

Abstract
Background
The mitogen-activated protein kinase (MAPK) cascade is an important intracellular mediator of angiotensin II (Ang II)-induced cell growth and differentiation. Here, we examined the effect of angiotensin II type 1 receptor (AT1) receptor blockade on renal injury and MAPK activity in Dahl salt-sensitive (DS) rats.
Methods
DS rats were maintained on a high (H: 8.0%NaCl, N = 8) or low (L: 0.3%NaCl, N = 7) salt diet, or H + candesartan cilexetil (10 to 15 mg/kg/day, N = 8). Urinary protein excretion (UproteinV), renal cortical collagen content, and glomerular injury (assessed by semiquantitative morphometric analysis) were determined after 4-week treatments. Plasma and kidney Ang II levels were measured by radioimmunoassay. Protein levels of AT1 and AT2 receptors in the renal cortical tissues were analyzed by Western-blotting analyses. MAPKs activities, including extracellular signal-regulated kinases (ERK) 1/2, c-Jun NH2-terminal kinases (JNK), p38 MAPK, and Big-MAPK-1 (BMK1), were measured by Western-blotting analyses or in vitro kinase assays.
Results
DS/H rats showed higher mean blood pressure (MBP), UproteinV, and renal cortical collagen content than DS/L rats. Increased ERK1/2, JNK, and BMK1 activities were observed in renal cortical tissues of DS/H rats (approximately 6.3-, 4.5-, and 2.5-fold, respectively), whereas p38 MAPK activity was unchanged. Plasma Ang II levels were significantly reduced in DS/H rats compared with DS/L rats, whereas kidney Ang II contents and AT1 receptor protein levels were similar. Candesartan did not alter MBP, but significantly reduced UproteinV and collagen content, and ameliorated progressive sclerotic and proliferative glomerular changes. Furthermore, candesartan decreased renal tissue Ang II contents (from 216 ± 19 to 46 ± 3 fmol/mL) and ERK1/2, JNK, and BMK1 activities (-45%, -60%, and -70%, respectively) in DS/H rats.
Conclusion
In DS hypertensive rats, some of the renoprotective effects of AT1 receptor blockade are accompanied by reductions in intrarenal Ang II contents and MAPK activity, which might not be mediated through arterial pressure changes.
Keywords: angiotensin II (Ang II), AT1 receptor, mitogen-activated protein kinase (MAPK), Dahl salt-sensitive (DS) rats, kidney
The Dahl salt-sensitive (DS) rat is a widely studied genetic model of salt-sensitive hypertension that develops renal damage characterized by glomerular injury [1-3]. Several studies have shown that plasma renin activity (PRA) and circulating angiotensin II (Ang II) levels are very low in these animals [4-6]. Furthermore, treatments with angiotensin-converting enzyme (ACE) inhibitors or Ang II receptor type 1 (AT1) receptor antagonists fail to prevent the development of hypertension [2, 3, 5, 7], suggesting that the systemic renin-angiotensin system is not a predominant mediator of salt-induced hypertension. However, recent studies have shown that ACE inhibitors or AT1 receptor antagonists ameliorated progressive sclerotic and proliferative glomerular changes [2, 3, 7], and increased the life expectancy of such animals [7, 8]. The data suggest that although the circulating renin-angiotensin system is suppressed, the intrarenal renin-angiotensin system plays a crucial role in the pathogenesis of glomerular injury in DS hypertensive rats. However, the precise mechanisms responsible for the renoprotective effects of AT1 receptor blockade remain unclear.
Glomerular diseases are commonly characterized by mesangial cell overgrowth and excessive accumulation of extracellular matrix proteins [9, 10, 11]. Ang II stimulates cellular hypertrophy and proliferation in mesangial cells through activation of multiple intracellular signaling pathways, including mitogen-activated protein kinases (MAPKs) [9, 11]. In cultured rat mesangial cells, Ang II activates extracellular signal-regulated kinases (ERK) 1/2 [12] and c-Jun NH2-terminal kinases (JNK) [13], which belong to MAPK subgroups. Another MAPK family member, p38 MAPK, was also activated by Ang II in these cells with high glucose [14]. Studies by Hamaguchi et al [15] provided in vivo evidence that Ang II infusion led to activation of glomerular ERK1/2 and JNK, followed by an increase in arterial pressure. It has also been shown in renal cortical slices that Ang II-induced stimulation of the collagen I α2 chain gene is blocked by a specific ERK/MAPK inhibitor or an AT1 receptor antagonist [16]. Collectively, these observations indicate that the MAPK cascade is an important mediator of Ang II-induced molecular actions in the kidney. Recently, Hamaguchi et al [17] showed that enhancement of glomerular ERK1/2 and JNK activities is associated with an increase in urinary protein excretion (UproteinV) in DS hypertensive rats. Although these data suggest that MAPK activation is involved in the progression of renal injury in these animals, the effects of AT1 receptor blockade on renal MAPK activity has not been determined.
The present study was designed to characterize the intrarenal renin-angiotensin system and MAPKs activities, and examine the effects of AT1 receptor blockade on renal injury as well as kidney tissue Ang II levels and MAPKs activities in DS hypertensive rats. We measured the activities of the classic MAPKs (ERK1/2, JNK, and p38 MAPK) and a new MAPK family member, Big MAPK-1 (BMK1) [18-20]. We obtained evidence that the intrarenal renin-angiotensin system is not suppressed in DS hypertensive rats, and that the renoprotective effects of AT1 receptor blockade are associated with reductions in kidney Ang II contents, and the activities of ERK1/2, JNK, and BMK1.
METHODS
Animal preparation
All experimental procedures were performed according to the guidelines for the care and use of animals established by the Kagawa Medical University. Male 6- or 7-week-old DS and Dahl salt-resistant (DR) rats (Seac Yoshitomi, Fukuoka, Japan) weighing 200 to 225 g at the beginning of the experiments were selected at random to receive high salt (H: 8% NaCl, Oriental Yeast, Tokyo, Japan) or low salt (L: 0.3% NaCl, Oriental Yeast) rat chow for 4 weeks. The numbers of animals used were as follows: 8, 7, 7, 7 for DS/H, DS/L, DR/H, and DR/L rats, respectively. In a separate experimental series, DS (N = 8) and DR rats (N = 7) were fed a high salt diet (8% NaCl) and treated with candesartan cilexetil (Takeda Pharmaceutical Industries, Ltd., Osaka, Japan) at a dose of 11 ± 1 mg/kg body weight per day. The dose of candesartan cilexetil was chosen on the basis of results from previous rat studies [21]. Candesartan cilexetil was dissolved in drinking water containing ethanol (0.05% to 0.075% v/v), polyethylene glycol 300 (0.05% to 0.075% v/v), and sodium bicarbonate (0.75 to 1.13 mmol/L), as described previously [22, 23]. In preliminary experiments, the effects of the vehicle on mean blood pressure (MBP), UproteinV, renal cortical collagen content, plasma and kidney Ang II levels, and renal cortical tissues MAPK activities were examined in other DR/H and DS/H rats (N = 5 each). The results showed that no parameters were altered by treatment with vehicle in both DR/H and DS/H rats (data not shown).
MBP was measured every week in conscious rats by tail-cuff plethysmography (BP-98A; Softron Co., Tokyo, Japan). Twenty-four-hour urine samples were collected one day before harvesting. Blood and kidney samples were harvested at the end of the fourth week. After decapitation, trunk blood was collected into chilled tubes containing an inhibitor mixture (5 mmol/L EDTA +20 μmol/L enalaprilat + 1.25 mmol/L O-phenanthroline + 10 μmol/L pepstatin) and processed for measurements of plasma Ang II [24, 25]. Blood was also collected into chilled tubes containing 5 mmol/L EDTA for measuring plasma renin activity (PRA) [25]. Just after removal of the kidneys, half of one kidney was homogenized in cold methanol and processed for measurements of kidney Ang II contents [24-26]. The other half of this kidney was fixed in 10% buffered paraformaldehyde for histologic examination. The remaining kidney was snap-frozen in liquid nitrogen and stored at -80°C until processing for protein extraction and analysis of collagen content.
Analysis of kidney samples for AT1 receptors and MAPKs
Protein levels of AT1 and AT2 receptors in the renal cortical tissues were analyzed by Western blotting using antibodies against AT1 and AT2 receptors (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), as previously described in detail [26, 27]. To check for equal loading, membranes were reprobed with an antibody against β-actin (Sigma Chemical Co., St. Louis, MO, USA). All values were normalized by arbitrarily setting the densitometry of DS/L rats to 1.0. Previously, we found that activation of ERK1/2 or p38 MAPK by an in-gel kinase assay with specific substrates and immunoblotting for phospho-ERK1/2 or phospho-p38 MAPK were highly correlated (R2 = 0.90) [18, 28, 29]. Therefore, we used immunoblotting with antibodies against phospho-ERK1/2 and phospho-p38 MAPK (Cell Signaling Technology Inc., Beverly, MA, USA) to evaluate ERK1/2 and p38 MAPK activation, as described previously [28, 29]. JNK activity was measured using a commercially available kit based on the phosphorylation of recombinant c-Jun. Immunoblotting was performed with antibodies against phospho-c-Jun (Cell Signaling Technology, Inc.) [28, 29]. BMK1 activity was measured by Western blotting analysis with a phospho-specific antibody for ERK5 (Cell Signaling Technology Inc.), as previously described [18, 19]. We also evaluated total ERK1/2, JNK, p38 MAPK, and BMK1protein expression using pan-ERK1/2, JNK, p38 MAPK, and BMK1 (ERK5) antibodies (Cell Signaling Technology Inc.). All values were normalized by arbitrarily setting the densitometry of DS/L rats to 1.0.
Histologic examination
Kidneys were fixed with 10% formalin (pH 7.4), embedded in paraffin, sectioned into 4-μm slices, and stained with Azan or hematoxylin-eosin (HE) reagents. The severity of glomerular injury score was evaluated using light microscopy according to previously described methods [2, 7, 30]. A minimum of 50 glomeruli was examined in each specimen. Scoring of mesangial matrix expansion was evaluated using the specimens with Azan staining as follows: 0 = no matrix expansion; 1 = minor; 2 = weak; 3 = moderate; and 4 = strong. Proliferative lesions were scored into 5 grades using specimens with HE staining as follows: 0 = no proliferation; 1 = minor (segmental lesion <25%); 2 = mild (25%<segmental lesion<50%); 3 = moderate (diffuse proliferation without severe sclerotic change); and 4 = severe (diffuse proliferation with nearly complete sclerosis).
Analytical procedures
UproteinV was determined using a protein assay kit (MicroTP-test, Wako, Japan). PRA and Ang II levels were measured as previously reported [24-26]. Urinary excretions of sodium (UNaV) and potassium (UKV) were measured using flame photometry (Hitachi 750; Tokyo, Japan) [31]. Renal cortical tissue collagen content was determined on the basis of the hydroxyproline concentration, as previously described [32].
Statistical analysis
The values are presented as mean ± SE. Statistical comparisons of the differences were performed using one-way or two-way analysis of variance combined with Newman-Keuls post-hoc test. P < 0.05 was considered statistically significant.
RESULTS
Blood pressure, kidney weight, UproteinV, UNaV, UKV, and renal cortical collagen accumulation
MBP was identical among the 5 groups at the beginning of the protocol. As shown in Figure 1A, DS/H rats progressively developed hypertension (MBP; 179 ± 5 mm Hg at 4 weeks). After 4 weeks of a high-salt diet, kidney weights and kidney weight-to-body weight ratios of DS/H rats were significantly higher than those of DR/L, DR/H, and DS/L rats (Table 1). UproteinV was significantly increased in DS/H rats compared with DS/L, DR/H, and DR/L rats (Fig. 1B). Candesartan did not alter MBP, kidney weight, or UproteinV in DR/H rats (Fig. 1 and Table 1). Furthermore, candesartan + DS/H rats did not exhibit significantly reduced MBP compared with untreated DS/H rats (168 ± 7 mm Hg at 4 weeks, Fig. 1A). However, candesartan treatment significantly reduced increases in kidney weights and UproteinV in DS/H rats, as shown in Table 1 and Figure 1B, respectively. The high-salt diet markedly increased UNaV and UKV in both DR and DS rats. Treatment with candesartan also tended to increase UNaV and UKV in both DR/H and DS/H rats, but these changes were not statistically significant (Table 1).
Fig. 1. (A) Mean blood pressure (MBP) profile in Dahl rats at 0, 1, 2, 3, and 4 weeks.
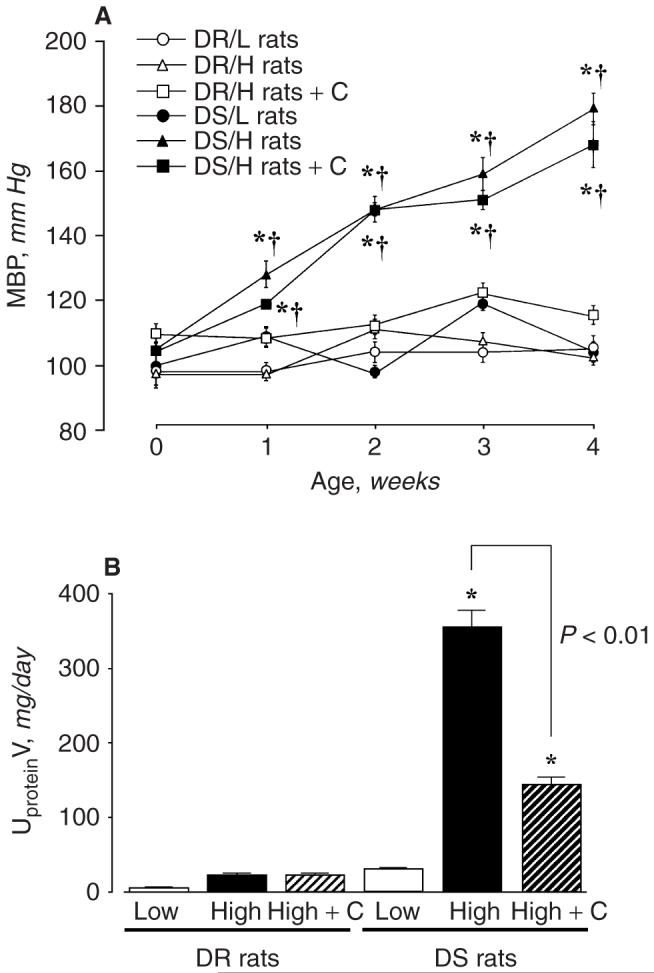
*P < 0.05 vs. baseline. †P < 0.05 vs. DS/L rats. (B) Urinary protein excretion in Dahl rats at 4 weeks. MBP and urinary protein excretions in DS/H rats were significantly higher than those in DS/L, DR/H, and DR/L rats. Candesartan treatment did not alter MBP but significantly decreased urinary protein excretion in DS/H rats. *P < 0.05 vs. DS/L rats. DR/L rats: Dahl salt-resistant rats fed a 0.3% NaCl diet; DR/H rats: Dahl salt-resistant rats fed an 8% NaCl diet; DS/L rats: Dahl salt-sensitive rats fed 0.3% NaCl diet; DS/H rats: Dahl salt-sensitive rats fed an 8% NaCl diet; C: candesartan.
Table 1.
Effects of 4 weeks of a high-salt diet and candesartan (C) on body weight (BW), kidney weight (KW), urinary excretions of sodium (UNaV) and potassium (UKV), and kidney collagen content in Dahl salt-resistant (DR) rats and Dahl salt-sensitive (DS)
| DR rats |
DS rats |
|||||
|---|---|---|---|---|---|---|
| Low salt (N = 7) | High salt (N = 7) | High salt + C (N = 7) | Low salt (N = 7) | High salt (N = 9) | High salt + C (N = 9) | |
| BW g | 321 ± 2 | 319 ± 4 | 313 ± 4 | 342 ± 3 | 334 ± 2 | 346 ± 2 |
| KW g | 1.16 ± 0.04 | 1.21 ± 0.01 | 1.11 ± 0.03 | 1.31 ± 0.03 | 1.91 ± 0.06a | 1.69 ± 0.03a,b |
| KW/BW % | 0.36 ± 0.01 | 0.38 ± 0.01 | 0.34 ± 0.01 | 0.38 ± 0.01 | 0.56 ± 0.02a | 0.49 ± 0.01a,b |
| UNaV mEq/day | 1.1 ± 0.3 | 16.7 ± 1.4a | 20.6 ± 1.8a | 0.8 ± 0.1 | 19.2 ± 1.8a | 23.7 ± 1.4a |
| UKV mEq/day | 1.1 ± 0.1 | 3.8 ± 0.1a | 4.1 ± 0.2a | 1.1 ± 0.1 | 4.0 ± 0.2a | 4.6 ± 0.1a |
| Kidney collagen content μg/mg dry weight | 12 ± 2 | 13 ± 1 | 10 ± 1 | 12 ± 3 | 24 ± 1a | 16 ± 1a,b |
Values are mean ± SE.
P < 0.05 vs. the same strain on a low salt diet.
P < 0.05, DS/a high salt diet vs. DS/a high salt diet + candesartan.
Increased collagen accumulation, characterized by Azan staining, was observed in DS/H rats (Fig. 2A). After 4 weeks of treatment with a high-salt diet, the hydroxyproline concentration in the renal cortical tissue of DS/Hrats was 25 ± 1 nmol/mg. The calculated cortical collagen content in DS/H rats was 24 ± 1 μg/mg, which was significantly higher than those of DS/L, DR/H, DR/H + candesartan, and DR/L rats (Table 1). In DS/H rats, candesartan treatment significantly decreased Azan staining in glomeruli (Fig. 2A), as well as collagen content in the renal cortex (Table 1).
Fig. 2. Photomicrographs of glomeruli in Dahl rats.

(A), Azan stain, and (B), HE stain, original magnification ×400, respectively. Glomerular scores of (C) glomerular matrix expansion and (D) proliferation. A minimum of 50 glomeruli was examined in each specimen. DS/H rats exhibited severely damaged glomeruli characterized by mesangial matrix expansion and cell proliferation. Candesartan markedly ameliorated these glomerular changes in DS/H rats. *P < 0.05 vs. DS/L rats. DR/L rats: Dahl salt-resistant rats fed a 0.3% NaCl diet; DR/H rats: Dahl salt-resistant rats fed an 8% NaCl diet; DS/L rats: Dahl salt-sensitive rats fed a 0.3% NaCl diet; DS/H rats: Dahl salt-sensitive rats fed an 8% NaCl diet. HE, hematoxylin-eosin; C, candesartan.
Histologic findings
The glomerular histologic findings with Azan and HE staining are illustrated in Figure 2. DR/L, DR/H, DR/H + candesartan, and DS/L rats showed normal glomeruli, whereas DS/H rats exhibited severely damaged glomeruli characterized by mesangial matrix expansion (Fig. 2A and C) and cell proliferation (Fig. 2B and D). As shown in Figure 2A-D, concurrent administration of candesartan markedly ameliorated these glomerular changes in DS/H rats.
PRA and Ang II levels in plasma and kidney
In DR rats, a high-salt diet significantly decreased PRA (DR/H, 0.7 ± 0.2 ng Ang I/mL/hr; DR/L, 4.7 ± 0.8 ng Ang I/mL/hr). DS/H rats also showed lower PRA compared with DS/L rats (DS/H, 0.6 ± 0.2 ng Ang I/mL/hr; DS/L, 3.7 ± 0.5 ng Ang I/mL/hr). Plasma Ang II levels were also reduced by a high-salt diet in both DR and DS rats (Fig. 3A and B). In addition, DR/H rats showed significantly lower kidney Ang II contents compared with those of DR rats fed a low-salt diet (194 ± 9 vs. 357 ± 54 fmol/g, Fig. 3C). However, a high-salt diet did not reduce the kidney Ang II contents in DS rats (DS/H rats: 214 ± 34 fmol/g, DS/L rats: 216 DR/H 18 fmol/g, Fig. 3D). In DR/H rats, candesartan treatment significantly increased PRA (8.1 ± 2.3 ng Ang I/mL/hr) and plasma Ang II concentrations (102 ± 8 fmol/g, Fig. 3A), but did not alter kidney Ang II contents (144 ± 16 fmol/g, Fig. 3C). Similarly, candesartan treatment significantly increased PRA (6.8 ± 2.2 ng Ang I/mL/hr) and plasma Ang II concentrations (68 ± 4 fmol/mL, Fig. 3B) in DS/H rats. However, kidney Ang II contents were markedly reduced by treatment with candesartan in DS/H rats (46 ± 3 fmol/g, Fig. 3D).
Fig. 3. Plasma angiotensin II concentrations (A and B) and kidney angiotensin II contents (C and D) in Dahl rats.
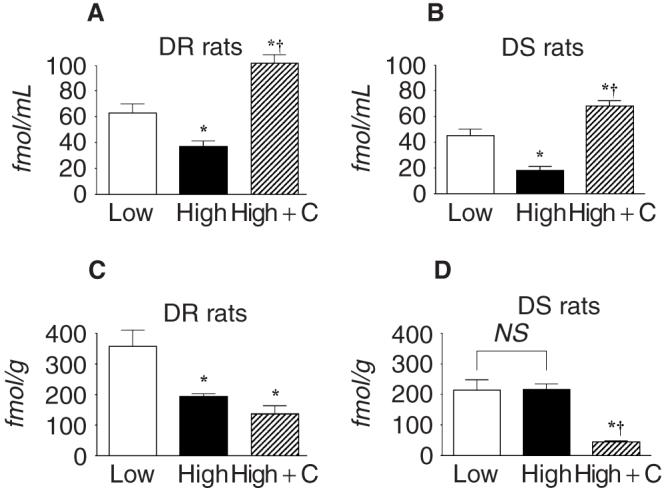
Plasma angiotensin II levels were reduced by a high salt diet in both DR and DS rats. DR/H rats showed significantly lower kidney angiotensin II contents compared with those of DR rats fed a low salt diet. However, a high salt diet did not reduce the kidney angiotensin II contents in DS rats. Candesartan treatment increased the plasma angiotensin II concentrations, but markedly decreased kidney angiotensin II contents in DS/H rats. *P < 0.05 vs. the same strain on a low salt diet. †P < 0.05: DR/H rats vs. DR/H + candesartan rats, or DS/H rats vs. DS/H + candesartan rats. DR rats, Dahl salt-resistant rats; DS rats, Dahl salt-sensitive rats; C, candesartan.
Renal cortical AT1 and AT2 receptors levels
Analyses of integrated densitometric values (IDV) showed that the ratios for AT1 receptors were significantly lower in the kidney samples of DR/H rats than those of DR/L rats. In DS rats, however, cortical tissue AT1 receptor levels were not suppressed by a high-salt diet (Fig. 4A). On the other hand, analyses of IDV showed that the ratios for AT2 receptors of kidney samples in DS rats were significantly increased (1.9 ± 0.1-fold) by a high-salt diet, whereas the levels in DR rats remained unaltered (Fig. 4B). As a control study to check for equal loading, membranes were reprobed with an antibody against β-actin. The results showed that IDV levels were unaltered among the groups.
Fig. 4. Renal cortical (A) angiotensin II type 1 receptor AT1 and (B) AT2 receptor protein levels in Dahl rats.
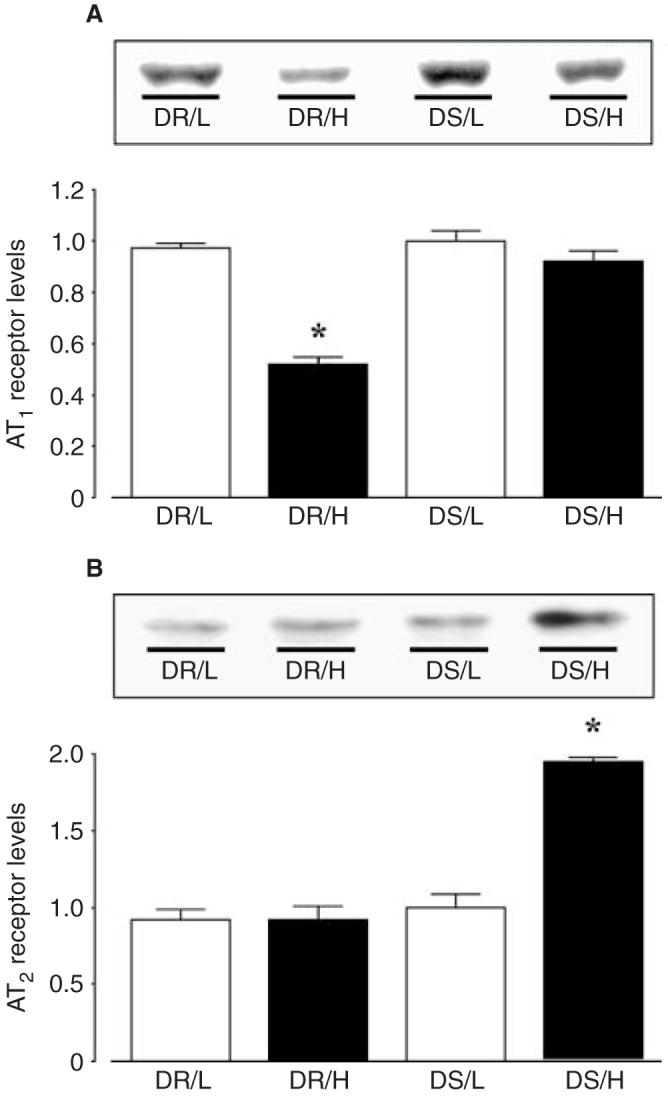
All values were normalized by arbitrarily setting the densitometry of DS/L rats to 1.0. DR rats fed a high salt diet showed significantly reduced cortical tissue AT1 receptor protein levels compared with DR/L rats. However, cortical tissue AT1 receptor levels in DS rats were unaffected by a high salt diet. On the other hand, cortical tissue AT2 receptor levels were significantly increased in DS/H rats. As a control study to check for equal loading, membranes were reprobed with an antibody against β-actin. The results showed that the densitometric values were unaltered among the groups (data not shown). *P < 0.05 vs. the same strain on a low salt diet. DR rats, Dahl salt-resistant rats; DS rats, Dahl salt-sensitive rats.
Renal cortical MAPKs activities
ERK1/2 activities in renal cortical tissues of DS/H rats were approximately 8-, 7-, and 6-fold higher than those of DR/L, DR/H, and DS/L rats, respectively (Fig. 5A). Furthermore, DS/H rats showed approximately 4-, 5-, and 4-fold higher renal cortical JNK activities compared with DR/L, DR/H, and DS/L rats, respectively (Fig. 5B). In DR/H rats, candesartan treatment altered neither ERK1/2 nor JNK activities (data not shown). However, candesartan treatment significantly reduced ERK1/2 and JNK activities by -45% and -60%, respectively, in DS/H rats (Fig. 5A and B). On the other hand, renal cortical p38 MAPK activities were not significantly different among all animals (Fig. 5C). BMK1 activities in renal cortical tissues of DS/H rats were approximately 2.5-fold higher than those of DR/L, DR/H, and DS/L rats (Fig. 5D). Candesartan did not alter BMK1 activities in DR/H rats, whereas increased BMK1 activities in DS/H rats were normalized by candesartan treatment, as shown in Figure 5D. No differences in the amounts of ERK1/2, JNK, p38 MAPK, or BMK1 were observed in samples by Western blotting analyses using pan-ERK1/2, JNK, p38 MAPK, and BMK1 (ERK 5) antibodies (data not shown).
Fig. 5. Renal cortical mitogen-activated protein kinases (MAPKs) activities in Dahl salt-sensitive (DS) rats.

Renal cortical tissues were harvested, lysed, and used for subsequent analyses. The activities of (A) extracellular signal-regulated kinases (ERK) 1/2, (B) c-Jun NH2-terminal kinases (JNK), (C) p38 MAPK, and (D) Big MAPK-1 (BMK1) were measured as described in Methods. All values were normalized by arbitrarily setting the densitometry of DS/L rats to 1.0. Densitometric analysis of the immunoreactive bands showed that a high salt diet markedly increased renal cortical ERK1/2, JNK, and BMK1 activities in DS rats, whereas p38 MAPK activity was unchanged. Candesartan (C) treatment significantly reduced ERK1/2, JNK, and BMK1 activities by -45%, -60%, and -70%, respectively, in DS-rats. On the other hand, no differences in the amounts of ERK1/2, JNK, p38 MAPK, and BMK1 were observed in samples by Western blotting analyses using pan-ERK1/2, JNK, p38 MAPK, and BMK1 (ERK5) antibodies (data not shown).
DISCUSSION
Otsuka et al [3] demonstrated increased glomerular activator protein-1 transcription activities in DS hypertensive rats. The authors also showed that AT1 receptor blockade ameliorated glomerular injury and markedly reduced activator protein-1 activation in these animals [2, 3]. These data indicate that locally produced Ang II in the kidney contributes to the stimulation of transcription and acts as a pivotal mediator of the pathogenesis of glomerular changes in DS hypertensive rats. The present study demonstrated that in DS rats, kidney Ang II contents and renal cortical AT1 receptor protein levels were not reduced by a high-salt diet. The results also demonstrated that the renal cortical activities of ERK1/2 and JNK, as well as a new MAPK family member, BMK1 [18-20], were significantly increased in DS hypertensive rats. Similar to the results of previous studies [2, 3, 5, 7], it was observed that AT1 receptor blockade with candesartan did not significantly alter MBP in DS hypertensive rats. However, candesartan markedly decreased kidney Ang II contents and MAPK activities, and ameliorated renal injury in these animals. These data suggest that in DS hypertensive rats, some of the renoprotective effects of AT1 receptor blockade are accompanied by reductions in intrarenal Ang II contents and MAPK activities.
Accumulating evidence supports the notion that Ang II is formed locally in the kidney [24-26, 33], and acts as a pivotal mediator of the pathogenesis of renal injury during the development of hypertension [9]. In agreement with previous studies [4-6], the present study showed that PRA and circulating Ang II levels were markedly reduced by a high-salt diet in both DR and DS rats. It was also observed that a high-salt diet significantly reduced kidney Ang II contents in DR rats; however, a high-salt diet did not reduce these levels in DS rats. These results indicate that in DS hypertensive rats, the intrarenal renin-angiotensin system is regulated in a manner distinct from the circulating renin-angiotensin system. It is possible that the failure to reduce kidney Ang II contents allows for the continued contribution of the intrarenal renin-angiotensin system to the progression of renal injury in DS hypertensive rats. However, the precise mechanisms responsible for maintenance of the kidney Ang II contents are not clear from the present study. Previous studies have shown reductions in total renin activity, renin release, and renin mRNA in the renal tissues of DS hypertensive rats [6]. Interestingly, we recently found that DS hypertensive rats showed markedly increased kidney angiotensinogen protein levels as well as urinary excretion of angiotensinogen [34]. Thus, these data suggest that in the kidneys of DS hypertensive rats, increases in angiotensinogen production could help maintain Ang II levels in the presence of renin suppression.
Kobori et al [35] demonstrated that chronic infusion of Ang II resulted in paradoxic increases in renal expression of angiotensinogen in rats. Other preliminary studies have also shown that increases in intrarenal angiotensinogen levels in Ang II-infused rats were prevented by treatment with an AT1 receptor antagonist, olmesartan (unpublished data, Kobori and Navar, 2003). We also investigated intrarenal angiotensinogen levels in Ang II-independent hypertension; our results showed that intrarenal Ang II levels were significantly reduced and intrarenal angiotensinogen levels were not increased in deoxycorticosterone-salt hypertensive rats [36]. Thus, these data indicate that AT1 receptor-mediated positive feedback on angiotensinogen production is present in the kidneys of Ang II-dependent hypertensive animals. The present results show that candesartan does not alter kidney Ang II contents in DR/H rats, but markedly reduces these levels in DS/H rats, suggesting a difference in AT1 receptor-mediated intrarenal Ang II regulation between DR/H and DS/H rats. Precise mechanisms responsible for candesartan-induced reductions in kidney Ang II contents in DS/H rats remain unclear. However, it can be speculated that augmented intrarenal angiotensinogen production is prevented by AT1 receptor blockade with candesartan in DS/H rats, leading to reductions in Ang II contents in the kidney.
AT1 receptor protein levels in renal cortical tissues were reduced by a high-salt diet in DR rats; however, these levels were not suppressed in DS rats. These data are consistent with those observed by Nakaya et al [22], who showed that mRNA expression of AT1A and AT1B receptor was similar between DS/L and DS/H rats. Previous studies have shown opposing changes in glomerular/vascular and tubular AT1 receptor mRNA in the kidneys in response to changes in sodium diet [37, 38]. Therefore, future studies will be required to clarify regional and segment-specific regulation of intrarenal AT1 receptor expression during the development of saltinduced hypertension. We also found that AT2 receptor protein levels were significantly increased in the renal cortical tissues of DS hypertensive rats. In vitro studies have shown that AT2 receptors suppress cell growth and induce apoptosis [39]. Although the functional roles of AT2 receptors in the kidneys remain to be elucidated [33], it can be speculated that the regulation of AT2 receptor expression in the kidneys might represent an adaptive mechanism to attenuate renal injury in DS hypertensive rats. Tea et al [40] showed that AT1 receptor blockade with valsartan induced apoptosis and reduced smooth muscle cell number in the aorta of spontaneously hypertensive rats (SHR), and that the effects of valsartan were prevented by a specific AT2 receptor antagonist, PD123319. These data suggest that AT2 receptor-mediated smooth muscle cell apoptosis contributes to the inhibitory effects of AT1 receptor antagonist on vascular hypertropic remodeling in cardiovascular disorders. Morrissey et al [41] examined the effects of PD123319 on renal fibrosis induced by ureteral obstruction in rats, and found that PD123319 exacerbated fibrosis of the tubulointerstitium in obstructive nephropathy; suggesting a potential antifibrotic effect of AT2 receptors in the kidney. Collectively, it is possible to speculate that at least some of the renoprotective effects of candesartan in DS rats are mediated through AT2 receptors. Effects of AT1 receptor blockade on angiotensin receptor levels in the kidney have been less consistent [42-44]. For example, treatment with the AT1 receptor antagonist 158,809 decreased both AT1 and AT2 receptor binding in kidneys of autoimmune nephritis rats [42]. In kidneys of strokeprone SHR, candesartan did not alter mRNA expression of AT1A and AT1B receptors, but significantly decreased that of AT2 receptor [43]. Bonnet et al [44] performed studies in diabetic SHR and found that AT1 receptor blockade with irbesartan significantly increased mRNA expression, binding, and immunostaining of AT1 receptor in the kidney, whereas those of AT2 receptor were not altered by irbesartan. It remains to be determined if these discrepant observations relate to differences in strains, methods used to assess angiotensin receptor expression, or blood pressure changes. Clearly, further studies are needed to determine the effects of candesartan on kidney AT1 and AT2 receptor expression in DS rats.
In agreement with previous studies [2, 3, 5, 7], the present results showed that candesartan did not decrease blood pressure in DS/H rats, even at a high dose. Nakaya et al [22] reported that prepubertal treatment (3 to 10 weeks) with candesartan caused partial attenuation of hypertension in DS/H rats, and suggested that early treatment with an AT1 receptor antagonist has multiple benefits, including the suppression of a vascular amplifier mechanism and central nervous function. Thus, it is possible that the age of the rats is critical for the antihypertensive effects of candesartan. In the present study, a high-salt diet markedly increased UNaV and UKV in both DR and DS rats. It was also observed that treatment with candesartan tended to increase UNaV and UKV in both DR/H and DS/H rats, but these changes were not statistically significant. For now, we cannot satisfactorily explain why candesartan did not increase UNaV and UKV in these animals. However, several studies have shown that UNaV and UKV are not altered by long-term administration of AT1 receptor antagonists or angiotensin-converting enzyme inhibitors in hypertensive high salt-treated animals, including SHR fed a high-salt diet [45] and deoxycorticosterone-salt hypertensive rats [46] and mice [47].
MAPKs are important mediators of the intracellular signal transduction pathways that are responsible for cell growth and differentiation [9, 27-29]. Studies have shown that ERK1/2 and JNK are activated by Ang II in cultured mesangial cells [10, 12, 13] and isolated glomeruli [15]. Thrarux et al [16] demonstrated that Ang II-induced stimulation of the collagen I α2 chain gene was blocked by a specific ERK inhibitor or AT1 receptor antagonist in the kidney. These data suggest that Ang II stimulates intrarenal collagen accumulation via AT1 receptor-mediated MAPKs activation. Consistent with previous studies [17], the present results showed that ERK1/2 and JNK were markedly activated in the renal cortical tissues of DS hypertensive rats. We also found that renal p38 MAPK activities were unchanged in these animals, suggesting different activation of each MAPK subfamily. In addition, the present study demonstrated that the activity of a new MAPK family member, BMK1, was also significantly increased in renal tissues of DS hypertensive rats. To our knowledge, this is the first study to show BMK1 activity in renal tissue. Recent studies have indicated that BMK1 is required for growth factor-induced cell proliferation in various cells [18, 20]. More recently, Takeishi et al [19] showed that BMK1 activity in heart tissue was increased during the development of cardiac hypertrophy. Although specific roles of the BMK1 signaling pathway in renal cells remain unclear, it can be speculated that BMK1 plays a role as a novel signal transduction pathway leading to renal injury in DS hypertensive rats. In the present study, AT1 receptor blockade resulted in amelioration of glomerular injury along with marked reductions in renal MAPKs activities in DS hypertensive rats, suggesting that renal injury is accompanied by AT1 receptor-mediated MAPKs activation, at least in part. Since AT1 receptor blockade with candesartan did not alter MBP in DS/H rats, the renal effects of candesartan may be mediated through mechanisms that are independent of arterial pressure.
CONCLUSION
The results of the present study indicate that the intrarenal renin-angiotensin system is not suppressed in DS hypertensive rats. The renoprotective effects of AT1 receptor blockade might be, at least partially, due to reductions in intrarenal Ang II levels and MAPKs activities, which are not dependent on arterial pressure changes.
ACKNOWLEDGMENTS
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Science and Culture of Japan, the Research Foundation for Pharmaceutical Sciences (to Akira Nishiyama), the Uehara Memorial Foundation (to Akira Nishiyama and Hiroyuki Kobori), the National Kidney Foundation (to Hiroyuki Kobori), and the Salt Science Research Foundation (to Youichi Abe). We gratefully thank Yukiko Nagai, Kayoko Miyata (Kagawa Medical University), and Eri Hiramoto (Okayama University Medical School) for excellent technical assistance. We are also grateful to Dr. L. Gabriel Navar (Department of Physiology, Tulane University Health Sciences Center) for critical reading of the manuscript and helpful suggestions, and to Takeda Pharmaceutical Industries, Ltd., for supplying candesartan cilexetil.
REFERENCES
- 1.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26:137–143. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 2.Otsuka F, Yamauchi T, Kataoka H, et al. Effects of chronic inhibition of ACE and AT1 receptors on glomerular injury in dahl salt-sensitive rats. Am J Physiol. 1998;274:R1797–R1806. doi: 10.1152/ajpregu.1998.274.6.R1797. [DOI] [PubMed] [Google Scholar]
- 3.Otsuka F, Ogura T, Yamauchi T, et al. Chronic treatment with angiotensin II type 1 receptor antagonist suppresses glomerular activator protein-1 activity in salt-sensitive hypertensive rats. Kidney Blood Press Res. 2000;23:35–41. doi: 10.1159/000025952. [DOI] [PubMed] [Google Scholar]
- 4.Zhao X, White R, Van Huysse J, Leenen FH. Cardiac hypertrophy and cardiac renin-angiotensin system in Dahl rats on high salt intake. J Hypertens. 2000;18:1319–1326. doi: 10.1097/00004872-200018090-00018. [DOI] [PubMed] [Google Scholar]
- 5.Sugimoto K, Fujiwara A, Takahashi I, et al. Effects of reninangiotensin system blockade and dietary salt intake on left ventricular hypertrophy in Dahl salt-sensitive rats. Hypertens Res. 1998;21:163–168. doi: 10.1291/hypres.21.163. [DOI] [PubMed] [Google Scholar]
- 6.Tank JE, Moe OW, Henrich WL. Abnormal regulation of proximal tubule renin mRNA in the Dahl/Rapp salt-sensitive rat. Kidney Int. 1998;54:1608–1616. doi: 10.1046/j.1523-1755.1998.00160.x. [DOI] [PubMed] [Google Scholar]
- 7.Kodama K, Adachi H, Sonoda J. Beneficial effects of long-term enalapril treatment and low-salt intake on survival rate of dahl salt-sensitive rats with established hypertension. J Pharmacol Exp Ther. 1997;283:625–629. [PubMed] [Google Scholar]
- 8.Kim S, Yoshiyama M, Izumi Y, et al. Effects of combination of ACE inhibitor and angiotensin receptor blocker on cardiac remodeling, cardiac function, and survival in rat heart failure. Circulation. 2001;103:148–154. doi: 10.1161/01.cir.103.1.148. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 10.Anderson PW, Zhang XY, Tian J, et al. Insulin and angiotensin II are additive in stimulating TGF-beta 1 and matrix mRNAs in mesangial cells. Kidney Int. 1996;50:745–753. doi: 10.1038/ki.1996.372. [DOI] [PubMed] [Google Scholar]
- 11.Wolf G, Haberstroh U, Neilson EG. Angiotensin II stimulates the proliferation and biosynthesis of type I collagen in cultured murine mesangial cells. Am J Pathol. 1992;140:95–107. [PMC free article] [PubMed] [Google Scholar]
- 12.Huwiler A, Stabel S, Fabbro D, Pfeilschifter J. Platelet-derived growth factor and angiotensin II stimulate the mitogen-activated protein kinase cascade in renal mesangial cells: Comparison of hypertrophic and hyperplastic agonists. Biochem J. 1995;305:777–784. doi: 10.1042/bj3050777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huwiler A, Van Rossum G, Wartmann M, Pfeilschifter J. Angiotensin II stimulation of the stress-activated protein kinases in renal mesangial cells is mediated by the angiotensin AT1 receptor subtype. Eur J Pharmacol. 1998;343:297–302. doi: 10.1016/s0014-2999(97)01542-2. [DOI] [PubMed] [Google Scholar]
- 14.Tsiani E, Lekas P, Fantus IG, et al. High glucose-enhanced activation of mesangial cell p38 MAPK by ET-1, ANG II, and platelet-derived growth factor. Am J Physiol Endocrinol Metab. 2002;282:E161–E169. doi: 10.1152/ajpendo.2002.282.1.E161. [DOI] [PubMed] [Google Scholar]
- 15.Hamaguchi A, Kim S, Yano M, et al. Activation of glomerular mitogen-activated protein kinases in angiotensin II-mediated hypertension. J Am Soc Nephrol. 1998;9:372–380. doi: 10.1681/ASN.V93372. [DOI] [PubMed] [Google Scholar]
- 16.Tharaux PL, Chatziantoniou C, Fakhouri F, Dussaule JC. Angiotensin II activates collagen I gene through a mechanism involving the MAP/ERK kinase pathway. Hypertension. 2000;36:330–336. doi: 10.1161/01.hyp.36.3.330. [DOI] [PubMed] [Google Scholar]
- 17.Hamaguchi A, Kim S, Izumi Y, Iwao H. Chronic activation of glomerular mitogen-activated protein kinases in Dahl salt-sensitive rats. J Am Soc Nephrol. 2000;11:39–46. doi: 10.1681/ASN.V11139. [DOI] [PubMed] [Google Scholar]
- 18.Suzaki Y, Yoshizumi M, Kagami S, et al. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: Potential role in cell survival following oxidative insults. J Biol Chem. 2002;277:9614–9621. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- 19.Takeishi Y, Huang Q, Abe J, et al. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: Comparison with acute mechanical stretch. J Mol Cell Cardiol. 2001;33:1637–1648. doi: 10.1006/jmcc.2001.1427. [DOI] [PubMed] [Google Scholar]
- 20.Berk Bc, Abe J, Min W, et al. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann N Y Acad Sci. 2001;947:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. discussion 109-111. [DOI] [PubMed] [Google Scholar]
- 21.Sasamura H, Shimizu-Hirota R, Nakaya H, Saruta T. Effects of AT1 receptor antagonist on proteoglycan gene expression in hypertensive rats. Hypertens Res. 2001;24:165–172. doi: 10.1291/hypres.24.165. [DOI] [PubMed] [Google Scholar]
- 22.Nakaya H, Sasamura H, Mifune M, et al. Prepubertal treatment with angiotensin receptor blocker causes partial attenuation of hypertension and renal damage in adult Dahl salt-sensitive rats. Nephron. 2002;91:710–718. doi: 10.1159/000065035. [DOI] [PubMed] [Google Scholar]
- 23.Mackenzie HS, Troy JL, Rennke HG, Brenner BM. TCV 116 prevents progressive renal injury in rats with extensive renal mass ablation. J Hypertens Suppl. 1994;12:S11–S16. [PubMed] [Google Scholar]
- 24.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension. 2002;39:129–134. doi: 10.1161/hy0102.100536. [DOI] [PubMed] [Google Scholar]
- 25.Fox J, Guan S, Hymel AA, Navar LG. Dietary Na and ACE inhibition effects on renal tissue angiotensin I and II and ACE activity in rats. Am J Physiol. 1992;262:F902–F909. doi: 10.1152/ajprenal.1992.262.5.F902. [DOI] [PubMed] [Google Scholar]
- 26.Harrison-Bernard LM, Zhuo J, Kobori H, et al. Intrarenal AT(1) receptor and ACE binding in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol. 2002;282:F19–F25. doi: 10.1152/ajprenal.00335.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kacimi R, Gerdes AM. Alterations in G protein and MAP kinase signaling pathways during cardiac remodeling in hypertension and heart failure. Hypertension. 2003;41:968–977. doi: 10.1161/01.HYP.0000062465.60601.CC. [DOI] [PubMed] [Google Scholar]
- 28.Ishizawa K, Yoshizumi M, Tsuchiya K, et al. Effects of losartan in combination with or without exercise on insulin resistance in Otsuka Long-Evans Tokushima Fatty rats. Eur J Pharmacol. 2001;430:359–367. doi: 10.1016/s0014-2999(01)01405-4. [DOI] [PubMed] [Google Scholar]
- 29.Kyaw M, Yoshizumi M, Tsuchiya K, et al. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res. 2001;24:251–261. doi: 10.1291/hypres.24.251. [DOI] [PubMed] [Google Scholar]
- 30.Hitomi H, Kiyomoto H, Hashimoto M, et al. A new approach for glomerular lesions: Evaluation of scanning acoustic microscopy (SAM) for experimental glomerular disease in rats. Ultrasound Med Biol. 2000;26:571–577. doi: 10.1016/s0301-5629(00)00146-0. [DOI] [PubMed] [Google Scholar]
- 31.Nishiyama A, Majid DS, Walker M, III, et al. Renal interstitial ATP responses to changes in arterial pressure during alterations in tubuloglomerular feedback activity. Hypertension. 2001;37:753–759. doi: 10.1161/01.hyp.37.2.753. [DOI] [PubMed] [Google Scholar]
- 32.Noma T, Mizushige K, Yao L, et al. Alteration in aortic wall stiffness and accumulation of collagen during the prediabetic stage of type II diabetes mellitus in rats. Jpn Circ J. 1999;63:988–993. doi: 10.1253/jcj.63.988. [DOI] [PubMed] [Google Scholar]
- 33.Navar LG, Nishiyama A. Intrarenal formation of angiotensin II. Contrib Nephrol. 2001;135:1–15. doi: 10.1159/000060154. [DOI] [PubMed] [Google Scholar]
- 34.Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension. 2003;41:592–597. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002;61:579–585. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension. Hypertension. 2003;41:42–49. doi: 10.1161/01.hyp.0000050102.90932.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amiri F, Garcia R. Differential regulation of renal glomerular and preglomerular vascular angiotensin II receptors. Am J Physiol. 1996;270:E810–E815. doi: 10.1152/ajpendo.1996.270.5.E810. [DOI] [PubMed] [Google Scholar]
- 38.Cheng HF, Becker BN, Burns KD, Harris RC. Angiotensin II upregulates type-1 angiotensin II receptors in renal proximal tubule. J Clin Invest. 1995;95:2012–2019. doi: 10.1172/JCI117886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: Regulation, signaling and function. Blood Press. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- 40.Tea BS, Der Sarkissian S, Touyz RM, et al. Proapoptotic and growth-inhibitory role of angiotensin II type 2 receptor in vascular smooth muscle cells of spontaneously hypertensive rats in vivo. Hypertension. 2000;35:1069–1073. doi: 10.1161/01.hyp.35.5.1069. [DOI] [PubMed] [Google Scholar]
- 41.Morrissey JJ, Klahr S. Effect of AT2 receptor blockade on the pathogenesis of renal fibrosis. Am J Physiol. 1999;276:F39–F45. doi: 10.1152/ajprenal.1999.276.1.F39. [DOI] [PubMed] [Google Scholar]
- 42.Uhlenius N, Miettinen A, Vuolteenaho O, Tikkanen I. Renoprotective mechanisms of angiotensin II antagonism in experimental chronic renal failure. Kidney Blood Press Res. 2002;25:71–79. doi: 10.1159/000063511. [DOI] [PubMed] [Google Scholar]
- 43.Nakaya H, Sasamura H, Kitamura Y, et al. Effects of angiotensin inhibitors on renal injury and angiotensin receptor expression in early hypertensive nephrosclerosis. Hypertens Res. 1999;22:303–312. doi: 10.1291/hypres.22.303. [DOI] [PubMed] [Google Scholar]
- 44.Bonnet F, Candido R, Carey RM, et al. Renal expression of angiotensin receptors in long-term diabetes and the effects of angiotensin type 1 receptor blockade. J Hypertens. 2002;20:1615–1624. doi: 10.1097/00004872-200208000-00025. [DOI] [PubMed] [Google Scholar]
- 45.Lassila M, Finckenberg P, Pere AK, et al. Comparison of enalapril and valsartan in cyclosporine A-induced hypertension and nephrotoxicity in spontaneously hypertensive rats on high-sodium diet. Br J Pharmacol. 2000;130:1339–1347. doi: 10.1038/sj.bjp.0703422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.French JF, Anderson BA, Downs TR, Dage RC. Dual inhibition of angiotensin-converting enzyme and neutral endopeptidase in rats with hypertension. J Cardiovasc Pharmacol. 1995;26:107–113. doi: 10.1097/00005344-199507000-00017. [DOI] [PubMed] [Google Scholar]
- 47.Peng H, Carretero OA, Alfie ME, et al. Effects of angiotensin-converting enzyme inhibitor and angiotensin type 1 receptor antagonist in deoxycorticosterone acetate-salt hypertensive mice lacking Ren-2 gene. Hypertension. 2001;37:974–980. doi: 10.1161/01.hyp.37.3.974. [DOI] [PubMed] [Google Scholar]