

Abstract
Survivin is a tumor-associated antigen (TAA) that has significant potential for use as a cancer vaccine target. To identify survivin epitopes that might serve as targets for CTL-mediated, anti-tumor responses, we evaluated a series of survivin peptides with predicted binding to mouse H2-Kb and human HLA-A*0201 antigens in peptide-loaded dendritic cell (DC) vaccines. H2-Kb-positive, C57BL/6 mice were vaccinated using syngeneic, peptide-loaded DC2.4 cells. Splenocytes from vaccinated mice were screened by flow cytometry for binding of dimeric H2-Kb:Ig to peptide-specific CD8+ T cells. Two survivin peptides (SVN57–64 and SVN82–89) generated specific CD8+ T cells. We chose to focus on the SVN57–64 peptide because that region of the molecule is 100% homologous to human survivin. A larger peptide (SVN53–67), containing multiple class I epitopes, and a potential class II ligand, was able to elicit both CD8+ CTL and CD4+ T cell help. We tested the SVN53–67 15-mer peptide in a therapeutic model using a peptide-loaded DC vaccine in C57BL/6 mice with survivin-expressing GL261 cerebral gliomas. This vaccine produced significant CTL responses and helper T cell-associated cytokine production, resulting in a significant prolongation of survival. The SVN53–67 vaccine was significantly more effective than the SVN57–64 core epitope as a cancer vaccine, emphasizing the potential benefit of incorporating multiple class I epitopes and associated cytokine support within a single peptide.
Keywords: Dendritic cell, Glioma, Peptide, Survivin, Tumor antigen, Vaccine
Introduction
Even with intensive treatment, malignant gliomas frequently recur as a result of continued growth of residual microscopic disease located beyond surgical resection margins [1, 2]. Surgery, radiation therapy and chemotherapy provide definite therapeutic benefit, but long-term survival is uncommon [3]. This situation has stimulated interest in exploring the usefulness of immunotherapy for patients with malignant gliomas.
Survivin is one of the most specific cancer antigens identified to date because it is expressed in a large percentage of tumors and is rarely detectable in normal tissue [4, 5]. Survivin expression occurs commonly in malignant gliomas [6–8] where its expression is associated with a poor prognosis [7, 9]. Also, because survivin is expressed by many different cancer types, its use as a tumor vaccine target may have broad implications for cancer vaccine therapy.
Epitopes of intracellular proteins, including those of survivin, appear on the surface of tumor cells in association with MHC I molecules. CD8+ CTLs are activated by tumor-derived peptides coupled to MHC class I antigens. The tumor antigen-MHC class I peptide complex binds to the antigen-specific T cell receptor on CD8+ cells and stimulates T cell activation. Thus, certain survivin epitopes that are correctly presented by tumor cells and are recognized by specific T cells may elicit potent antitumor responses [10]. Therefore, identification of survivin peptides that can bind to MHC class I molecules and elicit strong cellular immune responses is an important goal of anti-survivin cancer immunotherapy.
The generation of an effective and specific cell-mediated immune response may require the activation of both highly specific CD8+ lymphocyte subsets and CD4+ helper T cells. To activate a CD4+ T cell response, antigens must be presented to CD4+ T cells in conjunction with an MHC class II antigen. Once CD4+ cells have been activated, they proliferate and produce cytokines (e.g. IFN-γ, IL-2, and IL-4) that enhance the immune response. These cytokines are essential to provide a fully activated CD8+ antitumor CTL response.
The current study is based on our preliminary observation that a DNA vaccine against survivin inhibits the growth of malignant glioma in rats and mice [11, 12]. We previously demonstrated that a CTL response could be generated using the entire survivin molecule, as well as with specific domains of survivin, and that the responses generated from these could have a beneficial effect against cerebral gliomas [12]. In our current study, we set out to design a survivin peptide based upon a study of the immunogenicity of smaller peptides with predicted binding to both human and mouse MHC class I molecules. The purpose of this approach was to develop a survivin peptide that would be suitable for clinical application.
Here we hypothesized that a single peptide with multiple epitopes could be capable of eliciting an effective immune response to survivin epitopes presented on tumor cells. We also speculated that incorporation of CD4+ helper cell support of that immune response would promote a more robust antitumor response. We studied a peptide vaccine strategy based upon survivin amino acid sequences that are capable of stimulating CD8+ specific T cells against several different epitopes of survivin. In addition, we investigated a strategy using a larger survivin peptide containing a potential MHC class II ligand that is present in the survivin molecule to stimulate CD4+ cytokine-secreting helper T cells.
Materials and methods
Cell lines and culture conditions
GL261 murine glioma cells were grown on 100-mm tissue culture plates in complete Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum, 5,000 units penicillin/streptomycin, 50 μM 2-mercaptoethanol, 25 mM HEPES, and 1× non-essential amino acids at 37°C in 5% CO2 with media changes two to three times per week. The DC2.4 dendritic cell line was provided by Dr. Ken Rock (University of Massachusetts Medical School), and RMA-S cells were provided by Dr. Ron Germain (NIH).
Peptides
Peptide synthesis was performed using Fmoc chemistry and a solid support resin (Sigma-Genosys, The Woodlands, TX). Each peptide was stored at −20°C until use and initially diluted in DMSO. The following peptide sequences were synthesized SVN9–16 AWQPFLKD, SVN18–25 RISTFKNW, SVN39–46 AEAGFIHC, SVN53–67 DLAQCFFCFKELEGW, SVN55–64 AQCFFCFKEL, SVN56–64 QCFFCFKEL, SVN57–64 CFFCFKEL, SVN82–89 SGCAFLSV, SVN97–104 TLGEFLKL. A chicken ovalbumin (OVA258–265) peptide sequence SIINFEKL was used as a control.
Peptide binding
Computer predictions of peptide binding were determined utilizing the “SYFPEITHI” program [13]. The prediction is based on published motifs (pool sequencing, natural ligands) and takes into consideration the amino acids in the anchor and auxiliary anchor positions, as well as other frequent amino acids. Peptides were chosen for studies based upon strong binding predictions to both H2-Kb and HLA-A*0201. Direct binding of survivin peptides to H2-Kb molecules was quantitated by flow cytometry analysis of RMA-S cells [14]. RMA-S cells (deficient for expression of peptide loaded surface MHC class I) were cultured at 27°C for 24 h, after which 6 × 105 per 50 μl of PBS supplemented with 20% FCS were incubated with 50 μl of PBS containing 100 μM peptide 27°C for 1 h and transferred to 37°C for 3 h. After washing with PBS, cells were incubated with anti-H2-Kb mAb (AF6-88.5) (Biolegend) followed by goat anti-mouse PE conjugated (Fab′)2 (DAKO). The upregulation of H2-Kb molecules after binding with specific peptides was shown as mean fluorescence representative of increased binding characteristics of SVN peptides. OVA258–265 peptide was used as a positive control for H2-Kb binding. A random peptide (RND) without any known anchor motifs was used as a negative binding control.
Immunization of mice with peptide-loaded DCs
DC2.4 cells were maintained via in vitro cell culture. Prior to vaccination 1 × 107 cells were pulsed with 100 μg of peptide at 37°C for 2 h. Cells were washed and re-suspended in PBS. Four days after tumor implantation, 1 × 106 peptide-loaded DCs were injected subcutaneously into C57BL/6 mice. This inoculation was repeated as a booster vaccination 7 and 14 days later.
Splenocyte and T cell isolation
C57BL/6 mice were vaccinated with survivin peptide-loaded DC2.4 cells at weekly intervals for a total of three immunizations. Splenic tissue of vaccinated mice was harvested at endpoint sacrifice. Cells were teased from mouse spleens with sterile forceps and passed through a 70 μm filter (BD Falcon). Splenocytes were cultured in DMEM containing 10% fetal bovine serum, IL-2 (10 units/ml) and IL-7 (1 ng/ml). Medium was replenished on day 2. To isolate T cells, splenocytes were centrifuged for 5 min and the pellet was re-suspended in RBC lysis buffer (R&D Systems). Cells were washed twice and re-suspended in complete RPMI culture medium. Re-stimulation of splenocytes from immunized mice was performed in vitro for 5 days in media containing 10 μg/ml peptide.
Dimers
Survivin specific CD8+ T cells were detected utilizing BD DimerX Soluble Dimeric Mouse H2-Kb:Ig Fusion Protein (BD Biosciences Pharmingen). H2-Kb:Ig (0.5 μg) were passively loaded with survivin specific peptides at 160 molar excess overnight at 37°C. Peptide loaded H2-Kb:Ig complexes were prepared as a staining cocktail with anti-mouse PE conjugated (Fab′)2 (DAKO). Splenocytes were resuspended in FACS staining buffer (PBS, 1% FCS, 0.1% sodium azide) at 1 × 106 cells and treated with 20 μg rat anti-mouse CD16/CD32 FcgIII/II receptor block (BD Biosciences Pharmingen). Cells were doubly stained for CD8 using a FITC-conjugated rat anti-mouse CD8α mAb (GK1.2) (Biolegend). Flow cytometry with dimers was performed with 3 × 105 cells per sample. Cells were washed in FACS wash buffer containing 0.2% FBS and 0.5% sodium azide. Cells were incubated in antibodies and complexes for 1 h at 4°C, washed and prepared for FACS analysis. Final data were analyzed using FCS Express software. Analysis is based upon gating on CD8+ T cells only.
Tumor cell lysis by CTL
Cellular assays for specific T cell lysis of target were performed using the Live/Dead cell-mediated cytoxicity kit (Molecular Probes-Invitrogen). GL261 cells were trypsinized and suspended at 1 × 106 cells/ml followed by addition of 10 μl DiOC18 label (green fluorescence) for 20 min at 37°C. After labeling, cells were centrifuged, washed in PBS twice, and resuspended at 1 × 106 cells/ml of DMEM. Next, 1 × 104 GL261 DiOC18-labeled target cells were added to individual culture tubes. Splenocytes from vaccinated animals were added to the target cells in ratios ranging from 1:5 to 1:100 for 2 h at 37°C. Conditions were also set up to assay background levels, spontaneous membrane integrity loss and maximal lysis. Propidium Iodide was added to each tube to label cells with permeated cell membranes red. Cells were pelleted and used for FACS analysis. Percentage cytotoxicity was calculated using the formula: (experimental PI-DiOC18 cells − spontaneous)/(maximum PI-DiOC18 − spontaneous PI-DiOC18) × 100. Maximum release was determined by ethanol treatment of DiOC18 labeled target cells. Spontaneous release was measured by incubating PI-DiOC18 cells in the absence of effector cells. Final data were analyzed using FCS Express software. Analysis is based upon gating on GL261 cells to eliminate any background from effector cells.
Intracellular cytokine staining
Splenocytes from immunized mice were stimulated in vitro via survivin peptides. Cells were harvested and surface stained with anti-mouse CD4 PE/Cy5 (GK1.5) (Biolegend) for CD4 surface antigen, then fixed with paraformaldehyde to stabilize the cell membrane and permeabilized with saponin. Anti-mouse IL-2 FITC (JES6-5H4), anti-mouse IFNγ FITC (XMG1.2), anti-mouse IL-4 PE (11B11) (eBiosciences) were used to stain for intracellular cytokines at pre-titrated concentrations. Commercially prepared mouse in vitro activated splenocytes known to contain IL-2, IL-3, IL-4, IL-10, GM-CSF, IFN-γ expressing cells (eBiosciences) were used as positive control cells. Briefly, cells were incubate in the dark at room temperature for 20 min with mAb combinations, washed in permeabilization buffer, resuspended in staining buffer without saponin and prepared for FACS analysis. Final data was analyzed using FCS Express software. Analysis is based upon gating on CD4+ T cells only.
Intracerebral GL261 tumor cell injection and survival analysis
Male C57BL/6 mice were anesthetized with an intraperitoneal (i.p.) injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and fixed in a stereotactic head frame (David Kopf Instruments, Tujunga, CA). A midline scalp incision was made and the bregma was identified. Stereotactic coordinates were measured (2.0 mm lateral, and 1.2 mm anterior to the bregma) for implantation of cells into the deep frontal white matter. A burr hole was drilled at this point and 1 × 105 GL261 cells suspended in 2.5 μl of DMEM were injected through a Hamilton syringe with a fixed, 25-gauge needle at a depth of 3.0 mm relative to the dura mater. Injections were performed at 1 μl/min. The needle was withdrawn and the incision sutured. Kaplan–Meier survival plots were drawn and median survival times were determined for all groups. Survival differences were assessed for significance using the logrank Mantel–Cox method.
Results
H2-Kb binding of survivin epitopes
Table 1 shows predicted binding for each of the survivin peptides using SYFPEITHI analysis. Upregulation of H2-Kb molecules on the surface of murine RMA-S cells (deficient for expression of peptide loaded surface MHC class I) was used to determine actual peptide binding in vitro. Mean fluorescence represent increased binding characteristics of SVN peptides. Data shows the binding of 100 μM peptide at 37°C. The negative control peptide (random) does not contain anchor binding residues. The positive control (OVA258–265) peptide is a known immunogenic MHC class I ligand. As predicted by SYFPEITHI analysis and confirmed in actual cell binding assays (Fig. 1), SVN57–64 and SVN82–89 displayed the highest affinity for MHC-I. We continued to focus upon SVN57–64 based peptides, as this region of the survivin molecule is 100% homologous to human survivin.
Table 1.
Selected survivin peptide epitopes predicted by SYFPEITHI to bind MHC class I (H2-Kb) are shown
| Peptide position | Epitope | Predicted binding score | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| OVA258–265 | S | I | I | N | F | E | K | L | 25 |
| SVN57–64 | C | F | F | C | F | K | E | L | 20 |
| SVN82–89 | S | G | C | A | F | L | S | V | 18 |
| SVN97–104 | T | L | G | E | F | L | K | L | 22 |
| SVN18–25 | R | I | S | T | F | K | N | W | 13 |
| SVN39–46 | A | E | A | G | F | I | H | C | 12 |
| SVN9–16 | A | W | Q | P | F | L | K | D | 12 |
Positive control peptide (ovalbumin) represents a known immunogenic MHC class I ligand with a score indicating strong potential binding. Underlined amino acid residues represent MHC I anchor positions
Fig. 1.

H2-Kb binding of survivin peptides. Survivin peptide epitopes were used in MHC class I peptide binding assays. Mean fluorescence represents binding of SVN peptides to H2-Kb. Upregulation of H2-Kb molecules on the surface of murine RMA-S cells which are deficient in the expression of surface MHC class I molecules was used to determine actual survivin peptide binding in vitro. Data show the binding of 100 μM peptide at 37°C. Positive control peptide (ovalbumin) represents a known immunogenic MHC class I ligand. The negative control is an irrelevant peptide that does not bind MHC class I. Data represent mean fluorescence ± SEM of triplicate samples
CTL responses against GL261 cells
We tested splenocytes from mice immunized and boosted with peptide-loaded DC2.4 cells for their ability to kill survivin expressing GL261 glioma cells in culture (Fig. 2). CTL assays revealed significant cytotoxic effects from effector lymphocytes of SVN57–64 and SVN82–89 vaccinated mice against GL261 target cells, but not from lymphocytes of OVA258–265 peptide loaded DC vaccinated mice (not shown), consistent with the presence of specific anti-survivin CTL response. Although SVN82–89 produced a higher cytotoxic effect, we have chosen to focus on SVN57–64 based peptides, as this region of the survivin molecule is 100% homologous to human survivin and can avoid xenogeneic responses.
Fig. 2.

Cytotoxic T cell responses against GL261 glioma cells. CTL assays for specific T cell lysis of target were performed using the Live/Dead cell-mediated cytoxicity method using flow cytometry. Splenocytes from peptide-loaded, DC vaccinated animals were added to the target cells in ratios ranging from 60:1 to 100:1 and cytotoxicity was calculated as described in “Materials and methods”. Analysis is based upon gating on labeled GL261 cells to eliminate background from effector cells. Maximal cytotoxicity was simulated using ethanol-treated target cells. Spontaneous cytotoxicity represents GL261 target cells incubated in the absence of effector cells. Data represent mean percent specific lysis ± SEM of triplicate samples
Specific CD8+ T cells against naturally processed survivin epitopes
We analyzed splenocytes from mice immunized and boosted with full length survivin cDNA transfected DC2.4 cells for the presence of CD8+ T cells capable of recognizing the indicated peptide epitopes after re-stimulation in vitro (Fig. 3). FACS analysis utilizing specific peptide-loaded H2-Kb dimer IgG and gated on the CD8+ T cell population detected CD8+ T cells that were specific for SVN57–64 and SVN82–89 peptide, but not SVN18–25, SVN39–46, SVN9–16, SVN97–104 or OVA258–265 peptides. OVA peptide is shown as an irrelevant peptide control, splenocytes were not stimulated with OVA in vitro. The immune response derived from the survivin cDNA is an indicator of the natural processing of SVN57–64 and SVN82–89 peptide epitopes from the full length immunogen that primed this immune response in vivo.
Fig. 3.
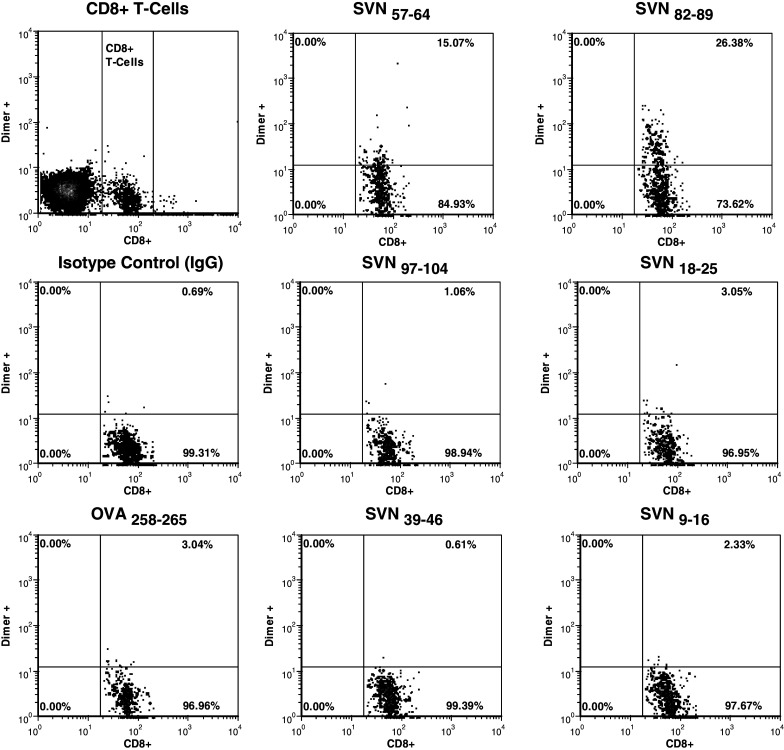
Specific CD8+ T cells against naturally processed survivin epitopes. Recombinant dimeric mouse H2-Kb:Ig molecules were loaded in vitro with survivin peptides and used to probe splenocytes from DC transfected with an expression vector containing the full length survivin cDNA that had been used to immunize mice. PE-labeled H2-Kb dimers were used to detect survivin-specific CD8+ T cells by flow cytometry. Splenocytes from vaccinated mice were doubly stained for CD8+ using a FITC conjugated rat anti-mouse CD8α mAb as described. Analysis is based upon gating on total CD8+ T cells. Ovalbumin serves as a control for nonspecific immune responses
Individually, SVN57–64 and SVN82–89 are effective immunogens in vivo
We analyzed splenocytes from mice immunized and boosted with peptide-loaded DC2.4 cells and re-stimulated with the same peptide in vitro, for the presence of CD8+ T cells capable of recognizing their respective immunizing peptide epitopes (Fig. 4). FACS analysis utilizing specific peptide-loaded H2-Kb dimer:IgG and gated on the CD8+ T cell population detected CD8+ T cells that were specific for SVN57–64 and SVN82–89 peptides from mice immunized with the same peptide, but without reactivity to empty dimers (IgG), or to dimers loaded with OVA258–265 peptides. OVA peptide is shown as a negative peptide control since splenocytes were not stimulated with OVA in vitro. Mice immunized with SVN57–64 were not reactive to SVN82–89 loaded dimers. Similarly, mice immunized with SVN82–89 were not reactive to SVN57–64, indicating non-crossreactivity. These results confirm that both SVN57–64 and SVN82–89 can effectively stimulate individual CD8+ T cell responses in vivo.
Fig. 4.

Specific CD8+ T cells against specific survivin peptides. Recombinant dimeric mouse H2-Kb:Ig molecules were loaded in vitro with survivin peptides and used to probe splenocytes from SVN57–64 or SVN82–89 peptide-loaded, DC-vaccinated mice. PE-labeled H2-Kb dimers were used to detect survivin-specific CD8+ T cells by flow cytometry. Splenocytes from vaccinated mice were doubly stained for CD8+ using a FITC-conjugated rat anti-mouse CD8α mAb as described. Analysis is based upon gating on total CD8+ T cells. Ovalbumin serves as a control for nonspecific immune responses. Data are representative of three separate experiments
Using SVN57–64 as the basis (core) for the design of a larger peptide capable of stimulating CD4+ T cell support, we designed a peptide with additional amino acids flanking SVN57–64. As a result, this produced a larger peptide that is predicted to bind human, and possibly murine, class II molecules, while retaining numerous internal MHC class I ligands in addition to those present in the core SVN57–64 peptide (Table 2). Of note, the 15-amino acid sequence of SVN53–67 contained in this peptide is also 100% homologous between mouse and human survivin proteins. Thus, SVN53–67 may be applicable as a peptide vaccine in both mouse and humans.
Table 2.
Amino acid sequence of the survivin peptide vaccine molecule containing potential class II epitope and core class I binding peptides used in vaccination studies
| AA49-ENEPDLAQCFFCFKELEGWEPDD-AA71 | |
|---|---|
| DLAQCFFCFKELEGW = SVN53–67 | Potential MHC class II ligand |
| AQCFFCFKEL = SVN55–64 | Potential MHC class I ligands (contained within SVN53–67) |
| QCFFCFKEL = SVN56–64 | |
| CFFCFKEL = SVN57–64 | |
Amino acids 49 through 71 represent a region of 100% homology between mouse and human survivin molecules
CTL responses against GL261 cells in response to SVN53–67 15-mer peptide vaccine
We tested splenocytes from mice immunized and boosted with SVN53–67 peptide-loaded DC2.4 cells for their ability to kill survivin expressing GL261 glioma cells in culture (Fig. 5). CTL assays revealed significant cytotoxic effects of lymphocytes from SVN53–67 peptide loaded DC vaccinated mice, but not from OVA258–265 loaded DC vaccinated mice. This demonstrates the presence of specific anti-survivin CTL in response to SVN53–67 based immunization.
Fig. 5.

SVN53–67 derived CTL responses against GL261. CTL assays for specific T cell lysis of target were performed using the Live/Dead cell-mediated cytoxicity method using flow cytometry. Splenocytes from peptide-loaded, DC vaccinated animals were added to the target cells in ratios ranging from 5:1 to 40:1 and cytotoxicity was calculated as described in “Materials and methods”. Analysis is based upon gating on labeled GL261 cells to eliminate background from effector cells. Maximal cytotoxicity was simulated using ethanol-treated target cells. Spontaneous cytotoxicity represents GL261 target cells incubated in the absence of effector cells. Data represent mean percent specific lysis ± SEM of triplicate samples
Stimulation of CD4+ T cells in response to SVN53–67 15-mer
Intracellular cytokine production was measured in CD4+ T cells by FACS following immunization and in vitro re-stimulation with survivin peptides (Fig. 6). The SVN53–67 (15-mer) strongly stimulated IL-4 and IFNγ production, but not IL-2 production. In contrast, the smaller survivin peptides, including SVN56–64, which contained the core SVN57–64 motif, failed to stimulate either IL-4 or IFNγ. These results indicated that the larger 15-mer peptide (SVN53–67) was indeed capable of stimulating CD4+ T cell based cytokine support while smaller peptides (SVN56–64 SVN57–64) did not.
Fig. 6.

Stimulation of CD4+ cells in response to SVN53–67 immunization. Splenocytes from immunized mice were stimulated in vitro using survivin peptides and intracellular cytokine production was measured in CD4+ cells by flow cytometry. Commercially prepared mouse in vitro activated splenocytes known to contain IL-2, IL-4, IFN-γ expressing cells were used as positive controls. Analysis is based upon gating on CD4+ T cells only. Data represent mean fluorescence ± SEM of triplicate samples
Survival of C57BL6 mice with intracerebral GL261 glioma implants treated with survivin peptide DC vaccines
C57BL/6 mice were immunized with SVN53–67, SVN57–64, SVN82–89, or OVA258–265 peptide loaded DC2.4 cells. Vaccinations began 4 days after tumor cell implantation and were repeated (boosted) every 7 days to simulate a therapeutic setting (Fig. 7). While the smaller survivin peptides, SVN57–64 and SVN82–89 produced strong CTL responses and specific CD8+ T cells in vitro, these peptides were not highly effective in vivo against murine survivin expressing GL261 cerebral gliomas (Table 3). In contrast, the SVN53–67 (15-mer) which contains multiple internal MHC class I ligands and at least one MHC class II ligand, was significantly more effective (>200% increase in survival) than any other smaller peptides in vivo. This result appears to be associated with the ability of the SVN53–67 15-mer peptide to initiate both CD4+ and CD8+ T cell responses.
Fig. 7.

Survival of C57BL6 mice with intracerebral GL261 glioma implants. Mice were implanted with GL261 cells and were treated with survivin peptide DC vaccines. C57BL/6 mice were immunized with SVN53–67, SVN57–64, SVN82–89, or OVA258–265 peptide loaded DC2.4 cells. Vaccinations began 4 days after tumor cell implantation and were repeated (boosted) every 7 days to simulate a therapeutic setting. Survival was plotted according to Kaplan–Meier methods. Long-term survivors were confirmed tumor-free by high field strength MRI (data not shown)
Table 3.
Survival data of C57BL6 mice with intracerebral GL261 glioma implants as depicted in Fig. 7
| Immunizing antigen | MeST | Range | n | P value (vs. control DC) | P value (vs. OVA) |
|---|---|---|---|---|---|
| Control DC | 19.5 | 18–25 | 10 | – | P = 0.79 |
| OVA258–265 | 21 | 19–22 | 7 | P = 0.79 | – |
| SVN82–89 | 25 | 21–32 | 10 | P < 0.001 | P < 0.001 |
| SVN57–64 | 25 | 19–35 | 14 | P < 0.001 | P < 0.001 |
| SVN53–67 | 57 | 43–120+ | 8 | P < 0.0001 | P < 0.0001 |
Mice were implanted with GL261 cells and were treated with survivin peptide DC vaccines. MeST represents median survival time of groups with P values determined by the logrank Mantel–Cox test for significance
Discussion
Survivin is an important tumor-associated antigen with restricted expression [4, 15, 16]. This characteristic makes it an excellent candidate target antigen for antitumor vaccines. MHC class I-restricted cytotoxic T cells directed against survivin peptides have been identified in patients with breast cancer, leukemia and melanoma [17]. Peptide epitopes from tumor-associated antigens (TAA), including survivin, can be recognized by cytotoxic T lymphocytes (CTL) in the context of MHC I molecules [10]. The human survivin protein contains HLA-A*0201 (HLA-A2) binding motifs. Andersen et al. [18] used HLA-A2-restricted peptides to test for specific T-cell reactivity in leukemia and melanoma patients. CTL responses against two survivin epitopes were detected in both patient groups, but no T cell reactivity was found in healthy control patients. Therefore, self-immunization with the survivin molecule appears to take place in some tumor patients. This raises the possibility that immunization with survivin epitopes could stimulate a memory response with therapeutic benefit for cancer patients.
Schmitz et al. [19] were the first to show that two survivin peptides (ELTLGEFLKL and TLPPAWQPFL) can bind to HLA-A*0201 and elicit CD8+ immune responses when presented by DC. Moreover, one of these peptides was shown to result from the natural intracellular processing of survivin. These studies demonstrated that survivin is recognizable by autologous T cells. In addition, the identification of HLA-A1, HLA-A2, HLA-A3, and HLA-A11-restricted survivin epitopes [10] suggests that it might be possible to target survivin-expressing tumor cells using survivin-based cancer vaccines for a large clinical population.
In the current study, we have shown that the survivin protein is processed into additional peptides that elicit a class I immune response capable of being re-stimulated by SVN57–64 and SVN82–89 peptides (Figs. 3, 4). This is a further indicator of the natural processing of survivin into potentially immunogenic epitopes, not previously described. The fact that immune response to SVN18–25, SVN39–46, SVN9–16, SVN97–104 peptides were not detected in these animals could be attributed to one or more of the following: (1) suboptimal class I binding by these peptides, (2) lack of adequate T cell receptor interaction with the peptides in the context of class I, and (3) lack of appropriate processing of the survivin protein.
Peptides processed from SVN53–67 are capable of binding HLA-A*0201 and are predicted to bind many additional common HLA molecules, including: A*03, A*1101, A*2402, A*26, A*6801, B*0702, B*08, B*1402, B*1501(B62), B*1510, B*18, B*2705, B*2709, B*3901, B*4402 and B*5101. Therefore, just as SVN53–67 is immunogenic in mice, it should be immunogenic in humans expressing the appropriate MHC-I haplotypes.
Immunotherapy has emerged as a potential treatment modality for cerebral gliomas because it is: (1) specific to tumor cells, (2) has the potential to eradicate infiltrating tumor cells after surgical debulking, and (3) may be capable of eliciting a long-lasting memory response to prevent tumor recurrence. Both immunological [20–22] and clinical responses [22, 23] have been observed in glioma patients immunized with (1) dendritic cells (DC) loaded with peptides eluted from the surface of allogeneic tumor cells [20]; (2) DC loaded with peptides eluted from autologous tumor cells [21]; (3) DC pulsed with tumor lysate from autologous tumor cells [22]; and (4) hybrids generated by fusing DC with autologous tumor cells [23]. In one study [22], patients with recurrent glioblastoma multiforme, who were immunized with tumor lysate-pulsed DCs, had increased survival (median = 133 weeks). Thus, DCs offer significant potential as an immunization vehicle to treat malignant gliomas.
More specifically, Zeis et al. [24] have shown that transfection of dendritic cells (DC) with survivin mRNA leads to resistance to tumor challenge. Xiang et al. [25] showed that a DNA-based survivin vaccine could produce a CTL response against Lewis lung carcinoma with regression of both primary tumor and pulmonary metastases. The mechanism of the antitumor immunity induced by this DNA vaccine involved MHC class I antigen-restricted CD8+ T cells. The survivin vaccine triggered tumor cell apoptosis, together with release of IFNγ. Similarly, others have shown that survivin DNA vaccines generate specific antitumor effects with increased lymphocyte infiltration at tumor sites [26, 27]. Recently a SVN85–93 peptide combined with a plasmid encoding murine interleukin-15 (IL-15) was shown to support robust, CTL-mediated, long-term, immunity in vivo when used as a tumor vaccine. This vaccine effectively protected BALB/c mice against fatal CT26 tumor challenge [28].
Another distinct survivin derived peptide epitope “SVN10” was used to derive T cell clones from cancer patients with multiple HLA-DR genotypes capable of recognizing naturally processed antigen [29]. In addition, an anti-DEC205-human survivin protein was able to stimulate CD4+ T cell responses better than approaches involving survivin plasmid DNA or survivin peptides with adjuvants, but was unable to induce CD8+ T cell immunity [30]. Each of these reports confirms the ability of survivin vaccines to generate CTL responses and the importance of cytokine support. Our study describes the first defined survivin peptide vaccine that is capable of stimulating a response to multiple CD8+ T cell epitopes and a CD4+ T cell cytokine response from a defined, single-peptide immunogen.
The effectiveness of a peptide vaccine may be enhanced by the capacity of DC to present both class I and class II associated epitopes. SVN53–67 is processed to expose different epitopes for CD8+ T cell recognition and is capable of stimulating CD4+ T helper cells as well. Thus, SVN53–67 represents a potential class II ligand in C57BL/6 mice. In our study, the smaller core peptides (SVN57–64 and SVN56–64) stimulated CD8+ responses and produced effective CTL in vitro. However, these same core peptides only produced modest prolongation of survival of mice with cerebral gliomas. In contrast, the SVN53–67 15-mer produced significant prolongation of survival in this model. Thus, the addition of CD4+ support may contribute to a more effective antitumor response in vivo.
Our study shows that the contribution of CD4 support may lead to a more effective peptide vaccine strategy. The enhanced survival demonstrated by SVN53–67 suggests that the addition of CD4+ helper T cell support associated with a specific survivin-derived CD8+ T cell response may lead to a more effective peptide antitumor vaccine. Other studies of ours indicate that SVN53–67 is capable of re-stimulating an autologous human CTL response capable of lysing tumor cells ex vivo (data not shown). In addition, SVN53–67 is predicted to function as a ligand for HLA-DRB1*0301 (DR17), HLA-DRB1*0401 (DR4Dw4), HLA-DRB1*0701, HLA-DRB1*1501 (DR2b) molecules, making it potentially capable of stimulating CD4 + support in humans. A peptide vaccination strategy combining CTL and CD4+ support provides one additional avenue to investigate for survivin-targeted, immunotherapeutic approaches for cerebral gliomas.
Acknowledgments
This work was supported by NIH 5R21 NS049309-02 (RAF), NIH 5P30 CA16056-29, the Linda Scime Fund of the Roswell Park Alliance Foundation, and the Glioblastoma Multiforme Grant awarded to MJC by the National Brain Tumor Foundation.
References
- 1.Giese A, Westphal M. Treatment of malignant glioma: a problem beyond the margins of resection. J Cancer Res Clin Oncol. 2001;127:217–225. doi: 10.1007/s004320000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halperin EC, Burger PC, Bullard DE. The fallacy of the localized supratentorial malignant glioma. Int J Radiat Oncol Biol Phys. 1988;15:505–509. doi: 10.1016/S0360-3016(98)90036-0. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;10:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 5.Adida C, Crotty PL, McGrath J, et al. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 6.Kajiwara Y, Yamasaki F, Hama S, et al. Expression of survivin in astrocytic tumors. Cancer. 2003;97:1077–1083. doi: 10.1002/cncr.11122. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki T, Lopes MB, Hankins GR, Helm GA. Expression of survivin an inhibitor of apoptosis protein in tumors of the nervous system. Acta Neuropathol (Berl) 2002;104:105–109. doi: 10.1007/s00401-002-0532-x. [DOI] [PubMed] [Google Scholar]
- 8.Chakravarti A, Noll E, Black P, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–1068. doi: 10.1200/JCO.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 9.Islam A, Kageyama H, Takada N, et al. A high expression of survivin mapped to 17q25 is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–23. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 10.Andersen MH, Pedersen LO, Capeller B, et al. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61:5964–5968. [PubMed] [Google Scholar]
- 11.Fenstermaker RA, Ciesielski MJ. Immunotherapeutic strategies for malignant gliomas. Cancer Control. 2004;11:181–191. doi: 10.1177/107327480401100306. [DOI] [PubMed] [Google Scholar]
- 12.Ciesielski MJ, Apfel L, Barone TA, Castro CA, Weiss TC, Fenstermaker RA. Antitumor effects of a xenogeneic survivin bone marrow derived dendritic cell vaccine against murine GL261 gliomas. Cancer Immunol Immunother. 2006;55:1491–1503. doi: 10.1007/s00262-006-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rammensee HG, Bachmann J, Emmerich NN, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 14.Kmieciak D, Bednarek I, Takiguchi M, Wasik TJ, Bratosiewicz J, Wierzbicki A, Teppler H, Pientka J, Hsu SH, Kaneko Y, Kozbor D. The effect of epitope variation on the profile of cytotoxic T lymphocyte responses to the HIV envelope glycoprotein. Int Immunol. 1998;10:1789–99. doi: 10.1093/intimm/10.12.1789. [DOI] [PubMed] [Google Scholar]
- 15.Gordan JD, Vonderheide RH. Universal tumor antigens as targets for immunotherapy. Cytotherapy. 2002;4:317–27. doi: 10.1080/146532402760271091. [DOI] [PubMed] [Google Scholar]
- 16.Altieri DC. Survivin and apoptosis control. Adv Cancer Res. 2003;88:31–52. doi: 10.1016/S0065-230X(03)88303-3. [DOI] [PubMed] [Google Scholar]
- 17.Altieri DC. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol Med. 2001;7:542–7. doi: 10.1016/S1471-4914(01)02243-2. [DOI] [PubMed] [Google Scholar]
- 18.Andersen MH, Pedersen LO, Becker JC, Straten P. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res. 2001;61:869–72. [PubMed] [Google Scholar]
- 19.Schmitz M, Diestelkoetter P, Weigle B, et al. Generation of survivin-specific CD8+ T effector cells by dendritic cells pulsed with protein or selected peptides. Cancer Res. 2000;60:4845–9. [PubMed] [Google Scholar]
- 20.Liau LM, Black KL, Martin NA, et al. Treatment of a glioblastoma patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class I-matched tumor peptides. Neurosurg Focus. 2000;9:e8. doi: 10.3171/foc.2000.9.6.9. [DOI] [PubMed] [Google Scholar]
- 21.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–7. [PubMed] [Google Scholar]
- 22.Yu JS, Liu G, Ying H, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–9. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 23.Kikuchi T, Akasaki Y, Irie M, et al. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337–44. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeis M, Siegel S, Wagner A, et al. A Generation of cytotoxic responses in mice and human individually against hematological malignancies using survivin-RNA-transfected dendritic cells. J Immunol. 2003;170:5391–7. doi: 10.4049/jimmunol.170.11.5391. [DOI] [PubMed] [Google Scholar]
- 25.Xiang R, Mizutani N, Luo Y, et al. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res. 2005;65:553–561. [PubMed] [Google Scholar]
- 26.Zhu K, Qin H, Cha SC, Neelapu SS, Overwijk W, Lizee GA, Abbruzzese JL, Hwu P, Radvanyi L, Kwak LW, Chang DZ. Survivin DNA vaccine generated specific antitumor effects in pancreatic carcinoma and lymphoma mouse models. Vaccine. 2007;25:7955–61. doi: 10.1016/j.vaccine.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 27.Nagaraj S, Pisarev V, Kinarsky L, Sherman S, Muro-Cacho C, Altieri DC, Gabrilovich DI. Dendritic cell-based full-length survivin vaccine in treatment of experimental tumors. J Immunother. 2007;30:169–79. doi: 10.1097/01.cji.0000211329.83890.ba. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Wang L, Wang H, Shang X, Niu W, Li J, Wu Y. A novel mimovirus vaccine containing survivin epitope with adjuvant IL–15 induces long-lasting cellular immunity and high antitumor efficiency. Mol Immunol. 2008;45:1674–1681. doi: 10.1016/j.molimm.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Piesche M, Hildebrandt Y, Zettl F, Chapuy B, Schmitz M, Wulf G, Trümper L, Schroers R. Identification of a promiscuous HLA DR-restricted T-cell epitope derived from the inhibitor of apoptosis protein survivin. Hum Immunol. 2007;68:572–6. doi: 10.1016/j.humimm.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Charalambous A, Oks M, Nchinda G, Yamazaki S, Steinman RM. Dendritic cell targeting of survivin protein in a xenogeneic form elicits strong CD4+ T cell immunity to mouse survivin. J Immunol. 2006;177:8410–21. doi: 10.4049/jimmunol.177.12.8410. [DOI] [PubMed] [Google Scholar]