

Abstract
Although germ cell formation has been relatively well understood in worms and insects, how germ cell-specific developmental programs are initiated is not clear. In Caenorhabditis elegans, translational activation of maternal nos-2 mRNA is the earliest known molecular event specific to the germline founder cell P4. Cis-elements in nos-2 3′UTR have been shown to mediate translational control, however the trans-acting proteins are not known. Here, we provide evidence that four maternal RNA-binding proteins, namely OMA-1, OMA-2, MEX-3 and SPN-4, bind nos-2 3′UTR to suppress its translation, and POS-1, another maternal RNA-binding protein, relieves this suppression in P4. The POS-1: SPN-4 ratio in P4 increases significantly over its precursor, P3; and POS-1 competes with SPN-4 for binding to nos-2 RNA in vitro. We propose temporal changes in the relative concentrations of POS-1 and SPN-4, through their effect on the translational status of maternal mRNAs such as nos-2, initiate germ cell-specific developmental programs in C. elegans.
Keywords: Caenorhabditis elegans, Translational control, RNA-binding protein, 3′UTR, Germ cells, Nanos
Introduction
Many maternally expressed genes play critical roles during early embryonic development. Since these genes are transcribed prior to fertilization, regulation at posttranscriptional stages is key for their proper functioning. Studies on many maternal mRNAs have revealed a central role for translation regulation in the proper coordination of various early events of embryogenesis. For example, pattern formation in Drosophila embryo depends on the translational control of maternal mRNAs such as oskar, nanos, caudal and hunchback ( Macdonald and Smibert, 1996; Dean et al., 2002). Similarly, the translational control of maternal mRNAs such as glp-1, apx-1 and pal-1 are essential for fate specification of certain blastomeres of Caenorhabditis elegans embryo (Evans and Hunter, 2005).
Genetic studies in flies point to the functioning of cascades of translational control during embryogenesis (Kuersten and Goodwin, 2003). For example, development of posterior structures depends on the restriction of hunchback translation to the anterior by Nanos (Sonoda and Wharton, 1999). Nanos translation, in turn, is restricted to the posterior by the action of Smaug and Oskar (Dahanukar et al., 1999; Smibert et al., 1999). Once again, translational control restricts Oskar to the posterior (Gunkel et al., 1998). However, baring a few examples of translational cascades characterized in Drosophila, their role during embryogenesis still remains largely unexplored. Protein factors involved in the translation of several maternal mRNAs are not known. Similarly, the target mRNAs for many maternal RNA-binding proteins have yet to be identified. Identification of these will be essential to obtain a complete picture of translational control in development.
The development of primordial germ cells (PGCs) in C. elegans is a good example of an embryonic process involving complex translational control. In this organism, the maternal components required for germ line development are sequestered to a single cell at the first embryonic division itself (Seydoux and Strome, 1999). However, the formation of PGCs is postponed to a later stage. This is because, as shown in Fig. 1, the posterior lineage, which preserves germline-specific maternal components, gives rise to various somatic lineages during the first four divisions. Therefore, the maternal mRNAs essential for the activation of germ cell-specific developmental programs must remain translationally quiescent through various developmental events from oocyte until the formation of the germline founder cell P4, which is born at the 28-cell stage. While the CCCH-type zinc finger protein PIE-1 has been shown as essential for RNA maintenance in germline blastomeres (Tenenhaus et al., 2001), it is not clear how the translational quiescence is maintained.
Fig. 1.
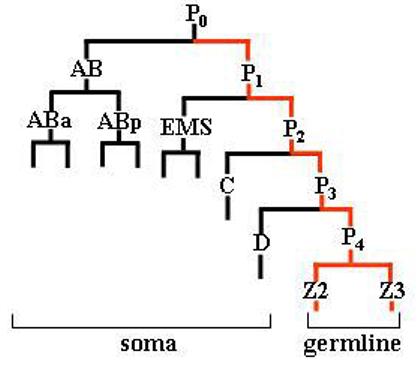
A line diagram showing abbreviated embryonic lineage (Sulston et al., 1983). The P lineage is shown in red.
The maternal mRNA encoded by nos-2, a C. elegans member of the nanos family of germ cell regulators, is currently the only known mRNA whose translation is specifically activated in P4 ( Subramaniam and Seydoux, 1999; D'Agostino et al., 2006). Earlier results have shown that the translation of nos-2 is repressed from oocytes until 28-cell embryo, and that this repression requires the functions of three distinct 3′UTR elements. It has also been shown that the CCCH-finger protein, POS-1, is essential for the activation of translation in P4 (D'Agostino et al., 2006). Here we report the identification of four additional maternal RNA-binding proteins, namely OMA-1, OMA-2, MEX-3 and SPN-4, which suppress nos-2 translation in successive stages: OMA-1 and OMA-2 – suppress in oocytes, MEX-3 – in early embryo and SPN-4 – in germline blastomeres. We find that these proteins suppress translation by directly binding to nos-2 3′UTR. Further, our results presented here suggest that POS-1 activates nos-2 translation in P4 by competing out SPN-4 for binding to nos-2 3′UTR. Thus, temporal changes in the concentration of these maternal RNA-binding proteins appear to mediate the PGC-specific activation of nos-2 translation.
Materials and methods
C. elegans strains
Worm strains were maintained as described (Brenner, 1974), except that all transgenic lines were kept at 25 °C to avoid silencing of transgene expression in the germline (Strome et al., 2001). Transgenes were introduced into unc-119(−) strain by biolistic bombardment as described (Praitis et al., 2001), with the following modifications: 1μm tungsten particles were used as the micro carrier with 1500-psi rupture discs. Mutant versions of the transgene were created by PCR and inserted into the plasmid, pKS111HisΔ5, which contains the GFP:H2B:nos-2 3′UTR (D'Agostino et al., 2006). The following strains were used:
EU769 - spn-4(or25) unc-42(e270) V/nT1[let-?(m435)] (IV;V)
JJ1014 - mex-3(zu155) dpy-5(e61)/hT1 I; pos-1(zu148) unc-42(e270)/hT1 V
JJ462 - +/nT1 IV; pos-1(zu148) unc-42(e270)/nT1 V
RNAi screen
A list of genes that encode putative RNA-binding proteins was prepared based on annotations available at www.wormbase.org. Of these, 131 were part of a library of RNAi clones (Table S1 in the supplementary material) (Kamath et al., 2003). Other target open reading frames (ORFs) were PCR-amplified, inserted into the RNAi feeding vector, L4440 and introduced into E. coli HT115. These E. coli clones were used for inducing RNAi by the feeding method (Timmons et al., 2001) in transgenic worms carrying pKS111HisΔ5.
Protein expression and purification
Full-length ORFs of mex-3 was PCR-amplified and inserted at the Bam HI site of pMAL-c4E, which expresses the inserted ORF as a fusion protein with the maltose-binding protein (MBP) (New England Biolabs). The ORF of spn-4 was cloned between Eco RI and Xho I sites, and the ORFs of oma-1 and oma-2 between Eco RI and Not I sites of pGEX-4T1 vector. The pos-1 ORF was inserted between Eco RI and Bam HI sites of pGEX-2T. The pGEX vectors express the cloned ORF as GST fusion protein (GE Lifesciences). Cloning techniques, including PCR, were carried out following standard protocols (Sambrook et al., 1989).
The transformants were grown in LB medium at 37 °C until 0.5 OD at 600 nm before induction with 0.05 mM IPTG for 2 hours at 16 °C. Cells were collected by centrifugation and lysed in lysis buffer (20 mM HEPES, pH 7.4 / 0.5 M NaCl / 5 mM DTT / 0.02 % Tween 20 / 0.1 mM PMSF) by incubation on ice with 0.5 mg / ml of lysozyme, followed by 3 rounds of freeze-thaw cycles. The lysates were treated with 20 μg / ml of DNase I and cleared by centrifugation. Fusion proteins were purified from clear supernatants by affinity chromatography using either amylose resin (MBP:MEX-3) or glutathione-agarose (GST fusions) following manufacturers' protocols (MBP – New England Biolabs; GST – GE Lifesciences). Purified proteins were concentrated by ultrafiltration, added with glycerol to a final concentration of 50 % and stored at −20 °C.
Electrophoretic mobility shift assay
Radiolabeled RNA fragments used for EMSA were prepared by in vitro transcription of DNA template using T7 RNA polymerase (Fermentas) with α-32P CTP (specific activity: 3000Ci / mmole) following standard protocols (Sambrook et al., 1989). Full-length transcripts were purified from urea gel and quantitated using a liquid scintillation counter. Template DNAs were generated by PCR amplification using appropriate primers from pKS111HisΔ5. The T7 promoter sequence was incorporated to DNA templates through the forward PCR primer. Required mutations were also introduced through PCR primers. A 360-bp cDNA fragment encoding the splicing factor (GenBank accession# AW828516) of Meloidogyne incognita, a parasitic nematode, was used as template for generating the non-specific unlabeled RNA. This RNA is not GC rich and, using the M fold RNA folding program (Zuker, 2003), we found that it does not form long stretches of stable double-stranded structures (data not shown). Unlabeled RNA was prepared in the same manner as above except that the α-32P CTP was replaced with CTP.
Binding reactions were carried out by incubating the appropriate RNA and protein in RNA-binding buffer (5 mM HEPES, pH 7.5 / 25 mM KCl / 2 mM MgCl2 / 1 mM EDTA / 2 mM DTT / 3.5 % glycerol / 0.25 mg / ml yeast tRNA) at room temperature (RT) for 20 minutes. RNA was denatured by first incubating at 75 °C for 10 min and then at 37 °C for further 10 min, before adding to the binding reactions. All lanes contained identical amounts of RNA and protein, except where indicated. For competitions, protein was incubated simultaneously with radiolabeled RNA and indicated amounts of unlabeled RNA. The reaction mixtures were eletrophoresed at +4 °C at 200 V on a 16 × 20 cm non-denaturing polyacrylamide gel in TBE buffer. The concentration of acrylamidebisacrylamide mix in these gels was 3.5% in the case of MEX-3, 6% in the case of OMA-1 and OMA-2 fusion proteins and 7.5% in the case of POS-1 and SPN-4 fusion proteins. Duration of electrophoresis varied, depending on the size of the RNA, from 4 to 20 hours. Following electrophoresis, the gel was dried and exposed to phosphor imager screen and imaged using a phosphor imager (Personal Molecular Imager FX, Bio-Rad). Intensity of radioactive bands were quantitated using the Quantity One software (Bio-Rad).
Pull down assay
This assay, similar to the affinity purification of fusion proteins described above, depends on the affinity of GST and MBP for their corresponding ligands, glutathione and amylose, respectively. For binding experiments with POS-1, glutathione-agarose beads were first washed three times in distilled water, followed by five times in RNA-binding buffer (RBB). Washed beads were incubated with GST::POS-1 at +4 °C for 20 minutes with gentle agitation. Protein-bound beads were incubated with RNA in RBB for 20 minutes at room temperature. After the incubation period, the beads were collected by brief centrifugation and washed 5 times with RBB. The GST::POS-1 protein was eluted from beads with 20mM glutathione and the bound RNA was separated by phenol:chloroform extraction. The RNA was then precipitated and separated on a 6% acrylamide gel containing 8M urea. The gel was dried and exposed to phosphor imager screen as described earlier. Binding experiments with MEX-3 were performed in a similar manner, except that amylose resin and maltose were used as the solid matrix and eluant, respectively.
Immunofluorescence
Embryos permeabilized by the freeze-crack method and fixed in formaldehyde were immunostained as described (Subramaniam and Seydoux, 1999). The following primary antibodies were used: anti-POS-1 (Tabara et al., 1999) and anti-SPN-4. Anti-SPN-4 antibodies were obtained by affinity purification of polyclonal antiserum of rabbits immunized with GST:SPN-4. Immunofluorescence, as well as the GFP fluorescence from embryos was imaged using a fluorescence microscope (Zeiss Axioskop) and CCD camera (Axiocam HRm). Immunofluorescence signal intensities were quantitated by measuring pixel density of deconvoluted Z-stack images using Axiovision software.
Results
Identification of proteins that control nos-2 translation
To identify proteins involved in the translational control of nos-2 mRNA, we carried out an RNAi-based screen of genes predicted to encode proteins with RNA-binding motifs. To facilitate the monitoring of NOS-2 expression, we performed the RNAi on transgenic worms that express the GFP:H2B reporter under the control of nos-2 3′UTR (see Materials and Methods for details). Expression pattern of GFP:H2B in embryos of these worms is reminiscent to that of the endogenous NOS-2 protein (D'Agostino et al., 2006). This screen identified four genes, namely oma-1, oma-2, mex-3 and spn-4, whose down regulation by RNAi resulted in misexpression of GFP:H2B (Fig. 2A and 2B). In the nonRNAi control embryos, GFP:H2B was not detected in any of the blastomeres until the 28-cell stage. Similar to endogenous NOS-2 expression, GFP:H2B first appeared at the 28-cell stage in the germline founder cell P4. In contrast, in the case of oma-1(RNAi) oma-2(RNAi) “double mutants”, significantly higher levels of GFP:H2B were first detected in oocytes. Since OMA-1 and OMA-2 are essential for oocyte maturation, their absence leads to oocyte arrest (Detwiler et al., 2001). To test whether the increased GFP:H2B expression was merely a result of accumulation of GFP in the arrested oocytes, we introduced the GFP:H2B:nos-2 3′UTR transgene into tra-2(q122) worms, another mutant in which the unmated hermaphrodites accumulate oocytes (Barton et al., 1987), and examined the expression of GFP:H2B in their oocytes. As shown in Fig. 2A, these oocytes did not show any increase in the level of GFP:H2B over wild-type control. In contrast, removal of OMA-1 and OMA-2 proteins in these worms by RNAi led to a dramatic increase in the level of GFP:H2B in their oocytes. Expression of GFP:H2B in oma-1(RNAi) and oma-2(RNAi) “single mutants” were almost at the level of non-RNAi control oocytes, which is probably a result of functional redundancy between these two nearly identical genes (Fig. 2A). We conclude OMA-1 and OMA-2 function redundantly to suppress nos-2 translation in the oocyte.
Fig. 2.
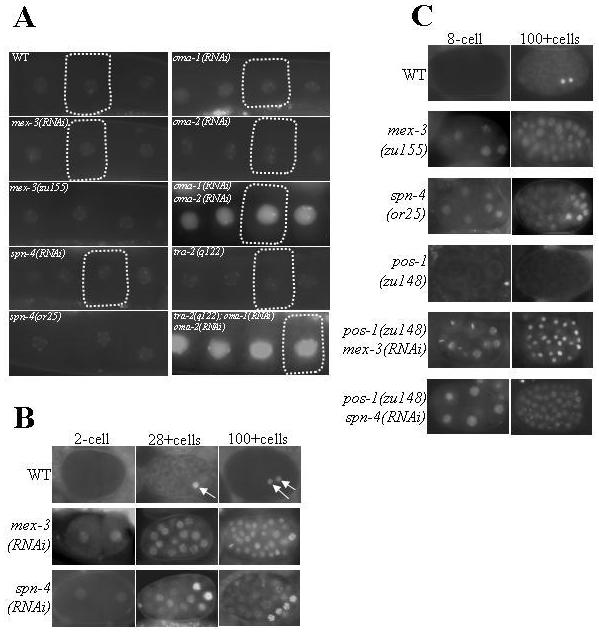
A and B: OMA-1, OMA-2, MEX-3 and SPN-4 are essential for the translation suppression of nos-2 mRNA. Distribution pattern of GFP:H2B expressed under the control of nos-2 3′UTR in oocytes (A; a single oocyte in each panel is outlined) and embryos (B) is shown. Genes disrupted by RNAi treatment are indicated in each panel; WT – non-RNAi control. To facilitate visualization, we expressed GFP as a fusion protein with the histone H2B, which concentrates fluorescence signal in nuclei. C: POS-1 acts as a de-repressor of nos-2 translation. Epistasis analysis of GFP:H2B expression among mex-3(−), spn-4(−) and pos-1(−) shown here reveals that POS-1 is not required for nos-2 translation in the absence of repressors such as MEX-3 and SPN-4.
In mex-3(RNAi) embryos, GFP:H2B was present in all the blastomeres of embryos starting from the 2-cell stage (Fig. 2B). We observed similar expression pattern in spn-4(RNAi) embryos as well, except that in these embryos GFP:H2B expression was significantly more pronounced in a few posterior blastomeres. Disruption of mex-3 or spn-4 by RNAi did not alter the background levels of GFP:H2B seen in the oocytes of control worms. We observed similar results in the genetic null alleles of these genes (Fig. 2A and 2C) (Draper et al., 1996; Gomes et al., 2001). From these results, we conclude mex-3 and spn-4 are essential for the suppression of nos-2 translation in the early embryo and probably not essential in oocyte.
POS-1 de-represses, rather than activates, nos-2 translation
Earlier we had shown that the CCCH-type zinc finger protein POS-1 is required for the activation of nos-2 translation in the primordial germ cells (PGCs) (D'Agostino et al., 2006). The POS-1 protein could function either by activating translation or by relieving the translational repression by repressors such as MEX-3 and SPN-4. To distinguish between these two possibilities, we determined the epistatic relationship among these genes. If POS-1 were to be required for the activation of nos-2 translation, then the ectopic GFP:H2B expression observed in embryos lacking MEX-3 or SPN-4 should be dependent on POS-1. On the other hand, if it functions as a derepressor, then GFP:H2B expression in mex-3(RNAi) or spn-4(RNAi) embryos would not be dependent on POS-1. To ensure complete absence of POS-1 protein, we used the null allele, pos-1(zu148) (Tabara et al., 1999), rather than pos-1(RNAi), in these epistasis analyses. The pattern of GFP:H2B observed in mex-3(RNAi) pos-1(zu148) embryos was similar to that of mex-3(RNAi) and that in spn-4(RNAi) pos-1(zu148) embryos was similar to that of spn-4(RNAi) (Fig. 2C), indicating that the POS-1 protein is not required for nos-2 translation in the absence of MEX-3 or SPN-4. We conclude that POS-1 derepresses, rather than activates, nos-2 translation.
All five proteins, OMA-1, OMA-2, MEX-3, SPN-4 and POS-1, directly bind to nos-2 3′UTR
To begin to investigate the mechanism(s) of the translational control, we tested whether any of the proteins identified by the RNAi screen, including POS-1, physically interact with nos-2 3′UTR. For this, we expressed these proteins as GST (OMA-1, OMA-2, SPN-4 and POS-1) or MBP (MEX-3) fusion in bacteria and purified using affinity chromatography. The purified recombinant proteins were then tested for their ability to bind radiolabeled 200-bp minimal nos-2 3′UTR RNA in electrophoretic mobility shift assay (EMSA). This 200-bp RNA has been shown earlier to be sufficient for the endogenous expression pattern of NOS-2 (D'Agostino et al., 2006). As shown in Fig. 3, all five proteins retarded the electrophoretic mobility of nos-2 RNA. In case of MEX-3, SPN-4, OMA-1 and OMA-2, incubation with 50-fold molar excess of unlabeled nos-2 3′UTR RNA completely abolished the shift of the radiolabeled RNA, whereas a similar molar excess of non-specific unlabeled RNA did not affect this mobility shift (Fig. 3A). In the case of POS-1, while the non-specific RNA did reduce the radioactive signal corresponding to mobility shift, its effect was far less than the unlabeled nos-2 3′UTR RNA (Fig. 3B). To validate these results further, we performed an alternative RNA-binding assay for POS-1 and MEX-3. This assay depends on the affinity of the fusion tags GST and MBP for their corresponding ligands covalently linked to a solid matrix (see Material and Methods for details). Even in this assay, 50-fold molar excess of unlabeled nos-2 3′UTR RNA significantly reduced the amount of radiolabeled RNA bound to POS-1 or MEX-3. In contrast, a similar molar excess of a non-specific unlabeled RNA did not alter binding of the radiolabeled RNA (Fig. 3C). We used PUF-8, another RNA-binding protein, as a negative control; this protein did not bind nos-2 3′UTR RNA in either of the in vitro binding assays (data not shown). Based on the above results, we conclude that the five proteins identified in our RNAi screen can specifically and directly bind to the nos-2 3′UTR in vitro. Since OMA-1 and OMA-2 share a high degree of sequence homology and their electrophoretic mobility shift patterns were identical, we tested only OMA-2 in the subsequent EMSAs.
Fig. 3.
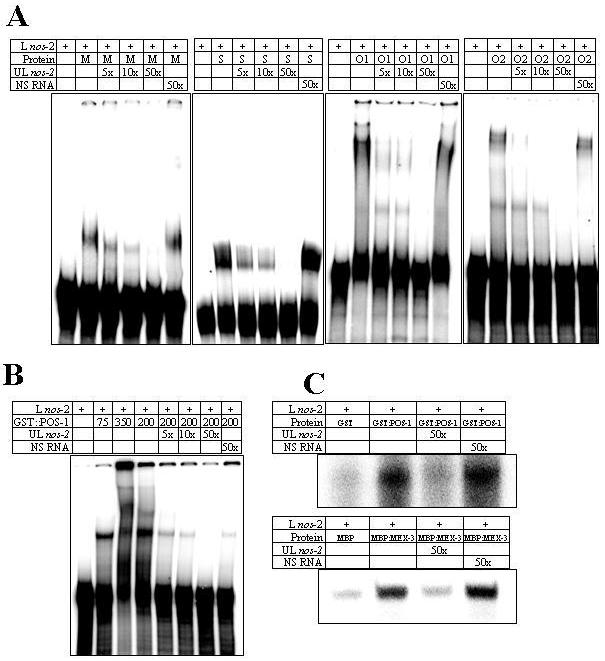
MEX-3, SPN-4, OMA-1, OMA-2 and POS-1 physically interact with nos-2 3′UTR. A) Electrophoretic mobility patterns of radiolabeled 200-bp nos-2 3′UTR RNA in the presence of MBP:MEX-3 (M), GST:SPN-4 (S), GST:OMA-1 (O1) and GST:OMA-2 (O2). L nos-2 – radiolabeled 200-bp nos-2 3′UTR; UL nos-2 – unlabeled nos-2 3′UTR; NS RNA – unlabeled non-specific RNA; and 5×, 10× and 50× – number of times molar excess over L nos-2. B) Electrophoretic mobility shift with GST:POS-1. Three different concentrations of GST-POS-1 were used: 75, 350 and 200 ng/μl. Comparison of lanes 2-4 indicates multimerization of this protein-RNA complex at higher protein concentrations. C) Binding of radiolabeled nos-2 3′UTR RNA to solid matrix in presence of the indicated components (see Materials and Methods for details).
We introduced a series of deletions in the 200-bp minimal nos-2 3′UTR and tested them in EMSA to identify the specific sequences that are responsible for the interaction. Deletion of any part of the minimal UTR abolished or significantly reduced the binding of MEX-3, SPN-4 and POS-1 (data not shown), indicating that the entire sequence of the minimal UTR may be essential for efficient interaction of these three proteins. Alternatively, it is also possible that the distance between different sequence elements within the UTR, rather than the whole of the minimal UTR sequence, is critical for proper interaction. To address this, we substituted 30-bp stretches with a non-specific sequence [(TG)15] of the same length and tested them in EMSA (Fig. 4). Of the five substitutions tested, SubB and SubC significantly reduced the mobility shift by MEX-3 and SPN-4 proteins (Fig. 4B and 4C), suggesting that the wild-type sequences of SubB and SubC are critical for the binding of these two proteins. These results were further confirmed by the pull down assay described above. In this assay, unlabeled SubC substitution did not compete with labeled wild-type RNA as efficiently as the unlabeled wild-type RNA for binding to MEX-3. Consistently, the binding of radiolabeled SubC substitution was poorer when compared the wild-type (Fig. 4F). Surprisingly, none of the substitutions had an appreciable effect on POS-1 binding (data not shown). In contrast, OMA-2 bound the region defined by SubD and SubE substitutions as efficiently as the 200-bp 3′UTR (Fig. 4D). Consistent with this, in the substitution analysis, only SubE significantly reduced OMA-2 binding (Fig. 4E). The binding of SubE substitution was significantly weaker in the pull down assay as well (Fig. 4F). These results indicate that the SubE region is sufficient for OMA-2 interaction with nos-2 3′UTR.
Fig. 4.
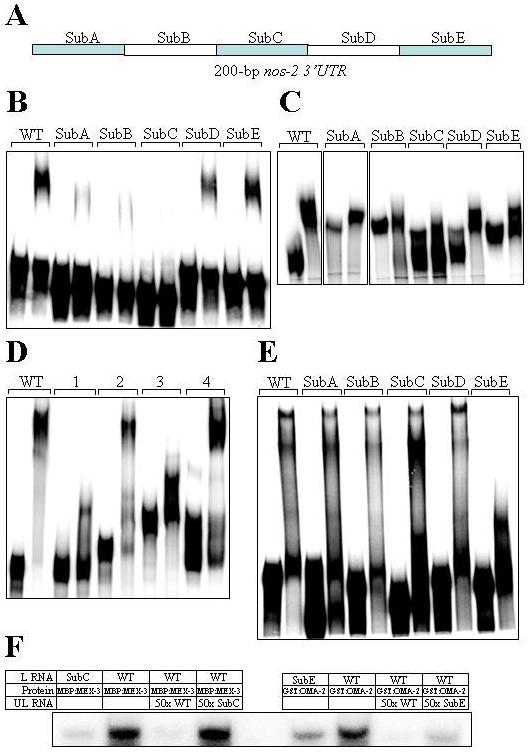
Determination of nos-2 3′UTR regions that are critical for interaction with the various proteins. A) Schematic illustration of the five regions of nos-2 3′UTR that were mutated by substitution. SubA begins immediately downstream of the stop codon. B-E) Electrophoretic mobility shifts of various mutant versions of radiolabeled nos-2 3′UTR by MBP:MEX-3 (B), GST:SPN-4 (C) and GST:OMA-2 (D and E). The first lane in each set is the mobility of RNA in the absence of protein. Radiolabeled RNA used in (D) contained the wild-type version of the following regions only: 1 – SubA-C; 2 – SubB-E; 3 – SubA-D and 4 – SubD-E, whereas those in other panels contained the 200-bp nos-2 3′UTR with the indicated regions substituted with (TG)15. WT in all panels indicate the wild-type version of the 200-bp nos-2 3′UTR. F) Binding of radiolabeled WT and mutant nos-2 3′UTR RNA to solid matrix in presence of the indicated components (see Materials and Methods for details). L RNA – radiolabeled RNA; UL RNA – unlabeled RNA.
Remarkably, SubB and SubC regions contain two 8-bp direct repeats (DR1 and DR2), which have been well conserved in the nos-2 3′UTR among the three Caenorhabditis species for which sequence information is available [Fig. 5A and (D'Agostino et al., 2006)]. To test whether these two repeats are essential for the RNA-protein interactions, we replaced these repeats with (TG)4 and tested in EMSA. Mutations in only one of either direct repeats reduced the mobility shift by MEX-3, which was further weakened when both repeats were simultaneously mutated (Fig. 5B). These results indicate that the two direct repeats are essential for the binding of MEX-3. In the case of SPN-4, while the double mutant and the DR1 mutant significantly reduced the shift, DR2 mutations did not affect the mobility shift, indicating a critical role for DR1 in SPN-4 binding (Fig. 5B). Together these results suggest that both MEX-3 and SPN-4 may bind the same region of nos-2 3′UTR.
Fig. 5.
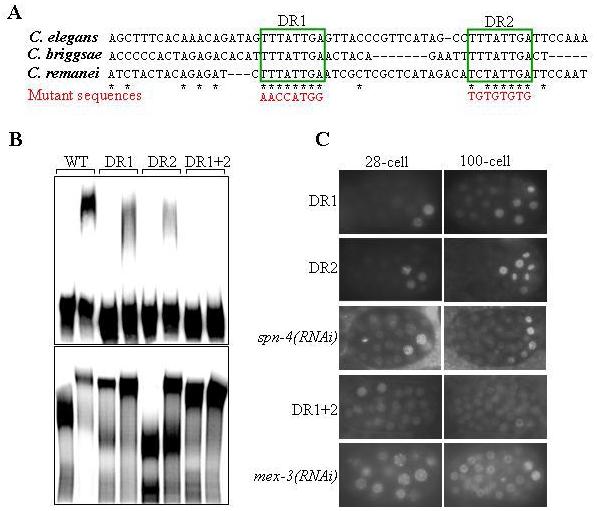
Binding to nos-2 3′UTR is essential for the translation suppression activity of MEX-3 and SPN-4.Two 8-bp direct repeats present in nos-2 3′UTR are critical for the binding of MEX-3 and SPN-4. (A) Alignment of the nos-2 3′UTR of the indicated species (D'Agostino et al., 2006). Only the region with two 8-bp direct repeats (DR1 and DR2; boxed) is shown. Stars indicate bases conserved in all three species. Sequences of mutations used in (B and C) are shown in red. B) Electrophoretic mobility shifts by MBP:MEX-3 (top) and GST:SPN-4 (bottom) of the various mutant versions of radiolabeled nos-2 3′UTR.
C) Expression pattern of GFP:H2B in embryos of transgenic worms carrying the GFP:H2B:nos-2 3′UTR transgene bearing the indicated mutations, or following spn-4(RNAi) or mex-3(RNAi). Note that the GFP:H2B distribution pattern in DR1 and DR2 is similar to that of spn-4(RNAi) and the pattern in DR1+DR2 is similar to that of mex-3(RNAi).
Binding to nos-2 3′UTR is essential for the translational suppression by MEX-3, SPN-4 and OMA-2
If the direct repeats DR1 and DR2 are required for the interactions with MEX-3 and SPN-4 proteins, and if these proteins controlled nos-2 translation by direct interaction with nos-2 3′UTR, then DR1 and DR2 mutations should have the same effect on nos-2 translation as that of the removal of these proteins. To test this, we prepared GFP:H2B:nos-2 3′UTR transgene constructs with the same DR1 and DR2 mutations used in the EMSA experiments and generated transgenic lines expressing the mutant constructs. Mutations in either one of the repeats led to weak GFP:H2B expression in all cells and stronger expression in a few cells at the posterior of the embryo – a pattern identical to the spn-4(RNAi) embryos. Although DR2 mutations did not affect the in vitro binding of SPN-4 to nos-2 3′UTR, these results indicate that both the direct repeats are probably critical for the interaction in vivo, where potential competitors are likely present (see below). In contrast, GFP:H2B expression was uniformly stronger in all cells of the embryo when both direct repeats were simultaneously mutated – a pattern strikingly similar to the removal of MEX-3 (Fig. 5C). This observation is remarkably consistent with the EMSA results described in the previous section, in which the double mutant RNA showed considerably weaker interaction with MEX-3 than either of the single mutants. In summary, the removal of MEX-3 and SPN-4 or mutations in the RNA sequence that is essential for their binding both have very similar effects on nos-2 translation. From these results, we conclude MEX-3 and SPN-4 suppress the translation of nos-2 mRNA by directly binding to nos-2 3′UTR.
As described earlier, SubE region is sufficient for OMA-2 interaction. Mutation of this region in the GFP:H2B:nos-2 3′UTR transgene did not express GFP:H2B in any stage during germ cell development (D'Agostino et al., 2006), which is contradictory to the effect of removal of OMA-1 and OMA-2 by RNAi. Since the SubE region contains a potential polyadenylation signal and the cleavage site, we reasoned that mutations that affect this entire region would probably interfere with the core translational machinery, leading to complete absence of translation. To reveal potential subdomains within SubE that might be critical for OMA-2 interaction more specifically, we generated RNA probes containing smaller substitutions (10-bp) of SubE and tested them in EMSA with GST:OMA-2. As shown in Fig. 6B, SubE-Δ27 more severely reduced the mobility shift than the other substitutions. Based on this, we generated a GFP:H2B:nos-2 3′UTR transgene construct carrying SubE-Δ27 substitution and introduced into worms. Quite remarkably, these transgenic worms strongly expressed GFP:H2B in oocytes that showed striking similarity to the expression pattern in oma-1(RNAi) oma-2(RNAi) (Fig. 6C). These results indicate that OMA-1 and OMA-2 suppress nos-2 translation in oocytes by directly binding to the SubE region of nos-2 3′UTR.
Fig. 6.
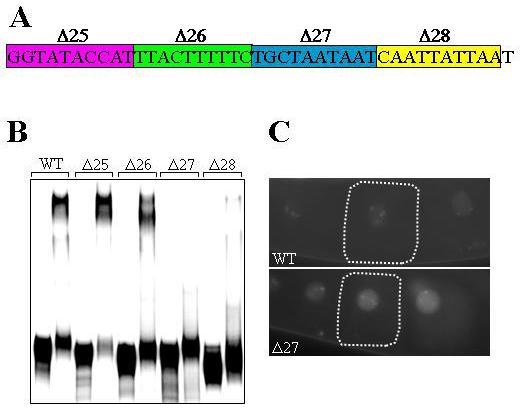
Interaction with nos-2 3′UTR is essential for the translation suppression activity of OMA-2. (A) Sequence of the SubE region. Sequences targeted by substitution analysis in EMSA are boxed and named. (B) Electrophoretic mobility shift by OMA-2 of radiolabeled nos-2 3′UTR bearing the indicated mutations. The first lane in each set is the mobility of RNA in the absence of protein. (C) Expression pattern of GFP:H2B in embryos of transgenic worms carrying the GFP:H2B:nos-2 3′UTR transgene with wild-type sequence (WT) or bearing Δ27 mutation.
POS-1 competes with SPN-4 for binding to nos-2 3′UTR
Two lines of evidence suggest a potential competition between SPN-4 and POS-1 for binding to nos-2 3′UTR. One, RNAi epistasis described earlier indicates that POS-1 acts to relieve the translational repression by SPN-4. Two, both these proteins are present in the germline blastomeres until P4 is born (Tabara et al., 1999; Ogura et al., 2003). Therefore, we decided to test this potential competition more directly. For this, we added both proteins simultaneously to the binding reactions and observed the changes in electrophoretic mobility shift patterns when the relative concentrations of these two proteins were varied. As shown in Fig. 7A, an increase in the POS-1 to SPN-4 ratio decreased the intensity of the band corresponding to the RNA-SPN-4 complex with a concomitant increase in the intensity of RNA-POS-1 complex. If binding of one protein was independent of the other, then a “super shift” resulting from the simultaneous binding of both proteins should have been observed. In contrast, we observed partitioning of RNA between the two proteins in a concentration-dependent manner, indicating that POS-1 and SPN-4 may indeed compete with each other for binding nos-2 3′UTR. Next, we quantified the fluorescence intensities of P3 and P4 cells in embryos immunostained with antibodies against POS-1 and SPN-4, and calculated the POS-1 to SPN-4 ratio in these cells. Significantly, this ratio in P4 was about 9-fold higher than P3 (Fig. 7B and C). Taken together, these results suggest that the higher POS-1 to SPN-4 ratio in P4 enables POS-1 to overcome the nos-2 translation repression by SPN-4.
Fig. 7.
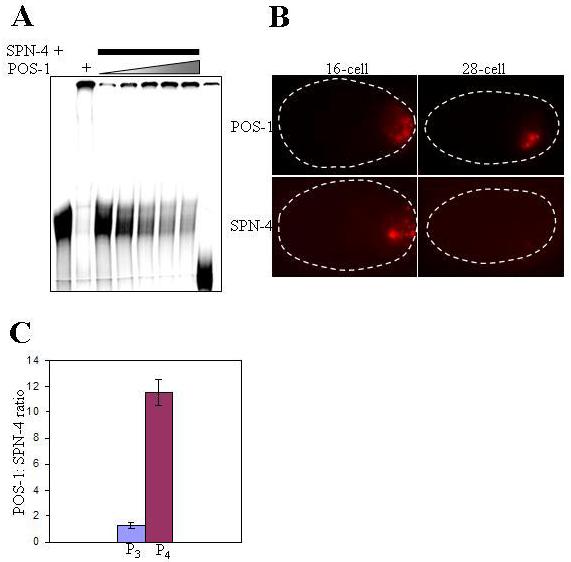
POS-1 competes with SPN-4 for binding to nos-2 3′UTR. (A) Electrophoretic mobility shift of the radiolabeled 200-bp nos-2 3′UTR incubated with GST:SPN-4 alone (lane 1), GST:POS-1 alone (lane 2) or with increasing concentration of GST:POS-1 at a constant concentration of GST:SPN-4 (lanes 3-7). No protein was added to RNA in lane 8. Lanes 1 and 3-7 contain 2 μl of GST:SPN-4 per lane. Amounts of GST:POS-1 in lane 2 are 4 μl and in lanes 3-7 are 2, 4, 6, 8 and 10 μl, respectively. (B) Representative examples of 16- and 28-cell embryos immunostained with anti-SPN-4 and anti-POS-1 antibodies (C) Bar graph showing average POS-1: SPN-4 ratios obtained by quantitation of immunofluorescence signals from 12 embryos for the indicated stages.
Discussion
We have identified four proteins, namely OMA-1, OMA-2, MEX-3 and SPN-4, which suppress nos-2 translation. While OMA-1 and OMA-2 suppress in the oocyte, MEX-3 and SPN-4 suppress in the embryo. Through a combination of genetic and biochemical experiments, we provide evidence that these proteins suppress nos-2 translation by directly interacting with its 3′UTR. Firstly, disruption of their expression by RNAi activates nos-2 translation prematurely. Secondly, these proteins interact specifically with nos-2 3′UTR in vitro. Finally, 3′UTR mutations that abolish in vitro interaction activate translation prematurely in vivo in a pattern that is remarkably similar to the removal of these proteins by RNAi. In addition, our epistatic analysis shows that the CCCH-finger protein POS-1, which is required for nos-2 translation in the germline founder cell P4 (D'Agostino et al., 2006), acts as a derepressor, rather than as an activator, of nos-2 translation. Results of our in vitro binding studies show that POS-1 and SPN-4 compete with each other for binding to the same region of nos-2 3′UTR. Significantly, POS-1: SPN-4 ratio increases about nine fold in P4 over its mother P3. Based on these results, we propose that the translational status of nos-2 in the embryonic germline depends on the relative concentrations of POS-1 and SPN-4. According to our model, POS-1: SPN-4 ratio is below the threshold required for translational activation in earlier stages, which subsequently increases above this threshold in P4, resulting in the activation nos-2 translation.
Translational repression in oocytes
Two closely related CCCH-finger proteins, OMA-1 and OMA-2, are expressed only in the female germline and are enriched in oocytes. Consistent with their expression pattern, these two proteins function in oocyte maturation. Although no direct downstream target has been reported, based on the presence of CCCH-type RNA-binding motifs, they have been proposed to regulate the translation of downstream target mRNAs (Detwiler et al., 2001). Our results show that the nos-2 mRNA is one of their direct targets. Firstly, simultaneous removal of OMA-1 and OMA-2 leads to nos-2 translation in oocytes. Secondly, both proteins interact in vitro with a short region of nos-2 3′UTR in a sequence-specific manner. Finally, a mutation in this region (Δ27) that severely reduces OMA-2 binding activates translation in vivo. In addition to these two proteins, at least one other protein must be involved in translation repression in oocytes, for mutations in a stem-loop at the 5′ region of nos-2 3′UTR abolishes this repression (D'Agostino et al., 2006) and neither of these proteins interact with the stem-loop. Surprisingly, MEX-3 and SPN-4, two other RNA-binding proteins that interact with nos-2 3′UTR and suppress translation in the embryo (see below), are unable to suppress nos-2 translation in the oocyte, although both are present in oocytes (Draper et al., 1996; Ogura et al., 2003).
Translational repression in the early embryo
Absence of GFP:H2B in the somatic blastomeres of transgenic embryos indicates that the translational repression mechanisms must operate in these cells till the 16-cell stage, by which nos-2 mRNA is degraded in these cells. At least four proteins seem to be involved in this repression: depletion of the two nearly identical CCCH-finger proteins MEX-5 and MEX-6 (Schubert et al., 2000; D'Agostino et al., 2006), the KH-domain protein MEX-3 or the RRM protein SPN-4 results in the activation of translation in all cells of the early embryo. Couple of observations indicate that the role of MEX-5 and MEX-6 on nos-2 translation is probably mediated via their effects on the expression of POS-1 and MEX-3: 1) Embryos lacking MEX-5 and MEX-6 express POS-1 ectopically in the anterior cells (Schubert et al., 2000) and this ectopic POS-1 is essential for the misexpression of GFP:H2B observed in these embryos (Schubert et al., 2000; D'Agostino et al., 2006). 2) The level of MEX-3 is significantly lower in embryos lacking MEX-6 (Huang et al., 2002). While MEX-3 directly interacts with nos-2 3′UTR, it is not clear whether MEX-5 or MEX-6 bind nos-2 3′UTR. We have earlier reported that the nos-2 mRNA degradation in the somatic cells is delayed in mex-5(RNAi) mex-6(RNAi) embryos (D'Agostino et al., 2006). A similar delay has been observed in mex-3(RNAi) embryos as well (M. Rana and K. Subramaniam, unpublished observations). In contrast, MEX-3 and SPN-4 appear to play a more direct role in the control of nos-2 translation. These proteins physically interact with nos-2 3′UTR in vitro, and mutations in the 3′UTR that disrupt this interaction show strikingly similar effects on translation in vivo as the RNAi depletion of these proteins, indicating that they probably interact with the 3′UTR in vivo and that this interaction is essential for the translation control of nos-2 mRNA.
At the 2-cell stage, both MEX-3 and SPN-4 appear to be essential to suppress nos-2 translation, for absence of either one leads to activation of GFP:H2B expression. Both these proteins are present in both cells of the 2-cell embryo and they have been shown earlier to physically interact (Huang et al., 2002). Therefore, it is possible they act together to suppress nos-2 translation at the 2-cell stage. However, in later stages, they appear to function independently. For instance, at the 4-cell stage, while MEX-3 is primarily present only in the anterior two cells (Draper et al., 1996), SPN-4 is restricted to the posterior two cells (Ogura et al., 2003). This spatial restriction of MEX-3 appears to restrict its translational repressor activity to the anterior cells, at least in the case of pal-1 mRNA (Huang et al., 2002). In a similar fashion, MEX-3 may repress nos-2 mRNA only in the anterior cells. However, since the accumulation of SPN-4 on P granules of mex-3(RNAi) embryos is significantly reduced (S. Jadhav and K. Subramaniam, unpublished observations), we think MEX-3 may have an indirect role on the repression of nos-2 translation in the posterior cells as well. This might explain the expression of GFP:H2B in all cells of mex-3(RNAi) embryos. Consistent with its spatial distribution pattern, SPN-4 appears to repress nos-2 translation primarily only in the posterior cells. In spn-4(RNAi) embryos, the levels of GFP:H2B was significantly higher in the posterior cells when compared to the anterior cells. Low levels of GFP:H2B seen in the anterior cells probably results from perdurance of the protein produced at the 2-cell stage of these embryos. Thus, these proteins appear to act together at the 2-cell stage and independently at later stages to repress nos-2 translation.
Activation of nos-2 translation in the germline founder cell
Activation of nos-2 translation in the germline founder cell P4 depends on the presence of POS-1: although nos-2 mRNA is present in pos-1(RNAi) embryos until the birth of PGCs, NOS-2 protein is not detected at any stage during embryogenesis (D'Agostino et al., 2006). However, POS-1 is not required for nos-2 translation in the absence of the repressors MEX-3 and SPN-4. Similarly, premature activation of translation caused by a 3′UTR mutation does not require POS-1 (D'Agostino et al., 2006). These observations clearly indicate that POS-1 functions as a derepressor, rather than as an activator, of nos-2 translation. Surprisingly, even though POS-1 protein is continuously present in the P lineage starting from the 2-cell stage (Tabara et al., 1999), it does not activate nos-2 translation until the 28-cell stage. One possible explanation for this is that POS-1 requires an unknown P4-specific factor for its derepressor activity. Alternatively, the ratio of POS-1 concentration to that of a repressor such as SPN-4 may determine the translational status and this ratio in P4 probably tilts in favor of derepression. Our results support the second model: 1) In vitro, both SPN-4 and POS-1 bind to nos-2 3′UTR, and POS-1 competes with SPN-4 for binding to nos-2 3′UTR in a concentration-dependent manner. 2) Quantitation of immunofluorescence signals indicate that POS-1: SPN-4 ratio increases in the P lineage. We propose that the POS-1: SPN-4 ratio increases in P4 above the threshold required for the activation of nos-2 translation. Genetic mutants or other means that alter this ratio will be essential to validate this model.
Translation regulation by MEX-3, SPN-4 and POS-1
All three proteins, MEX-3, SPN-4 and POS-1 regulate the translation of a few other maternal mRNAs. The target mRNAs identified so far are pal-1 – negatively regulated by MEX-3 and SPN4 (Hunter and Kenyon, 1996; Huang et al., 2002); glp-1 – negatively regulated by POS-1 and positively regulated by SPN-4 (Ogura et al., 2003); skn-1 – negatively regulated by SPN-4 (Gomes et al., 2001); and apx-1 – positively regulated by POS-1 (Tabara et al., 1999). In addition, MEX-3 suppresses the translation of rme-2 in the germline stem cells of adult gonad (Ciosk et al., 2004). Of these, the translational regulation by direct binding of the 3′UTR has been demonstrated only in the case of glp-1 mRNA. SPN-4 and POS-1 proteins bind at different sites within glp-1 3′UTR and mediate opposite effects on translation. While SPN-4 binds the temporal control region (TCR) and promotes translation, POS-1 binds the spatial control region (SCR) and suppresses translation. Asymmetric distribution of the two proteins in the 2-cell embryo – SPN-4 is present in both cells, but POS-1 is restricted to the posterior cell – ensures restriction of glp-1 translation to the anterior (Ogura et al., 2003).
Comparison of the translation control of glp-1 and nos-2 mRNAs reveal striking diversity in the translation regulation mediated by these two proteins. Although the relative concentration of SPN-4 and POS-1 is critical for the translation of both these mRNAs, the final outcome is opposite: a higher POS-1: SPN-4 ratio suppresses glp-1(Ogura et al., 2003), but activates nos-2. It is not clear at the moment how they promote translation of one mRNA while inhibiting the translation of another. Some clues emerge from the comparison of the 3′UTR sequences of glp-1 and nos-2. There are some important differences between these two 3′UTRs. Firstly, both SPN-4 and POS-1 bind distinct and relatively short regions of the glp-1 3′UTR. In contrast, they require the entire 200 bp of nos-2 3′UTR for maximal binding. Secondly, the two 8-bp direct repeats, which are critical for SPN-4 binding of nos-2 3′UTR, are not present in the SPN-4 binding element (TCR) of glp-1 3′UTR. Finally, 3′UTRs of the two mRNAs do not share any significant similarity at the sequence or secondary structure level. Based on these observations, we propose the final outcome of translation regulation depends on the type of 3′UTR sequence these proteins bind. Binding of one specific 3′UTR sequence could lead to association with an additional protein factor that might positively influence the translation machinery, while the binding of a different RNA sequence could lead to association with a different protein factor that might negatively influence the translational machinery. Identification of protein partners of SPN-4 and POS-1, and additional target mRNAs with which these two proteins directly interact, will be helpful to test this hypothesis.
Significantly, MEX-3, SPN-4 and POS-1 have been shown to interact among them (Huang et al., 2002; Ogura et al., 2003). In addition, these three proteins and the nos-2 mRNA associate with P granules (Draper et al., 1996; Subramaniam and Seydoux, 1999; Tabara et al., 1999; Ogura et al., 2003). Presently, it is not clear whether these interactions play any role on the translation control of nos-2 or any other mRNA. Experiments focused on determining the importance of these interactions will be an interesting challenge and help us understand the mechanism(s) by which these proteins differently influence the translation of different target mRNAs. Such an understanding will help us explain the role of P granule-like structures present in the germ cells of many organisms.
Translation regulation of nanos gene family members
Members of the nanos gene family are the evolutionarily conserved regulators of germ cell development (Kobayashi et al., 1996; Forbes and Lehmann, 1998; Subramaniam and Seydoux, 1999; Koprunner et al., 2001; Tsuda et al., 2003). In addition to their function, even the basic aspects of the regulation of their expression have been conserved: 1) The Drosophila, C. elegans and zebra fish members are controlled at the translation level by mechanisms that require 3′UTR. 2) In both Drosophila and C. elegans, translation repression in oocytes and embryos are mediated by two distinct regions of the 3′UTR (Forrest et al., 2004; D'Agostino et al., 2006). However, there is at least one major difference between the translation regulation of Drosophila nanos and C. elegans nos-2. The protein factors that control these two mRNAs do not share either sequence or functional (other than the regulation of nanos) similarity. The worm proteins OMA-1 and OMA-2, which bind nos-2 3′UTR and suppress translation in oocytes, are CCCH-type zinc finger proteins and are essential for oocyte maturation (Detwiler et al., 2001). In contrast, the fly protein Glorund, which binds nanos 3′UTR and suppress translation in oocytes, is an hnRNP family protein and does not appear to be essential for oocyte maturation (Kalifa et al., 2006). Similarly, the fly protein Smaug, which represses nanos translation in embryos, is essential for nuclear divisions (Dahanukar et al., 1999). In contrast, MEX-3 and SPN-4, which repress the worm nos-2 in the embryo, do not resemble Smaug at the sequence level and are not involved in cell division. Consistently, the cis-elements of the two 3′UTRs as well do not share sequence similarity. These differences possibly reflect the fundamental difference in the process of embryogenesis in these two species. The fly zygote undergoes a series of nuclear divisions and forms a multinucleate syncytium. During the ensuing cellularization, the first cells to form are the PGCs, known in the fly as pole cells. In contrast, the worm zygote does not form a syncytium. Instead, it undergoes an asymmetric cell division generating a larger anterior cell called AB and a smaller posterior cell called P1. Although P1 inherits the maternally synthesized germ cell components, unlike the fly pole cells, P1 is not a PGC. As mentioned earlier, the P lineage produces one somatic daughter at each of first four rounds of cell division before becoming committed to PGC fate (Fig. 1). Therefore, the developmental contexts in which PGCs arise in these two species are different. Consequently, the RNA-binding proteins available at these different contexts for the translation control of nanos mRNA may not be similar. In addition, at least some of the mechanistic details as well may have diverged. For instance, although Smaug mediates translation repression by blocking translation initiation (Nelson et al., 2004), it also promotes mRNA degradation by recruiting deadenylation complex (Semotok et al., 2005). While such a mechanism may operate in the somatic blastomeres of worm embryo, an additional mechanism that does not involve RNA degradation is essential in the P lineage to suppress translation, for nos-2 mRNA is preserved in this lineage until the birth of PGCs.
Acknowledgements
We would like to thank Julie Ahringer for generously providing 131 RNAi clones; Jim Priess for the anti-POS-1 antibody; and the C. elegans Genetic Consortium for the stains EU769, JJ1014 and JJ462. This work was supported by a Senior Research Fellowship to KS awarded by the Wellcome Trust, London, UK.
References
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton MK, Schedl TB, Kimble JE. Gain-of-function mutations of fem-3, a sex-determination gene in Caenorhabditis elegans. Genetics. 1987;115:107–119. doi: 10.1093/genetics/115.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosk R, DePalma M, Priess JR. ATX-2, the C. elegans ortholog of ataxin 2, functions in translational regulation in the germline. Development. 2004;131:4831–41. doi: 10.1242/dev.01352. [DOI] [PubMed] [Google Scholar]
- D'Agostino I, Merritt C, Chen PL, Seydoux G, Subramaniam K. Translational repression restricts expression of the C. elegans Nanos homolog NOS-2 to the embryonic germline. Dev. Biol. 2006;292:244–252. doi: 10.1016/j.ydbio.2005.11.046. [DOI] [PubMed] [Google Scholar]
- Dahanukar A, Walker JA, Wharton RP. Smaug, a novel RNA-binding protein that operates a translational switch in Drosophila. Mol. Cell. 1999;4:209–18. doi: 10.1016/s1097-2765(00)80368-8. [DOI] [PubMed] [Google Scholar]
- Dean KA, Aggarwal AK, Wharton RP. Translational repressors in Drosophila. Trends Genet. 2002;18:572–7. doi: 10.1016/s0168-9525(02)02792-0. [DOI] [PubMed] [Google Scholar]
- Detwiler MR, Reuben M, Li X, Rogers E, Lin R. Two zinc finger proteins, OMA-1 and OMA-2, are redundantly required for oocyte maturation in C. elegans. Dev. Cell. 2001;1:187–99. doi: 10.1016/s1534-5807(01)00026-0. [DOI] [PubMed] [Google Scholar]
- Draper BW, Mello CC, Bowerman B, Hardin J, Priess JR. MEX-3 is a KH domain protein that regulates blastomere identity in early C. elegans embryos. Cell. 1996;87:205–16. doi: 10.1016/s0092-8674(00)81339-2. [DOI] [PubMed] [Google Scholar]
- Evans TC, Hunter CP. Translational control of maternal RNAs. In: Wormbook, editor. The C. elegans Research Community, Wormbook. 2005. doi/10.1895/wormbook.1.3.4.1, http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes A, Lehmann R. Nanos and Pumilio have critical roles in the development and function of Drosophila germline stem cells. Development. 1998;125:679–690. doi: 10.1242/dev.125.4.679. [DOI] [PubMed] [Google Scholar]
- Forrest KM, Clark IE, Jain RA, Gavis ER. Temporal complexity within a translational control element in the nanos mRNA. Development. 2004;131:5849–57. doi: 10.1242/dev.01460. [DOI] [PubMed] [Google Scholar]
- Gomes JE, Encalada SE, Swan KA, Shelton CA, Carter JC, Bowerman B. The maternal gene spn-4 encodes a predicted RRM protein required for mitotic spindle orientation and cell fate patterning in early C. elegans embryos. Development. 2001;128:4301–14. doi: 10.1242/dev.128.21.4301. [DOI] [PubMed] [Google Scholar]
- Gunkel N, Yano T, Markussen FH, Olsen LC, Ephrussi A. Localization-dependent translation requires a functional interaction between the 5′ and 3′ ends of oskar mRNA. Genes Dev. 1998;12:1652–64. doi: 10.1101/gad.12.11.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang NN, Mootz DE, Walhout AJ, Vidal M, Hunter CP. MEX-3 interacting proteins link cell polarity to asymmetric gene expression in Caenorhabditis elegans. Development. 2002;129:747–59. doi: 10.1242/dev.129.3.747. [DOI] [PubMed] [Google Scholar]
- Hunter CP, Kenyon C. Spatial and temporal controls target pal-1 blastomere-specification activity to a single blastomere lineage in C. elegans embryos. Cell. 1996;87:217–26. doi: 10.1016/s0092-8674(00)81340-9. [DOI] [PubMed] [Google Scholar]
- Kalifa Y, Huang T, Rosen LN, Chatterjee S, Gavis ER. Glorund, a Drosophila hnRNP F/H homolog, is an ovarian repressor of nanos translation. Dev. Cell. 2006;10:291–301. doi: 10.1016/j.devcel.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Yamada M, Asaoka M, Kitamura T. Essential role for the posterior morphogen nanos for germline development in Drosophila. Nature. 1996;380:708–711. doi: 10.1038/380708a0. [DOI] [PubMed] [Google Scholar]
- Koprunner M, Thisse C, Thisse B, Raz E. A zebrafish nanos-related gene is essential for the development of primordial germ cells. Genes Dev. 2001;15:2877–85. doi: 10.1101/gad.212401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuersten S, Goodwin EB. The power of the 3′ UTR: translational control and development. Nat. Rev. Genet. 2003;4:626–37. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- Macdonald PM, Smibert CA. Translational regulation of maternal mRNAs. Curr. Opin. Genet. Dev. 1996;6:403–7. doi: 10.1016/s0959-437x(96)80060-8. [DOI] [PubMed] [Google Scholar]
- Nelson MR, Leidal AM, Smibert CA. Drosophila Cup is an eIF4E-binding protein that functions in Smaug-mediated translational repression. Embo J. 2004;23:150–9. doi: 10.1038/sj.emboj.7600026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K, Kishimoto N, Mitani S, Gengyo-Ando K, Kohara Y. Translational control of maternal glp-1 mRNA by POS-1 and its interacting protein SPN-4 in Caenorhabditis elegans. Development. 2003;130:2495–503. doi: 10.1242/dev.00469. [DOI] [PubMed] [Google Scholar]
- Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–26. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor. New York: Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning, a Laboratory Manual. [Google Scholar]
- Schubert CM, Lin R, de Vries CJ, Plasterk RH, Priess JR. MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol. Cell. 2000;5:671–82. doi: 10.1016/s1097-2765(00)80246-4. [DOI] [PubMed] [Google Scholar]
- Semotok JL, Cooperstock RL, Pinder BD, Vari HK, Lipshitz HD, Smibert CA. Smaug recruits the CCR4/POP2/NOT deadenylase complex to trigger maternal transcript localization in the early Drosophila embryo. Curr. Biol. 2005;15:284–94. doi: 10.1016/j.cub.2005.01.048. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Strome S. Launching the germline in Caenorhabditis elegans: regulation of gene expression in early germ cells. Development. 1999;126:3275–83. doi: 10.1242/dev.126.15.3275. [DOI] [PubMed] [Google Scholar]
- Smibert CA, Lie YS, Shillinglaw W, Henzel WJ, Macdonald PM. Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro. RNA. 1999;5:1535–47. doi: 10.1017/s1355838299991392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Wharton RP. Recruitment of Nanos to hunchback mRNA by Pumilio. Genes Dev. 1999;13:2704–12. doi: 10.1101/gad.13.20.2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Powers J, Dunn M, Reese K, Malone CJ, White J, Seydoux G, Saxton W. Spindle dynamics and the role of gamma-tubulin in early Caenorhabditis elegans embryos. Mol. Biol. Cell. 2001;12:1751–64. doi: 10.1091/mbc.12.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam K, Seydoux G. nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development. 1999;126:4861–4871. doi: 10.1242/dev.126.21.4861. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Tabara H, Hill RJ, Mello CC, Priess JR, Kohara Y. pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans. Development. 1999;126:1–11. doi: 10.1242/dev.126.1.1. [DOI] [PubMed] [Google Scholar]
- Tenenhaus C, Subramaniam K, Dunn MA, Seydoux G. PIE-1 is a bifunctional protein that regulates maternal and zygotic gene expression in the embryonic germ line of Caenorhabditis elegans. Genes Dev. 2001;15:1031–40. doi: 10.1101/gad.876201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–12. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, Kobayashi S, Saga Y. Conserved role of Nanos proteins in germ cell development. Science. 2003;301:1239–41. doi: 10.1126/science.1085222. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]