NIH fellowship (T32-DK007044) to Dr. Eduardo Fricovsky.
Supported by: NIH HL-43617 and HL-67922
Footnotes
No conflicts of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258–65. doi: 10.1016/j.jaad.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez AR, Rogers RS, 3rd, Sheridan PJ. Tetracycline and other tetracycline-derivative staining of the teeth and oral cavity. Int J Dermatol. 2004;43:709–15. doi: 10.1111/j.1365-4632.2004.02108.x. [DOI] [PubMed] [Google Scholar]
- 3.Yang Q, Nakkula RJ, Walters JD. Accumulation of ciprofloxacin and minocycline by cultured human gingival fibroblasts. J Dent Res. 2002;81:836–40. doi: 10.1177/154405910208101208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holman BL, Idoine J, Fliegel CP, et al. Detection and localization of experimental myocardial infarction with 99m Tc-tetracycline. J Nucl Med. 1973;14:595–9. [PubMed] [Google Scholar]
- 5.Holman BL, Zweiman FG. Time course of 99mTc(Sn)-tetracycline uptake in experimental acute myocardial infarction. J Nucl Med. 1975;16:1144–6. [PubMed] [Google Scholar]
- 6.Peterson JT. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail Rev. 2004;9:63–79. doi: 10.1023/B:HREV.0000011395.11179.af. [DOI] [PubMed] [Google Scholar]
- 7.Jordan J, Fernandez-Gomez FJ, Ramos M, Ikuta I, Aguirre N, Galindo MF. Minocycline and cytoprotection: shedding new light on a shadowy controversy. Curr Drug Deliv. 2007;4:225–31. doi: 10.2174/156720107781023938. [DOI] [PubMed] [Google Scholar]
- 8.Bastos LF, Merlo LA, Rocha LT, Coelho MM. Characterization of the antinociceptive and anti-inflammatory activities of doxycycline and minocycline in different experimental models. Eur J Pharmacol. 2007;576:171–9. doi: 10.1016/j.ejphar.2007.07.049. [DOI] [PubMed] [Google Scholar]
- 9.Garcia RA, Pantazatos DP, Gessner CR, Go KV, Woods VL, Jr, Villarreal FJ. Molecular interactions between matrilysin and the matrix metalloproteinase inhibitor doxycycline investigated by deuterium exchange mass spectrometry. Mol Pharmacol. 2005;67:1128–36. doi: 10.1124/mol.104.006346. [DOI] [PubMed] [Google Scholar]
- 10.Scarabelli TM, Stephanou A, Pasini E, et al. Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to smac/DIABLO, and reduces the mitochondrial leakage of cytochrome C and smac/DIABLO. J Am Coll Cardiol. 2004;43:865–74. doi: 10.1016/j.jacc.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 11.Garcia RA, Brown KL, Pavelec RS, Go KV, Covell JW, Villarreal FJ. Abnormal cardiac wall motion and early matrix metalloproteinase activity. Am J Physiol Heart Circ Physiol. 2005;288:H1080–7. doi: 10.1152/ajpheart.00860.2004. [DOI] [PubMed] [Google Scholar]
- 12.Senft AP, Dalton TP, Shertzer HG. Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde. Anal Biochem. 2000;280:80–6. doi: 10.1006/abio.2000.4498. [DOI] [PubMed] [Google Scholar]
- 13.Villarreal FJ, Kim NN, Ungab GD, Printz MP, Dillmann WH. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88:2849–61. doi: 10.1161/01.cir.88.6.2849. [DOI] [PubMed] [Google Scholar]
- 14.Walters JD, Nakkula RJ, Maney P. Modulation of gingival fibroblast minocycline accumulation by biological mediators. J Dent Res. 2005;84:320–3. doi: 10.1177/154405910508400405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang SQ, Ding B, Guo ZG, Li YX. Inhibitory effect of antisense oligodeoxynucleotide to p44/p42 MAPK on angiotensin II-induced hypertrophic response in cultured neonatal rat cardiac myocyte. Acta Pharmacol Sin. 2004;25:41–6. [PubMed] [Google Scholar]
- 16.Hall D. Rapid fluorimetric assay of minocycline in plasma or serum: comparison with microbiological assay. Br J Clin Pharmacol. 1977;4:57–60. doi: 10.1111/j.1365-2125.1977.tb00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villarreal FJ, Griffin M, Omens J, Dillmann W, Nguyen J, Covell J. Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation. 2003;108:1487–92. doi: 10.1161/01.CIR.0000089090.05757.34. [DOI] [PubMed] [Google Scholar]
- 18.Griffin MO, Jinno M, Miles LA, Villarreal FJ. Reduction of myocardial infarct size by doxycycline: a role for plasmin inhibition. Mol Cell Biochem. 2005;270:1–11. doi: 10.1007/s11010-005-2540-3. [DOI] [PubMed] [Google Scholar]
- 19.Sutton TA, Kelly KJ, Mang HE, Plotkin Z, Sandoval RM, Dagher PC. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2005;288:F91–7. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- 20.Rempe S, Hayden JM, Robbins RA, Hoyt JC. Tetracyclines and pulmonary inflammation. Endocr Metab Immune Disord Drug Targets. 2007;7:232–6. doi: 10.2174/187153007782794344. [DOI] [PubMed] [Google Scholar]
- 21.Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist. 2005;11:308–22. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- 22.Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744–51. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- 23.Domercq M, Matute C. Neuroprotection by tetracyclines. Trends Pharmacol Sci. 2004;25:609–12. doi: 10.1016/j.tips.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Lampl Y, Boaz M, Gilad R, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–10. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 25.Holman BL. Radionuclide methods in the evaluation of myocardial ischemia and infarction. Circulation. 1976;53:I112–9. [PubMed] [Google Scholar]
- 26.Baker PJ, Evans RT, Coburn RA, Genco RJ. Tetracycline and its derivatives strongly bind to and are released from the tooth surface in active form. J Periodontol. 1983;54:580–5. doi: 10.1902/jop.1983.54.10.580. [DOI] [PubMed] [Google Scholar]
- 27.Walters JD. Characterization of minocycline transport by human neutrophils. J Periodontol. 2006;77:1964–8. doi: 10.1902/jop.2006.060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson ML. Chemical and biological dynamics of tetracyclines. Adv Dent Res. 1998;12:5–11. doi: 10.1177/08959374980120011901. [DOI] [PubMed] [Google Scholar]
- 29.Paemen L, Martens E, Norga K, et al. The gelatinase inhibitory activity of tetracyclines and chemically modified tetracycline analogues as measured by a novel microtiter assay for inhibitors. Biochem Pharmacol. 1996;52:105–11. doi: 10.1016/0006-2952(96)00168-2. [DOI] [PubMed] [Google Scholar]
- 30.Ryan ME, Usman A, Ramamurthy NS, Golub LM, Greenwald RA. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem. 2001;8:305–16. doi: 10.2174/0929867013373598. [DOI] [PubMed] [Google Scholar]
- 31.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106:1543–9. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 32.Lee CZ, Yao JS, Huang Y, et al. Dose-response effect of tetracyclines on cerebral matrix metalloproteinase-9 after vascular endothelial growth factor hyperstimulation. J Cereb Blood Flow Metab. 2006;26:1157–64. doi: 10.1038/sj.jcbfm.9600268. [DOI] [PubMed] [Google Scholar]
- 33.Gu Z, Kaul M, Yan B, et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–90. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 34.Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem. 2005;94:819–27. doi: 10.1111/j.1471-4159.2005.03219.x. [DOI] [PubMed] [Google Scholar]
- 35.Leon H, Baczko I, Sawicki G, Light PE, Schulz R. Inhibition of matrix metalloproteinases prevents peroxynitrite-induced contractile dysfunction in the isolated cardiac myocyte. Br J Pharmacol. 2008;153:676–83. doi: 10.1038/sj.bjp.0707621. [DOI] [PMC free article] [PubMed] [Google Scholar]