

Abstract
Karenia brevis is a toxic marine dinoflagellate endemic to the Gulf of Mexico. Blooms of this harmful alga cause fish kills, marine mammal mortalities and neurotoxic shellfish poisonings. These harmful effects are attributed to a suite of polyketide secondary metabolites known as the brevetoxins. The carbon framework of all polyketides is assembled by a polyketide synthase (PKS). Previously, PKS encoding genes were amplified from K. brevis culture and their similarity to a PKS gene from the closely related protist, Cryptosporidium parvum, suggested that these genes originate from the dinoflagellate. However, K. brevis has not been grown axenically. The associated bacteria might be the source of the toxins or the PKS genes. Herein we report the localization of PKS encoding genes by a combination of flow cytometry/PCR and fluorescence in situ hybridization (FISH). Two genes localized exclusively to K. brevis cells while a third localized to both K. brevis and associated bacteria. While these genes have not yet been linked to toxin production, the work describes the first definitive evidence of resident PKS genes in any dinoflagellate.
Keywords: Karenia brevis, Brevetoxin, Polyketide synthase, Dinoflagellate, Fluorescence in situ hybridization
1. Introduction
The planktonic marine dinoflagellate Karenia brevis blooms annually in the Gulf of Mexico and is most prevalent along the west coast of Florida. Historically, blooms have occurred primarily during the fall and winter months. However, over recent years the Florida red tide specifically and HABs (harmful algal blooms) in general appear to be more frequent, persistent and widespread (Chretiennot-Dinet, 2001; Hallegraeff, 1993). Associated with these blooms are massive fish kills, marine mammal mortalities, human poisonings due to the consumption of tainted shellfish and complaints of respiratory irritations among beach-goers (Kirkpatrick et al., 2004; Van Dolah et al., 2002). These effects are caused by the neurotoxic brevetoxins. The brevetoxins are a suite of polyether ladder type compounds which have two parent backbone structures, brevetoxin A and brevetoxin B, each with several side chain variants (Fig. 1). Brevetoxins act at the voltage-gated sodium channel by prolonging and lowering the threshold for channel opening, resulting in a dose-dependant depolarization of excitable membranes (Jeglitsch et al., 1998). The brevetoxins are representative of a larger family of dinoflagellate-derived polyketide toxins that pose a threat to human health through the consumption of tainted seafood. These include ciguatoxin, okadaic acid and the related dinophysistoxins, pectenotoxins, yessotoxin and the azaspiracids, to name just a few.
Fig. 1.
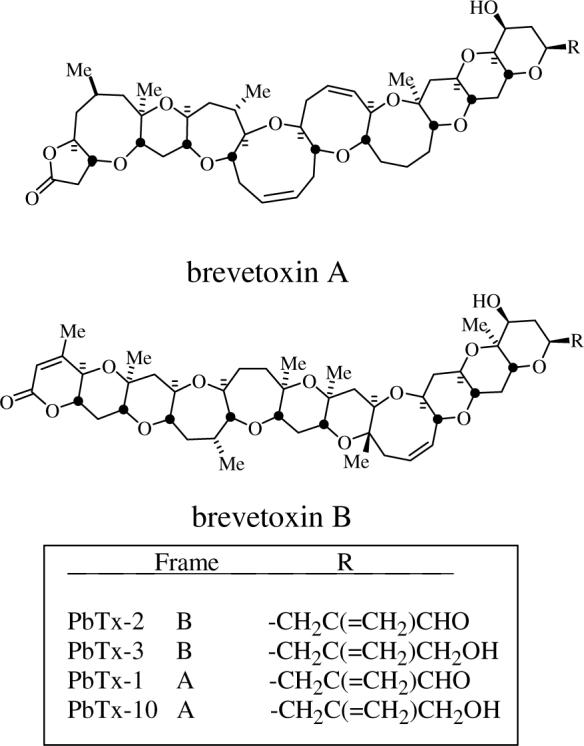
Structures of the polyether brevetoxins.
Brevetoxin production may not be exclusive to K. brevis. Several raphidophytes, including Chatonella anti-qua, Chatonella marina, Fibrocapsa japonica and Hetero-sigma akashiwo have all been reported to produce brevetoxin B in culture (Khan et al., 1995, 1996a,b, 1997). Furthermore, brevetoxins have been isolated from a fish-killing bloom in which the predominant organism was Chatonella verruculosa (Bourdelais et al., 2002). Raphidophytes are unarmored, photosynthetic marine eukaryotes which belong to the Stramenopiles, and are phylogenetically distinct from dinoflagellates, which fall within the Alveolates. The biogenic origin of the brevetoxins was established by two groups independently using stable isotope incorporation experiments (Chou and Shimizu, 1987; Lee et al., 1986, 1989). The head-to-tail incorporation of acetate units confirmed the polyketide origins of these compounds.
Over the past decade, numerous polyketide biosynthetic pathways have been cloned and expressed in heterologous hosts, offering unique opportunities for overexpression and/or manipulation (Pfeifer and Khosla, 2001). Three fundamental types of polyketide synthases have emerged (Shen, 2003). Type I are large multifunctional enzymes having several functional domains located within a single protein. Type II PKSs are multiprotein complexes of several individual enzymes found only in bacteria and typically produce aromatic polyketides. Type III PKSs are found only in plants, utilize unusual starter units and act on acyl Co-A thioesters. The complex structures of dinoflagellate-derived polyketides suggest that they are produced by type I modular synthases.
However, no biosynthetic pathway for a secondary metabolite from a dinoflagellate has been identified at the genomic level. The numerous challenges associated with molecular genetic studies of dinoflagellates (Plum-ley, 1997) no doubt serve as a deterrent to many researchers. Particularly daunting is the extraordinary size of the dinoflagellate genome (Rizzo et al., 1982). Furthermore, many polyketide toxin-producing dinoflagellates have not been maintained in culture axenically, raising the question of whether the ultimate origins of these secondary metabolites are dinoflagellates or their associated bacteria. A bacterial origin for dinoflagellate polyketide toxins has been the subject of much speculation (Rausch de Traubenberg and Lassus, 1991). However, no polyketide toxin-producing bacteria have been isolated from a dinoflagellate to date.
Recently, we reported the amplification of type I, but not type II, polyketide synthase genes from non-axenic cultures of marine dinoflagellates (Snyder et al., 2003). Using a degenerate primer set for the β-ketosynthase domain of type I PKS encoding genes, a product of the anticipated size was amplified by reverse transcriptase PCR (RT-PCR) from the Wilson strain of K. brevis. Four of the fifteen sequenced amplicons (AS1−1L, AT1−6L, AT2−10L and AT2−15) were PKS related. The amino acid sequences were subjected to a phylogenetic analysis, and three (AT1−6L, AT2−10L and AT2−15) branched into a clade with PKS encoding genes from the related protist Cryptosporidium parvum, a member of the Coccidia which is a sister clade to the Dinophyceae within the Alveolata (Zhu et al., 2002).
In part based on this analysis and the apparent methylation of the DNA, we surmised that these three sequences originated from K. brevis rather than from associated bacteria. Herein we report that based on PCR screening of dinoflagellate and bacterial fractions as well as fluorescence in situ hybridization, two of the four PKS amplicons localize exclusively to K. brevis, while a third localizes to K. brevis and a small population of the associated bacteria.
2. Results and discussion
2.1. Amplification of PKS genes from other K. brevis strains with specific primers
If the amplified PKS encoding genes were derived from K. brevis, presumably they should be detectable in all available isolates of the dinoflagellate. In addition, the bacterial consortia present in these isolates of K. brevis were analyzed by DGGE of the 16S rRNA gene to verify the variation of the bacterial population present in isolates derived from distinct geographical and temporal origins. The results of the DGGE analysis are shown in Fig. 2. The cultures CCMP2281 and JR1 are the same isolate and both are reportedly axenic. While the DGGE indicates that JR1 possesses a significantly less complex consortia of associated bacteria, the claim of axenicity should be viewed with some caution. We have in fact amplified and sequenced bacterial 16S ribosomal genes from CCMP2281. The DGGE analysis of JR1 indicates the presence of one major and two minor bands. The major band is also present in JR and CCMP2228, but is absent from CCMP2229 and CCMP718. CCMP2229 and CCMP718 both have one predominant associated bacteria (which are not the same). These bacteria are not present in CCMP2228, JR and JR1. Thus, while a few individual strains do share some common bands, there is not a single band that is common to all strains (Fig. 2). We therefore sought first to establish the presence of these PKS genes in all available isolates of K. brevis and their absence in associated bacteria or other marine protists. Six different cultures (or five distinct isolates) of K. brevis were examined by PCR or RT-PCR for the presence and expression of PKS genes. All strains tested positive for the presence of brevetoxin by ELISA (Naar et al., 2002). In addition to K. brevis isolates, flow sorted bacterial consortia from K. brevis CCMP718 and CCMP2229, five unique heterotrophic bacterial isolates from CCMP718 (data not shown) and a collection of 10 other species of marine protist, were screened by PCR using genomic templates.
Fig. 2.
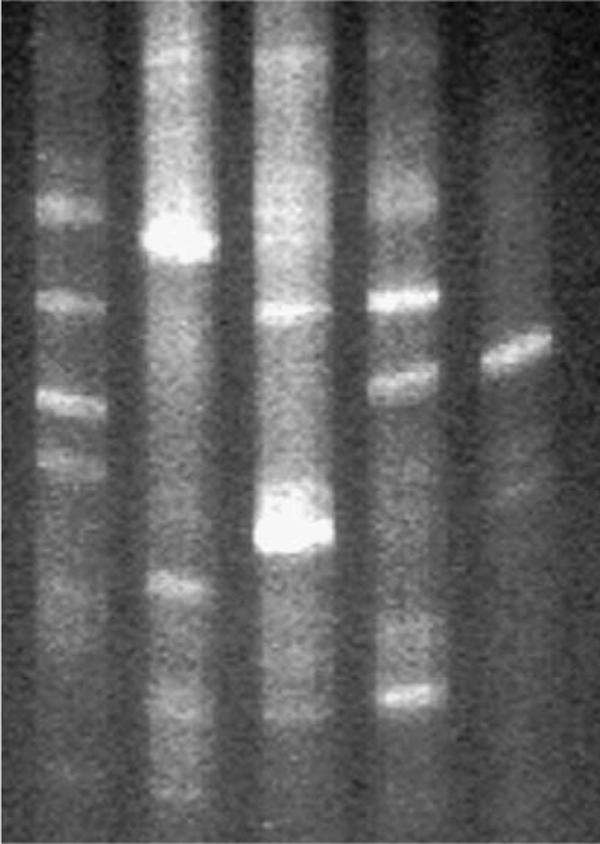
DGGE analysis of 16S ribosomal gene amplified from different strains of K. brevis. The order of the lanes are (from left to right) CCMP2228, CCMP2229, CCMP 718, JR, JR-1 (see Section 4 for experimental details).
Two sets of sequence-specific primers were designed from each of the four PKS related amplicons (1L a/b, 10L a/b, 6L a/b, 15 a/b, Table 1). Both sets of primers designed for AT2−10L yielded products of the expected size and Tm from all five isolates of K. brevis tested (CCMP718, 2228, 2229, 2281, JR and NOAA1) either by PCR using a genomic DNA template or by RTPCR (Table 2). One primer set, specific for sequence AT2−15 (15a), failed to yield a product from CCMP718 or CCMP2228 using a genomic template or from CCMP718 cDNA. Nonetheless, a product of the expected size was amplified from all strains tested using the second AT2−15 primer set (15b) with both the genomic DNA or cDNA templates. Using a genomic DNA template, products were obtained from CCMP2228, JR and NOAA1, and primer set 6La. No product was obtained using CCMP718 or 2281 even though 2281 and JR are ostensibly the same strain and the amplicon was originally derived from CCMP718 using degenerate PKS primers. No product was obtained using the 6Lb primer set and genomic DNA. Surprisingly, these two primer sets (6La and 6Lb) yielded products of the anticipated size in all RT-PCRs with the single exception of CCMP2228 and primer set 6La. Neither primer set targeting AS1−1L yielded a product from either RT-PCR or genomic PCR, using any of the five K. brevis isolates including the Wilson strain (data not shown). This suggested to us that the original product (AS1−1L) was amplified from a bacterial contaminant, which is consistent with the placement of this sequence in the phylogenetic analysis (Snyder et al., 2003). The identities of the products amplified from genomic DNA were confirmed either by sequencing (indicated in Table 2) or were checked against the product amplified from a recombinant plasmid, which carried the target sequence, for identical restriction patterns using the enzymes HincII and SphI (AT2−10L), HincII (AT2−15) or SphI (AT1−6L). Each restriction site was predicted to occur only once in the amplified product. Melting curves of products amplified from cDNA were compared to those products amplified from the corresponding recombinant plasmid. Restriction patterns of all amplified products were consistent with the expected product with the following exception. Digestion of the products from the 10La primer set using HincII revealed two distinct banding patterns. Products amplified from CCMP2228, 2281 and NOAA1 gave the predicted band at ∼275 bp when digested with HincII which was also consistent with the positive control amplified from the recombinant plasmid. Products amplified from CCMP718, 2229 and JR each gave smaller bands at ∼200 bp. Sequencing of a representative for each of the 10La products and subsequent sequence analysis revealed a C → T point mutation which resulted in a second HincII digestion site in the CCMP718, 2229 and JR group, located ∼70 bp downstream of the previously identified HincII site.
Table 1.
Sequence-specific PKS primers
| Primer set | Forward primer-3′ position | Reverse primer—3′ position | 5′ → 3′ sequence | Product size (bp) | Tm (°C) |
|---|---|---|---|---|---|
| 1La | AS11L-300 | CAGCTTGTTCATCTTCACTTGTTGCCACTCAT | 66 | ||
| AS11L-471 | TTCACCGCGCACATACCCATCTG | 203 | 63 | ||
| 1Lb | AS11L-357 | CGACTGCGCAATTGCTGGTGGTGTA | 66 | ||
| AS11L-504 | TTGCAGATTCGAAAGGATGGTGAC | 172 | 59 | ||
| 10La | AT210L-214 | GGACAGGCAACCACCCATCAATCT | 62 | ||
| AT210L-540 | GCCGCGCAACGTACAAGTCACA | 349 | 62 | ||
| 10Lb | AT210L-302 | CGATTGGCAACGCAGAACTCA | 58 | ||
| AT210L-471 | CAAGCGCCATGGCACAAACAC | 190 | 60 | ||
| 15a | AT215−42 | ACCCGCAGCAACGTCTCCTCCTC | 64 | ||
| AT215−410 | GGTGACAGCATGCCAGCCTTACAA | 391 | 62 | ||
| 15b | AT215−87 | GAATGCTGCTGGTGTCCTCAAGAAG | 60 | ||
| AT215−308 | GCCAAGCGCACAGCAACAAGACC | 246 | 64 | ||
| 6La | AT16L-135 | CGTTGGCCAAGACAAGTGCG | 59 | ||
| AT16L-236 | CAGCCCGAGTGCATACGAGAT | 121 | 56 | ||
| 6Lb | AT16L-263 | GGCCGAGCTGCACAATGGATAC | 60 | ||
| AT16L-531 | AGAACCGCCAAGACTCACTAATG | 290 | 54 |
Table 2.
PKS PCR survey with K. brevis strains, other dinoflagellate, haptophyte, raphidophyte and bacterial templates
| Microorganism | Primer set |
|||||
|---|---|---|---|---|---|---|
| 10La | 10Lb | 15a | 15b | 6La | 6Lb | |
| K. brevis 718 (Wilson strain) | +*/+ | +/+ | −/− | +/+ | −/+ | −/+ |
| K. brevis 2228 | +*/+ | +/+ | −/+ | +/+ | +/− | −/+ |
| K. brevis 2229 | +/+ | +/+ | +/+ | +/+ | −/+ | −/+ |
| K. brevis 2281 (JR1) | +/+ | +/+ | +/+ | +*/+ | −/+ | −/+ |
| K. brevis JR | +/+ | +/+ | +/+ | +/+ | +/+* | −/+ |
| K. brevis NOAA1 | + | +/+ | + | + | + | −/+ |
| A. cartarae 1314 | −/− | −/− | −/− | −/− | +* | −/− |
| A. operculatum 120 | −/− | −/− | −/− | −/− | − | −/− |
| C. subsalsa 217 | −/− | −/− | −/− | −/− | − | −/− |
| H. akashiwo 1870 | −/− | −/− | −/− | −/− | − | −/− |
| P. hoffmannianum 683 | −/− | −/− | −/− | −/− | − | −/− |
| P. lima (Baden) | −/− | −/− | −/− | −/− | − | −/− |
| P. micans 1591 | −/− | −/− | −/− | −/− | − | −/− |
| P. parvum (Yasumoto) | −/− | −/− | −/− | −/− | − | −/− |
| Prymnesium sp. 1212 | −/− | −/− | −/− | −/− | − | −/− |
| Symbiodinium sp. 831 | −/− | −/− | −/− | −/− | − | −/− |
| K. brevis 718 sorted bacteria | − | − | − | − | +* | + |
| K. brevis 2229 sorted bacteria | − | − | − | − | + | + |
For each primer set, the results are shown as PCR (genomic template) or PCR(genomic)/RTPCR.
Sequenced.
2.2. Screening for PKS genes from other marine eukaryotes with specific primers
Sequence-specific primers based on each of the four sequences AS1−1L, AT1−6L, AT2−10L and AT2−15 were tested against cDNA and genomic DNA from a variety of dinoflagellates, raphidophytes and haptophytes (Table 2). The primer sets based on the AT2−10L and AT2−15 sequences failed to yield a product from any organism other than K. brevis, either from DNA or cDNA. However, the primer set (6La) based on AT1−6L consistently yielded a product of the anticipated size from Amphidinium carterae. Sequencing indicated that this product matched AT1−6L exactly.
2.3. Flow sorting PCR and DGGE
K. brevis strains (CCMP718 and CCMP2229) were subjected to flow cytometric cell sorting to separate dinoflagellate cells from associated bacteria. Multi-parametric sort gate logic (Fig. 3) based on a combination of forward scatter, side scatter, pulse width and chlorophyll autofluorescence, was used to discriminate the bacterial and dinoflagellate populations from each other. Each targeted population (bacteria or dinoflagellate) was aseptically collected in sterile microfuge tubes for nucleic acid extraction and analysis and brevetoxin ELISA.
Fig. 3.
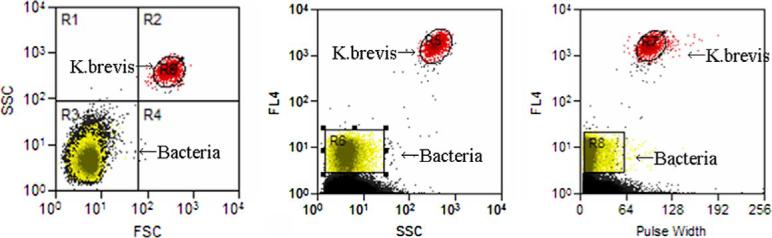
Flow cytometric cell sorting gate logic for separation of bacterial and 6 dinoflagellate populations from a culture of Karenia brevis CCMP718. Dinoflagellate population (red) sorted by gates R9 + R5 + R7, and bacterial population (yellow) sorted by gates R3 + R6 + R8. SSC = side scatter, FSC = forward scatter, FL4 = Chlorophyll autofluorescence detection by 670/30 nm 670/30 nm bandpass. All excitation at 488 nm.
DNA was extracted from pre-sorted culture, as well as the dinoflagellate and bacterial fractions, and subsequently used in PCRs using degenerate PKS, and universal 18S rRNA, and 16S rRNA primer sets (Fig. 4A). The presorted material and the sorted dinoflagellate fraction yielded all three of the expected products, indicating that the bacteria were not successfully removed from K. brevis. However, the bacterial fraction yielded only PKS and 16S products, and failed to yield an 18S product. Therefore, we were able to obtain dinoflagellate-free bacteria. To ensure that the sorted bacteria were representative of the bacterial consortium associated with K. brevis, DGGE analysis was performed on the 16S ribosomal gene products amplified from CCMP718. While there are differences in relative intensities of the bands, the sorted dinoflagellate fraction and the sorted bacterial fraction appear to have the same number of bands. (Fig. 4B). In an effort to identify previously amplified PKS genes, the PKS product amplified from the bacterial fraction from CCMP718 using degenerate primers was cloned and 40 inserts were sorted into four groups by RFLP analysis using seven restriction enzymes (KpnI, BamHI, XbaI, XhoI, HpaII, RsaI and SphI). A representative from each group (K. brevis: bac(3), bac(23), bac(28) and bac(30)) was sequenced and none of the sequences was found to match any of those previously amplified, including AT1−6L. However, one of the bacterial sequences K brev_bac(30), clustered with AT1−6L, AT2−10L and AT2−15 in the phylogenetic tree (vide infra). Dinoflagellate and bacterial fractions were analyzed by brevetoxin ELISA. While the culture was strongly positive for brevetoxins, the bacterial fraction was negative and the dinoflagellate fraction tested weakly positive, but below the limit of quantitation (Fig. 5A). This suggested that brevetoxin is mostly present in the bulk media. Subsequent analysis of culture media from which K. brevis cells were gravity filtered supported this assumption (Fig. 5B).
Fig. 4.
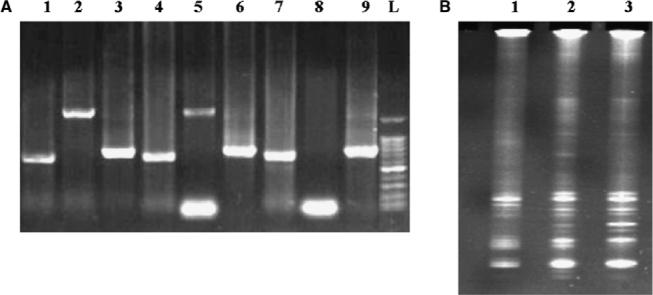
PCR screening of flow-sorted bacterial and dinoflagellate fractions. (A) Screening for the presence of PKS, 18S rRNA and 16S rRNA genes from CCMP718. Products are in numerical order corresponding to PKS, 18S and 16S, respectively, for each of the following groups: 1−3 presorted K. brevis, 4−6 K. brevis fraction and 7−9 bacterial fraction. L refers to a 100 bp ladder (Promega). (B) DGGE analysis of 16S rRNA gene amplified from individual fractions. Lane 1, presorted K. brevis. Lane 2, K. brevis sorted fraction. Lane 3, sorted bacterial fraction.
Fig. 5.

(A) ■ sorted KB cells;  sorted bacteria; □ whole KB culture,
sorted bacteria; □ whole KB culture,  PbTx-3 standards. (B) □ KB filtrate,
PbTx-3 standards. (B) □ KB filtrate,  KB filtered cells,
KB filtered cells,  PbTx-3 standards. Numbers below the bars represent the number of K. brevis cells extracted, or an equivalent volume of culture.
PbTx-3 standards. Numbers below the bars represent the number of K. brevis cells extracted, or an equivalent volume of culture.
Sequence-specific primers for PKS sequences AS1−1L, AT1−6L, AT2−10L and AT2−15 were tested against the sorted bacterial fractions from CCMP2229 and CCMP718 (Table 2). Although AT2−10L and AT2−15 were amplified from the total genomic DNA and genomic DNA from the sorted dinoflagellate fraction, neither was amplified from the bacterial fractions. One primer set (6La) yielded a product from the sorted bacterial fractions from both isolates of K. brevis. The 121-bp product from each strain was sequenced and found to match the AT1−6L sequence exactly.
The consistent amplification of the two PKS gene fragments (AT2−10L and AT2−15) from all strains of K. brevis and the absence of product from any other source, including flow sorted bacteria, bacterial isolates or any other protist, indicated that these two genes are clearly exclusive to K. brevis. However, the situation with AT1−6L appeared to be somewhat ambiguous. The sporadic amplification fragment from total genomic DNA from K. brevis culture, its presence in flow sorted bacteria and at least one other dinoflagellate, suggested that this sequence is derived from a bacterium which may be associated with marine eukaryotes. The amplification of this gene fragment from flow sorted bacteria from CCMP718 and CCMP2229 was somewhat surprising since this primer set failed to yield a product from these two strains when using total genomic DNA as the template, although cDNA from most isolates did yield a product. We speculate that flow sorting the bacterial consortium or the use of a cDNA template yields similar, positive result because each provides a template enriched in the target sequence when compared to total genomic DNA, as sorting removes dinoflagellate DNA and using a cDNA template eliminates unexpressed genes or structural DNA.
2.4. Phylogenetic analysis
Sequence and phylogenetic analysis was performed in order to infer a proper context for both dinoflagellate and the bacterial PKS/FAS-like gene sequences. Overall, although FAS sequences tend to show slightly higher similarities, KS sequences, even at the amino acid level, exhibit high sequence divergences (range = 0.04−0.86) which make definitive phylogenetic reconstructions tenuous using any algorithm (Kong et al., 2004; Lopez, 2003). Nevertheless, the amino acid based distance KS/FAS phylogeny in Fig. 6 yielded several expected groupings and had the same basic topology as the MP tree. For example, bacterial and eukaryotic PUFA (polyunsaturated fatty acid) PKS sequences (Metz et al., 2001) retrieved from GenBank, often formed a consistent clade with relatively high bootstrap support, and exhibited high divergences with dinoflagellate PKS sequences. Confirmed KS sequences from terrestrial sources were included as another reference and showed high bootstrap support for their own grouping. Other anonymous marine bacterial or fungal FAS sequences (e.g. K155, K618 and JL085) designated as outgroups appeared distinct from both PUFA and the K. brevis sequences within the single, major KS clade (which had 100% bootstrap support).
Fig. 6.
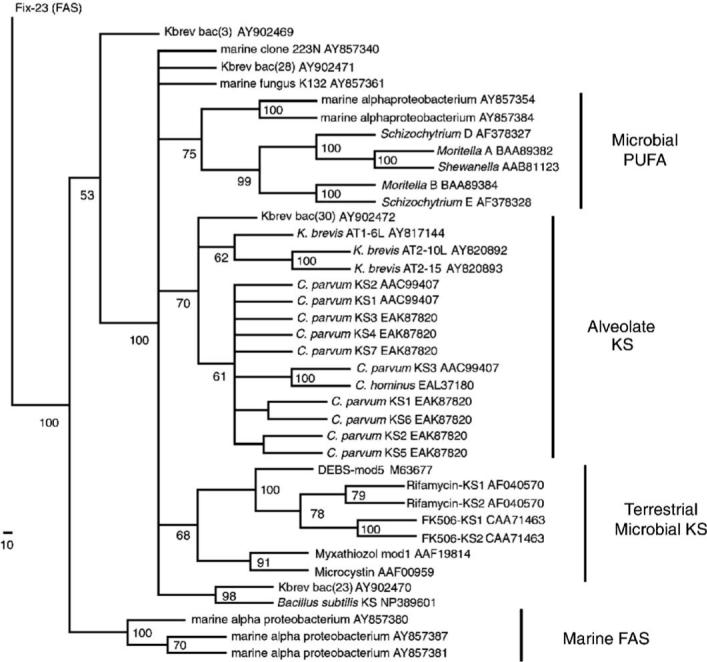
Minimum evolution distance phylogeny of KS and FAS sequences generated with the neighbor-joining algorithm and mean pairwise amino acid distances. A total number of 350 amino acid residues from 39 PKS or FAS sequences were analyzed in PAUP* for this reconstruction. Only reference sequences are shown with their corresponding GenBank accession number. 500 bootstrap replicates were performed with the final bootstrap percentages (>50%) shown at the nodes, with well-supported clades indicated by name. K132, clone 223N, Kbrev_bac(28), Kbrev_bac(23) and Bacillus sequences are part of the major KS clade. The longest branch length of 0.34 was between the bifurcating node and 19E-Schizochytrium. The minimum evolution score of this tree was 8.46769, while the corresponding MP tree had a consistency index of 0.5845. The scale bar in the left corner represents the number of amino acid residue changes along each branch.
Phylogenetic analysis indicated that alveolate KS sequences from C. parvum, Cryptosporidium hominus and K. brevis formed a single, moderately supported group in spite of relatively high mean pairwise distances. However, maximum parsimony analysis yielded a similar topology, and bootstrap support for this alveolate clade increased to about 95% when the Kbrev_bac(30) sequence was omitted from the analysis.
Also similar to previous results (Snyder et al., 2003), K. brevis AT2−10L and AT2−15 exhibited high sequence similarity (0.755) to the exclusion of the more distant AT1−6L KS sequence. However, there were interesting differences in the KS active site motif even among these sequences: AT2−15 (CSGGL), AT2−10 (CSSGL) and AT1−6 (CSSSL). Furthermore, AT1−6L did not show any strong affnity for any bacterial sequences, in spite of its amplification from sorted bacterial fractions. As a side note, the C. parvum sequences used here (AAC99407) had been originally designated as “FASs” (Zhu et al., 2000), but the current results indicate that these may be more closely related to PKS than FAS.
While K. brevis AT2−10L and AT2−15 KS exhibited high similarity with each other and to the C. parvum sequences to a lesser extent, the novel K. brevis associated bacterial sequences (Kbrev_bac) also displayed interesting results by their dispersion in several different parts of the tree. For example, Kbrev_bac(23) showed affnity with a Bacillus subtilis KS fragment (0.49 mean divergence), and Kbrev_bac(3) appeared to outgroup with FAS sequences. The sequence from Kbrev_bac(28) did not show clear affnity with any distinct KS clades, but rather high identity (∼0.97) to an unidentified Prorocentrum mexicanum symbiont (Mofft and Neilan, 2003) after a BLASTP search. Although bacterial Kbrev_ bac(30) clustered with the three K. brevis sequences in several reconstructions, it did not show exceptionally high amino acid sequence conservation to any K. brevis, may have contributed to the overall lower bootstrap percentage (70%) for the alveolate KS group, and was only similar with some C. parvum sequences by having an alanine residue in the KS active site. Furthermore, a separate analysis of DNA base composition, another criteria for analyzing possible “horizontal gene transfers” (McInerney, 1998), did not yield unequivocal evidence of recent PKS gene transfers between K. brevis and its bacterial associates, such as Kbrev_bac(30) (Lopez, 2003).
2.5. Fluorescence in situ hybridization
To cross-validate the PCR results, FISH was used to detect the presence of specific AT1−6L or AT2−10L PKS target sequences in individual intact dinoflagellate and bacteria cells from the K. brevis culture CCMP718 (Fig. 7). The fluorochrome CY3 was chosen as the fluorescent label for the oligonucleotide probes, for a number of reasons, in spite of the fact that the emission signature for this dye is similar to the autofluorescence signature of chlorophyll. CY3 has been successfully used for FISH protocols with other algae, including dinoflagellates (Yokouchi et al., 2003). It is relatively easy to decolorize cells and reduce the background levels of chlorophyll autofluorescence to a degree that does not interfere with visualization of the probe. However, our primary reason for the choice of CY3 is that after fixation with paraformaldehyde (or other fixatives such as glutaraldehyde, etc.), Karenia cells showed a very strong green autofluorescence throughout the cell when illuminated with blue light and a strong blue autofluorescence with UV excitation. Our attempts to decolorize this autofluorescence were unsuccessful and it masks the emission by all the common 488 nm excitation fluors we have tried, including FAM, FITC, and AlexaFluor-488; as well as most common UV probe fluorochromes.
Fig. 7.
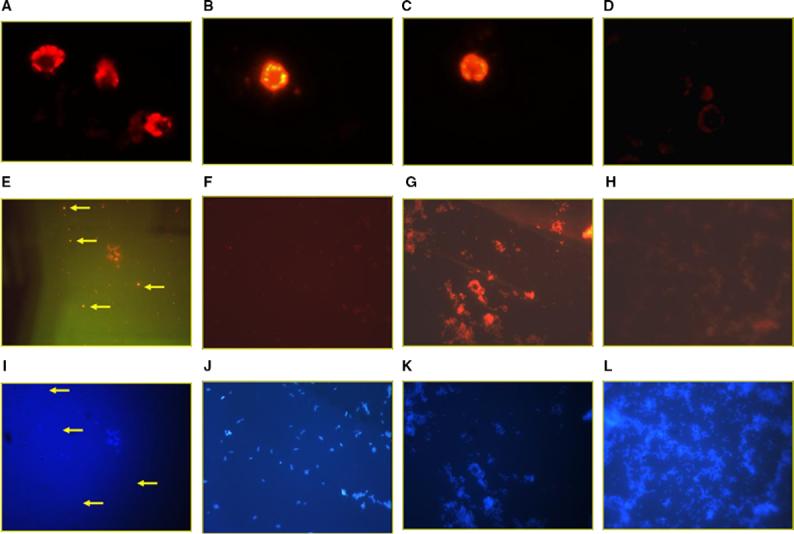
Fluorescent in situ hybridization of K. brevis and its flow-sorted bacterial fraction. (A–D) FISH with whole fixed cells of the dinoflagellate K. brevis CCMP718, all at 400× total magnification and 2 s camera integration time. (E, F) PI-PCR/FISH with whole fixed cells of the bacterial population sorted by flow cytometry from K. brevis CCMP718, all at 1000· total magnification and 5 s camera integration time. (I–L) DAPI counterstain of the same images showed above in (E–F), respectively to show total bacterial population of the field of view, all at 1000× total magnification and 0.13 s camera integration time. CY3-labeled FISH probes were as follows: (A) K. brevis with AT1−6L PKS probe. (B) K. brevis with AT2−10L PKS probe. (C) K. brevis with universal 18S rRNA probe. (D) K. brevis with negative control “no-bind” probe. (E) Bacterial fraction with AT1−6L PKS probe. Arrows indicate putative positive cells. (F) Bacterial fraction with AT2−10L PKS probe. (G) Bacterial fraction with universal 16S rRNA probe. (H) Bacterial fraction with negative control “no-bind” probe.
Sorted dinoflagellate cells were fixed, permeabilized and probed by FISH with fluorescent-labeled oligonucleotide probes for: (1) AT1−6L PKS gene sequences; (2) AT2−10L PKS gene sequences; (3) universal eukaryotic 18S rRNA gene sequences and (4) negative control (using a “no-bind” probe of no significant similarity to known sequences).
In the case of sorted bacterial cell populations, the bacterial cells were fixed, permeabilized and probed by a combination of prokaryotic in situ PCR and fluores-cent in situ hybridization (PI-PCR/FISH), using fluores-cent-labeled oligonucleotide probes for: (1) AT1−6L PKS gene sequences; (2) AT2−10L gene sequences; (3) universal bacterial 16S rRNA gene sequences and (4) negative control (“no-bind” probe as above). Results from positive and negative control probes were as expected, with positive control probes indicating that the intact cells were capable of being labeled by FISH, and negative controls indicating that such labeling was not due to mere physical characteristics of the fixed cells or entrapment of the probe. The AT2−10L PKS probe exclusively labeled K. brevis, whereas the PKS probe for the AT1−6L sequence labeled both K. brevis cells and also labeled a small coccoid-type cell of bacterial size from the sorted bacterial fraction (Fig. 7). This AT1−6L hybridizing cell type appears to be in relatively low abundance with respect to the total bacterial population found in this K. brevis CCMP718 culture.
The positive FISH labeling with the AT1−6L probe for both dinoflagellate cells and some members of the bacterial population would appear to be consistent with the PCR results which find amplification of AT1−6L sequences from the sorted bacterial population of this culture. Likewise, the positive PKS probe results for the AT2−10L sequence appears consistent with the PCR results, in that this probe labeled the K. brevis cells, but did not label any cells from the sorted bacterial population. Thus both the PCR and the FISH independently support the conclusion that at least the AT2−10L PKS sequence is of dinoflagellate origin and is not found in the associated bacterial population.
In the case of whole cell labeling of K. brevis, some variation was seen in the localization of the probe within the cells, depending on the probe used. However, we must point out that the process of fixation, permeabilization and decolorization used here resulted in severe physical deformation of these dinoflagellate cells, as the cytoplasm and intracellular structures dehydrate and collapse around the nucleus. The resulting cellular morphology after such manipulations is highly variable even from the same batch of fixed unialgal culture. Also these cells were neither DNase nor RNase treated, although in other FISH experiments where nuclease control treatments were used (data not shown), it was determined that the fluorescent signal seen from these probes was predominately due to RNA. In general, the AT1−6L PKS probe appears to give a hybridization signal predominately from the cytoplasm collapsed around the nucleus, but the nucleus itself appears relatively dark. On the other hand, the AT2−10L PKS probe and the 18S rRNA probe both show signal throughout the cell, although the AT2−10L probe did show some increase in signal intensity from the cytoplasm periphery. This would seem to imply that the AT1−6L PKS probe signal observed in K. brevis is from endosymbiotic bacteria whereas the AT2−10L signal is from nucleic acid of the Karenia cell itself. While these results do not rule out the possibility that endosymbiotic bacteria caused (in part or whole) the AT2−10L probe signal, no labeling of the sorted bacterial population or of exogenous bacteria in the algal population was observed. Nonetheless, it is unwise at this point to over-interpret the localization pattern of the FISH labeling within the K. brevis cells, due to the severe and random deformation of the Karenia morphology caused by the fixation and permeabilization steps. For this work, FISH results were simply scored for suffcient retention of the probe at a particular stringency by the Karenia cells, and that the signal intensity was significantly greater than background and significantly greater than the no-bind negative control probe under similar conditions.
3. Conclusions
While the brevetoxins have been studied for more than 30 years, aside from the identification of precursors, little is known about their biosynthesis. To date, no gene or enzyme has been identified that is associated with the biosynthesis of a polyketide from any dinoflagellate. Further complicating the subject is the presence of associated bacteria in the unialgal cultures which have not been successfully eliminated. These uncultured bacterial symbionts can carry their own PKS biosynthetic repertoire. Determining the ultimate source of brevetoxin will require more extensive studies, such as isolating a complete PKS operon for comparative analyses. However, the large size of the dinoflagellate genome (Rizzo et al., 1982) and the lack of a general dinoflagellate transformation system discourage such studies. Nonetheless, advances in the genetics of polyketide biosynthesis have paved the way for studies of brevetoxin biosynthesis at the molecular level. This study expands on the discovery of putative PKS genes from K. brevis that are closely related to PKS genes from the protist C. parvum. One of these PKS encoding genes (AT2−10L) has been localized to K. brevis, both by fluorescence in situ hybridization and PCR screening of flow-cytometric sorted K. brevis associated bacteria. While not specifically analyzed by FISH, the AT2−15 sequence is likely also derived from K. brevis based on the PCR results, the high homology to AT2−10L (80% identical), and resulting phylogeny. Although K. brevis, like many dinoflagellates, produces significant amounts of polyunsaturated fatty acids, these genes are unrelated to PKSs responsible for PUFA production in other marine eukaryotes. While neither AT2−10L or AT2−15 have been linked to toxin production per se, this work does provide the first evidence of resident PKS genes in K. brevis or any other dinoflagellate and strengthens the hypothesis that the ultimate origin of the brevetoxins is indeed K. brevis.
4. Experimental
4.1. General procedures
4.1.1. Isolation of RNA and DNA
Dinoflagellate cells were isolated by centrifugation (3000g for 5 min, 20 °C). DNA was purified using a Bio101 FastDNA kit (Qbiogene) and FastPrep instrument (Qbiogene) according to the manufacturer's instructions. Total RNA was purified by a modified guanidine thiocyanate method described for Gonyaulax polyhedra (Bae and Hastings, 1994), or using a Bio101 FastRNA kit (Qbiogene). The total RNA was treated with DNase (Promega) to remove any residual DNA. Nucleic acids were quantified spectrophotometrically using a lQuant (BioTek Instruments, Inc).
4.1.2. Cloning and sequencing of amplified fragments
Amplified DNA fragments were spun-purified from 1.2% agarose gels with Ultrafree-DA Millipore columns according to the manufacturer's instructions. Purified bands were cloned with the TOPO-TA kit and competent Escherichia coli (INVαF’ or TOP10) cells (Invitrogen) according to the manufacturer's instructions. Both strands were sequenced in house on an ABI automated sequencer (Perkin Elmer) using the M13R and T7 primers or sequencing was performed by MWG Biotech using T3 or T7 primers. Translated DNA sequences were compared to protein databases over the Internet using the program Basic Local Alignment Search Tool (BLAST) (McGinnis and Madden, 2004).
4.1.3. RFLP analysis
The plasmid alkaline lysis minipreparation procedure was performed as described previously (Sambrook et al., 2001). For PKS products amplified from sorted bacteria, two of fifty microliters of each plasmid preparation were digested with each of seven restriction enzymes: KpnI, BamHI, XbaI, XhoI, HpaII, RsaI and SphI (NEB). For 16S rRNA products amplified from cultured bacteria, a 1.5 kbp 16S product was amplified from 48 bacterial isolates using 20 ng DNA each and Taq polymerase (NEB) in a 50 μl reaction. Five microliters of each PCR was used for each of the four digestions using the enzymes HindIII, Sau3AI, HpaII and RsaI (NEB). Digestions were performed in a 20 μl reaction volume at the manufacturer's recommended temperature for one to three hours.
4.2. Dinoflagellate strains and culturing conditions
K. brevis (strains CCMP718, 2228, 2229, 2281, time and location of isolation may be found on the CCMP website: http://ccmp.bigelow.org/index.php), A. carterae (CCMP1314), Amphidinium operculatum (CCMP120), Chatonella subsalsa (CCMP217), H. akashiwo (CCMP1870), Prorocentrum hoffmannianum (CCMP683), Prorocentrum micans (CCMP1591), Prymnesium parvum (CCMP708) and Symbiodinium sp. (CCMP831) were obtained from the Provasoli-Guillard Center for the Culture of Marine Phytoplankton (CCMP) at the Bigelow Laboratory for Ocean Sciences, West Boothbay Harbor, ME. K. brevis strains JR, JR1 (which are identical to CCMP2281. CCMP2281 and JR1 are reportedly axenic) and NOAA1 (also known as Charlotte Harbor (Loret et al., 2002)) were obtained from the US EPA Gulf Ecology Division Laboratory in Gulf Breeze, Florida. Prorocentrum lima was obtained from the collection of Daniel Baden while at the University of Miami. P. parvum was obtained from the laboratory of Takeshi Yasumoto (Sendai, Japan). Cultures were maintained in variety of media: RE medium (K. brevis strains CCMP718, 2228, 2229, 2281, JR); f/2-Si A. carterae (CCMP1314) and A. operculatum (CCMP120)); Li-Si (C. subsalsa (CCMP217), P. micans (CCMP1591), and H. akashiwo (CCMP1870)); Prov 50 (P. hoffmannianum (CCMP683), P. parvum (CCMP708), P. parvum (Yasumoto), Prorocentrum lima, Symbiodinium sp. (CCMP831)). Cultures were maintained at 20−22 °C under constant illumination either from Cool White or Grow-Lux wide spectrum lamps (Guillard, 1975).
4.3. Flow cytometry and cell sorting
All flow cytometry analysis and cell sorting was performed on a DakoCytomation MoFlo high-speed cell sorter, equipped with a water-cooled tunable Argon Laser, 635 nm diode laser, sterile two-way and Cyclone single-cell sorting module, sterile sorting chamber with biohazard aerosol containment and evacuation module, cell-orienting nozzles, and a custom double-range pulse width analog-to-digital converter board. For the work reported here, all excitation was done at 488 nm, and sorting decisions were based on a multi-parameter combination of side scatter (SSC), forward scatter (FSC), pulse width and chlorophyll autofluorescence (FL4), as shown in Fig. 3. Cells were sorted under aseptic conditions into sterile microfuge tubes at a sheath pressure of 30 psi, with a sample differential of 2.5 psi, using the “sort-purify-1-drop” mode. Emission filters for FL4 (chlorophyll detection) = 670/30 nm. Purity of sorts was also monitored at time of sorting by epifluorescent microscopic observation of SYTO-9 stained subsamples of the sorted populations. Typically, 106−7 K. brevis cells were sorted along with bacterial cells corresponding to the same volume of K. brevis culture, or 107−8 bacterial cells.
4.4. PCR and RT-PCR
The DNA-free RNA was reverse-transcribed by MMLV reverse transcriptase (Promega) using random hexamers according to the manufacturer's instructions. The following PCR conditions were typical: template cDNA (20 ng) or DNA (100 ng), 0.5 μM each primer, 3mM MgCl2, 1.25 U Taq (NEB or Genechoice) or Pfu (Stratagene). Degenerate PKS primers (forward: 5′-MGIGARGCIYTICARATGGAYCCICARCARMG-3′, reverse: 5′-GGRTCNCCIARYTGIGTICCIGTICCRTGIGC-3′), specific PKS primers (Table 1), universal bacterial 16S rRNA primers (24f: 5′-GGAGAGTTTGATCMTGGCT-3′, 1493r: 5′-ACGGYTACCTTGTTACGACTT-3′) (Prokic et al., 1998) and universal eukaryotic 18S rRNA primers (forward: 5′-GGTTGATCCTGCCAGTAGTCATATGCTTG-3′,reverse:5′-GA TCCTTCCGCAGGTTCACCTACGGAAACC-3′). The following standard PCR protocol was used to perform amplification reactions: 95 °C/5′, then 95 °C/20″, Tanneal/20″, 72 °C/t × 30−40 cycles and a final 72 °C/5′ extension. For the sequence-specific PKS primers, Tanneal = 60 °C and textend = 20 s with 39 cycles. Genomic PCR and RT-PCR were performed on a Mastercycler gradient thermal cycler (Brinkmann, Eppendorf). Real-time RT-PCR was performed on a DNA Engine Opticon thermal cycler (MJ Research) with IQ SYBR Green Supermix reagent (Bio-Rad) according to the manufacturer's instructions. Melting curve analyses were performed between 65.0 and 90.0 °C; reading every 0.2 °C and holding for 1 s between reads.
4.5. Sequence and phylogenetic analysis
Previously described PKS and FAS sequences were retrieved from GenBank as reference sequences, such as DEBS-6 for erythromycin (M63677), rifamycin (AF040570), FAS Fix-23 – (X64131), myxothiazol MtaF from Stigmatella aurantiaca (AAF19814), McyD from Microcystis aeruginosa (AAF00959), FK506 from Streptomyces (CAA71463), pksM from B. subtilis (NP_389601), C. parvum (AAC99407; EAK87820 and T31307) and C. hominus (EAL37180); eicosapentaenoic acid biosynthetic (polyunsaturated fatty acid synthase – PUFA) sequences stemmed from Schizochytrium (AF378327−9), Moritella marina (BAA89382−4) and Shewanella (AAB81123). Novel marine microbe-derived PKS and FAS sequences were also included in phylogenetic analyses for reference and comparison: marine fungal strain K132 (AY857361), and eubacterial strains JL085 (AY857380), K155 (AY857387), K618 (AY857381), JL048 (AY857384), JL081 (AY857354) and 223N (AY857340) (Also see the HBOI Marine Microbial Database – http://www.hboi.edu/dbmr/dbmr_hbmmd.html)(Gunasekera et al., 2005). Protein sequences were aligned with ClustalX version 1.81 (Thompson et al., 1997), using the Gonnett series as the protein weight matrix. Default gap opening and gap extension penalties of 10.0 and 0.2 were adopted because conserved domains were aligned most accurately with few gaps. In the absence of a complete three-dimensional PKS crystal structure, these motifs assisted in the evaluation of the final multiple sequence alignment. Minor corrections on highly divergent regions were performed manually. Several regions were excluded from phylogenetic analysis due to ambiguous alignments or gap placements. Base composition and dGC content of DNA codon positions were determined using the program GCUA: General Codon Usage Analysis program (McInerney, 1998). Phylogenetic analyses of amino acid sequences using either maximum parsimony (MP) or minimum evolution (ME) neighbor-joining optimality criteria were conducted with PAUP* version 4.0b10 (Swofford, 2002) using the heuristic search option and the TBR branch-swapping algorithm. MP utilized random addition of taxa with ten repetitions. The ME analysis (Rzhetsky and Nei, 1992) was also carried out with PAUP* 4.0b10 using mean character distances. Robustness of the ME analyses was tested by bootstrapping with at least 500 replicates for each run (Felsenstein, 1985).
4.6. DGGE analysis of 16S rRNA gene
The 16S rRNA gene was amplified from 50 ng of genomic DNA purified from sorted bacteria, using the universal prokaryotic primer set (24f, 1393r). Cycling conditions were an initial 95 °C/5′, then 95 °C/30″, 55 °C/30″,72 °C/1.5′ × 30 cycles and a final 72 °C/10′. The 1.5 kbp product was purified using the PCR Kleen Kit (Bio-Rad). One microliter of the purified 16S rRNA PCR was used as the template for a nested PCR using the primer set (GC-clamp forward, 5′-CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGAACGCGAAGAACCTTAC-3′, reverse, 5′-CGGTGTGTACAAGGCCCGGGAACG-3′, E. coli positions 984 and 1378, respectively) (Heuer et al., 1997). These primers were also used directly (1 μM each) with genomic DNA (100 ng) from K. brevis isolates (CCMP718, CCMP2228, CCMP2229, CCMP2281, JR) for comparison of bacterial populations. Cycling conditions: initial 95 °C/5′, then 95 °C/30″, 60 °C(−1 degree/cycle)/2′, 72 °C/2′ × 30 cycles and a final 72 °C/10′ extension. The resulting 450 bp product was purified using the PCR Kleen Kit (Bio-Rad). All PCRs performed in preparation for the DGGE analysis consisted of 50 μl final volume reactions and contained 2.5 U turbo Pfu DNA polymerase (Stratagene), 1× cloned Pfu reaction buffer (1.5 mM Mg2+ final) (Stratagene), 7% DMSO, 0.1 mmol each dNTP, 0.25 μM each primer.
Thirty microliters of each purified reaction (450 bp product) were mixed with five microliters of 3× loading dye, prepared from the standard 6× Blue/Orange loading dye (Promega) by dilution with an equal volume of 50% sterile glycerol. Twelve microliters of each sample (purified PCR product plus dye) were loaded onto the DGGE gel (8% polyacrylamide gel prepared with a gradient from 40% to 47.5% of urea/formamide). To enhance band sharpness, the top of the gel was overlaid with a comb-length layer of polyacrylamide containing no denaturant. After loading at room temperature the DCode gel apparatus (Bio-Rad) was placed into the pre-heated running buffer (1× TAE, 60 °C) and electrophoresed at a constant 150 V for 5.5 h.
4.7. Brevetoxin ELISA
K. brevis cells from each of five strains (CCMP718, 2228, 2229, 2281 and JR) were counted using a Coulter Multisizer II (Coulter Scientific Instruments). The cultures (1.25 × 106 cells) were extracted with chloroform (20 ml), centrifuged and the organic layer was dried in vacuo. The dried crude extract was resuspended in methanol (1 ml), extracted with petroleum ether, centrifuged and the methanol layer was dried in vacuo. The crude extract was resuspended in 200 μl of methanol and dilutions were prepared according to the manufacturer's (HabLab, UNC Wilmington) instructions. Similarly prepared crude extracts from the flow sorted K. brevis CCMP718 dinoflagellate fraction (3 × 105 cells) and bacterial fraction (6 × 105 cells) were tested for the presence of brevetoxin by ELISA.
4.8. Isolation of heterotrophic bacteria from K. brevis
Bacteria were subcultured from K. brevis CCMP718 by inoculating marine agar (Difco) plates with aliquots (20 μl) of dinoflagellate culture that was diluted 10-fold. The plates were incubated at ambient temperature for three days. Fifty single colonies displaying a variety of morphologies were plated three times to ensure homogeneity of the isolates.
4.9. FISH and PI-PCR/FISH labeling of whole cells
All solutions and buffers were treated with diethyl pyrocarbonate (DEPC) and sterilized to prevent any potential nuclease contamination of samples. Dense dinoflagellate or bacterial suspensions, harvested by cell sorting (approx. 107 cells ml−1), were fixed overnight with 4% paraformaldehyde at 4 °C, then washed with 1:1 Ethanol:1× PBS (Sinigalliano et al., 2003). For dinoflagellates, fixed cells were subsequently decolorized in 70% ethanol (Simon et al., 2000) with a slight modification: cells were washed in 2 mL of 70% EtOH, with changes of fresh EtOH every hour over a period of 4−5 h. This decolorization pre-treatment removed most of the chlorophyll and thus decreased autofluorescence background. Successive washes in 70% EtOH over a period of hours instead of days rapidly decreased the chlorophyll concentration significantly to the point where CY3-labeled probes could be visualized clearly against the reduced autofluorescent background. The fixed and decolorized cells were resuspended in 1:1 Ethanol:1× PBS, and then these cells were permeabilized by incubating them in the 50% EtOH at 80 °C for 20 min. In the case of bacterial cells, samples were treated the same except that no decolorization was done. After permeabilization, the pre-treated cells (10 μL) were spotted on gelatin subbed slides (0.3% gelatin with 0.03% chromium potassium sulphate), dried, then serially dehydrated by successive immersion of slides in 53% EtOH, 78% EtOH, and 98% EtOH, then allowed to dry. For bacterial samples, the cell spots on the slides were treated with lysozyme, washed, then the target signal was amplified prior to FISH by prokaryotic in situ PCR (PI-PCR) as previously described (Sinigalliano et al., 2003), except that primers specific for AT1−6L PKS genes, AT2−10L PKS genes, or universal 16S rRNA genes were used. FISH was then carried out with the cells immobilized on slides as described below. Frame-Seal™ incubation chambers (MJ Research, Inc. Watertown, MA, USA) were mounted around the periphery of the cell spot to serve as a reservoir for the hybridization mixture. The cell spots within the chambers were then overlain with a hybridization solution of 5X SSC with 0.1% Tween20, containing the respective CY3-labeled oligonucleotide probe at 5 μM final concentration (see below for sequence characteristics of probes used), then sealed with a plastic cover slip. Slides were incubated in a PTC-200 Thermal Cycler equipped with a Slide Chambers™ dual block alpha unit, and heated at 95 °C for 5 min, then held the at 55 °C hybridization temperature for 3 h. After hybridization, the chambers were removed from the slides and the slides were then washed three times for 10 min each in 50 mL of 0.5× SSC prewarmed to 55 °C. Slides were then counter-stained with DAPI or Hoechst-33342 (1 μg mL −1)and examined by epifluorescence microscopy (Leica DMLB microscope equipped for phase contrast and epifluorescence with a 100 W mercury lamp, and standard DAPI, rhodamine, and FITC filter sets). Images were acquired and digitized with a cooled color low-light CCD camera (Hammamatsu Model C5810, Hammamatsu Corp, Japan) and analyzed with the software package ImagePro Plus (Media Cybernetics, Silver Springs, MD, USA). The following probes were used in the FISH: The two PKS probes were AT2−10L 5′-[Cy3]-CGCCATGGCACAAACA-3′ and AT1−6L 5′-[Cy3]-TGATCTGGGAGCCGCAA-3′. The universal 18S rRNA probe used was 5′-[Cy3]-ACCAGACTTGCCCTCC-3′ (Yokouchi et al., 2003), and the universal 16S rRNA probe was 5′-[Cy3]-GWATTACCGCGGCKGCTG-3′ (Karner and Fuhrman, 1997). The negative control “No-Bind” probe was 5′-[Cy3]-CCTAGTGACGCCGTCGAC-3′ (Karner and Fuhrman, 1997). The following primers were used for the in situ PCR: For AT2−10L, the forward primer was 5′-CGCAACGTACAAGTCACATCTG-3′ and the reverse primer was 5′-CACCTGATGGCCGCTGTAAG-3′. For AT1−6L, the 6Lb set was used (Table 1). The primer pair for 16S amplification were universal 16S rRNA forward primer 27f 5′-AGAGTTTGATCMTGGCTCAG-3′ and universal 16S rRNA reverse primer 1392r 5′-ACGGGCGGTGTGTRC-3′. GenBank Accession Nos.: AS1−1L [AY820891], AT1−6L [AY817144], AT2− 10L [AY820892], AT2−15 [AY820893], Kbrev_bac(3) [AY902469], Kbrev_bac(23) [AY902470], Kbrev_ bac(28) [AY902471] and Kbrev_bac(30) [AY902472].
Acknowledgments
This work was supported by the following: NIH/ NIEHS S11 ES11181 and P30 ES 05705, NIH/NIGMS S06 GM008205 and R25 GM061347 to R.V.S. and USDA CSREES 2001-02471. Portions of this material is based upon work supported by the National Science Foundation under Grant No. 9974984 to J.V.L. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the sponsors.
References
- Bae YM, Hastings JW. Cloning, sequencing and expression of dinoflagellate luciferase DNA from a marine alga, Gonyaulax polyedra. Biochim. Biophys. Acta. 1994;1219:449–456. doi: 10.1016/0167-4781(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Bourdelais AJ, Tomas CR, Naar J, Kubanek J, Baden DG. New fish-killing alga in Coastal Delaware produces neurotoxins. Environ. Health Perspect. 2002;110:465–470. doi: 10.1289/ehp.02110465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HN, Shimizu Y. Biosynthesis of brevetoxins. Evidence for the mixed origin of the backbone carbon chain and possible involvement of dicarboxylic acids. J. Am. Chem. Soc. 1987;109:2184–2185. [Google Scholar]
- Chretiennot-Dinet M. Global increase of algal blooms, toxic events, casual species introductions and biodiversity. Oceanis. 2001;24:223–238. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Guillard RRL. Culture of phytoplankton for feeding marine invertebrates. Cult. Mar. Invertebr. Anim., [Proc. Conf.] 1975:29–60. [Google Scholar]
- Gunasekera A, Sfanos KA, McCarthy PJ, Lopez JV. HBMMD: an enhanced database of the microorganisms associated with deeper water marine invertebrates. Appl. Microbiol. Biotechnol. 2005;66:373–376. doi: 10.1007/s00253-004-1763-7. [DOI] [PubMed] [Google Scholar]
- Hallegraeff GM. A review of harmful algal blooms and their apparent global increase. Phycologia. 1993;32:79–99. [Google Scholar]
- Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gelelectrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 1997;63:3233–3241. doi: 10.1128/aem.63.8.3233-3241.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeglitsch G, Rein K, Baden DG, Adams DA. Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J. Pharmacol. Exp. Ther. 1998;284:516–525. [PubMed] [Google Scholar]
- Karner M, Fuhrman JA. Determination of active marine bacterioplankton: a comparison of universal 16S rRNA probes, autoradiography, and nucleoid staining. Appl. Environ. Microbiol. 1997;63:1208–1213. doi: 10.1128/aem.63.4.1208-1213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Ahmed MS, Arakawa O, Onoue Y. Properties of neurotoxins separated from a harmful red tide organism Chattonella marina. Isr. J. Aquacult./Bamidgeh. 1995;47:137–141. [Google Scholar]
- Khan S, Arakawa O, Onoue Y. Neurotoxin production by a chloromonad Fibrocapsa japonica (Raphidophyceae). J. World Aquacult. Soc. 1996a;27:254–263. [Google Scholar]
- Khan S, Arakawa O, Onoue Y. A toxicological study of the marine phytoflagellate, Chattonella antiqua (Raphidophyceae). Phycologia. 1996b;35:239–244. [Google Scholar]
- Khan S, Arakawa O, Onoue Y. Neurotoxins in a toxic red tide of Heterosigma akashiwo (Raphidophyceae) in Kagoshima Bay, Japan. Aquacult. Res. 1997;28:9–14. [Google Scholar]
- Kirkpatrick B, Fleming LE, Squicciarini D, Backer LC, Clark R, Abraham W, Benson J, Cheng YS, Johnson D, Pierce R. Literature review of Florida red tide: implications for human health effects. Harmful Algae. 2004;3:99–115. doi: 10.1016/j.hal.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Leebens-Mack J, Ni W, dePamphilis CW, Ma H. Highly heterogeneous rates of evolution in the SKP1 gene family in plants and animals: functional and evolutionary implications. Mol. Biol. Evol. 2004;21:117–128. doi: 10.1093/molbev/msh001. [DOI] [PubMed] [Google Scholar]
- Lee MS, Repeta DJ, Nakanishi K, Zagorski MG. Biosynthetic origins and assignments of carbon 13NMR peaks of brevetoxin B. J. Am. Chem. Soc. 1986;108:7855–7856. doi: 10.1021/ja00284a072. [DOI] [PubMed] [Google Scholar]
- Lee MS, Qin GW, Nakanishi K, Zagorski MG. Biosynthetic studies of the brevetoxins: a group of potent neurotoxins produced by dinoflagellates Gymnodinium breve. J. Am. Chem. Soc. 1989;111:6234–6241. [Google Scholar]
- Lopez JV. Naturally mosaic secondary metabolite operons: variability and putative horizontal transfer of discrete catalytic domains in the epothilone polyketide synthase locus. Mol. Genet. Genom. 2003;270:420–431. doi: 10.1007/s00438-003-0937-9. [DOI] [PubMed] [Google Scholar]
- Loret P, Tengs T, Villareal TA, Singler H, Richardson B, Mcguire P, Morton S, Busman M, Campbell L. No difference found in ribosomal DNA sequences from physiologically diverse clones of Karenia brevis (Dinophyceae) from the Gulf of Mexico. J. Plankton Res. 2002;24:735–739. [Google Scholar]
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney JO. GCUA: General codon usage analyses. Bioinform. Appl. Note. 1998;14:372–373. doi: 10.1093/bioinformatics/14.4.372. [DOI] [PubMed] [Google Scholar]
- Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, Yazawa K, Knauf V, Browse J. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science. 2001;293:290–293. doi: 10.1126/science.1059593. [DOI] [PubMed] [Google Scholar]
- Moffit MC, Neilan BA. Evolutionary affiliations within the superfamily of ketosynthases reflect complex pathway associations. J. Mol. Evol. 2003;56:446–457. doi: 10.1007/s00239-002-2415-0. [DOI] [PubMed] [Google Scholar]
- Naar J, Bourdelais A, Tomas C, Kubanek J, Whitney PL, Flewelling L, Steidinger K, Lancaster J, Baden DG. A competitive ELISA to detect brevetoxins from Karenia brevis (formerly Gymnodinium breve) in seawater, shellfish, and mammalian body fluid. Environ. Health Perspect. 2002;110:179–185. doi: 10.1289/ehp.02110179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer BA, Khosla C. Biosynthesis of polyketides in heterologous hosts. Microbiol. Mol. Biol. Rev. 2001;65:106–118. doi: 10.1128/MMBR.65.1.106-118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumley FG. Marine algal toxins: biochemistry, genetics, and molecular biology. Limnol. Oceanogr. 1997;42:1252–1264. [Google Scholar]
- Prokic I, Bruemmer F, Brigge T, Goertz HD, Gerdts G, Schuett C, Elbraechter M, Mueller WEG. Bacteria of the genus Roseobacter associated with the toxic dinoflagellate Prorocentrum lima. Protist. 1998;149:347–357. doi: 10.1016/S1434-4610(98)70041-0. [DOI] [PubMed] [Google Scholar]
- Rausch de Traubenberg C, Lassus P. Dinoflagellate toxicity: are marine bacteria involved. Evidence from the literature. Marine Microbial Food Webs. 1991;5:205–226. [Google Scholar]
- Rizzo PJ, Jones M, Ray SM. Isolation and properties of isolated nuclei from the Florida red tide dinoflagellate Gymnodinium breve (Davis). J. Protozool. 1982;29:217–222. doi: 10.1111/j.1550-7408.1982.tb04014.x. [DOI] [PubMed] [Google Scholar]
- Rzhetsky A, Nei M. Statistical properties of the ordinary least-squares, generalized least squares, and minimum-evolution methods of phylogenetic inference. J. Mol. Evol. 1992;35:367–375. doi: 10.1007/BF00161174. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- Simon N, Campbell L, Ornolfsdottir E, Groben R, Guillou L, Lange M, Medlin LK. Oligonucleotide probes for the identification of three algal groups by dot blot and fluorescent whole-cell hybridization. J. Eukaryot. Microbiol. 2000;47:76–84. doi: 10.1111/j.1550-7408.2000.tb00014.x. [DOI] [PubMed] [Google Scholar]
- Sinigalliano CD, Jones RD, Kuhn DN, Guerrero MA. Visualization of the cbbL gene in healthy and starved chemoautotrophic nitrifying bacteria. Curr. Microbiol. 2003;47:77–83. doi: 10.1007/s00284-002-3941-0. [DOI] [PubMed] [Google Scholar]
- Snyder RV, Gibbs PDL, Palacios A, Abiy L, Dickey R, Lopez JV, Rein KS. Polyketide synthase genes from marine dinoflagellates. Mar. Biotechnol. 2003;5:1–12. doi: 10.1007/s10126-002-0077-y. [DOI] [PubMed] [Google Scholar]
- Swofford DL. Sinauer; Sunderland, MA: 2002. PAUP* Phylogenetic Analysis Using Parsimony (*and other methods). Version 4. [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dolah FM, Doucette GJ, Gulland FMD, Rowles TL, Bossart GD. Impacts of algal toxins on marine mammals. In: Vos JG, Bossart GD, Bossart G, Fournier M, O'Shea T, editors. Toxicology of Marine Mammals. Taylor & Francis, Inc.; 2002. pp. 247–269. [Google Scholar]
- Yokouchi H, Takeyama H, Miyashita H, Maruyama T, Matsunaga T. In situ identification of symbiotic dinoflagellates, the genus Symbiodinium with fluorescence-labeled rRNA-targeted oligonucleotide probes. J. Microbiol. Methods. 2003;53:327–334. doi: 10.1016/s0167-7012(02)00250-6. [DOI] [PubMed] [Google Scholar]
- Zhu G, LaGier MJ, Stejskal F, Millership JJ, Cai X, Keithly JS. Cryptosporidium parvum: the first protist known to encode a putative polyketide synthase. Gene. 2002;298:79–89. doi: 10.1016/s0378-1119(02)00931-9. [DOI] [PubMed] [Google Scholar]
- Zhu G, Marchewka MJ, Woods KM, Upton SJ, Keithly JS. Molecular analysis of a Type I fatty acid synthase in Cryptosporidium parvum. Mol. Biochem. Parasitol. 2000;105:253–260. doi: 10.1016/s0166-6851(99)00183-8. [DOI] [PubMed] [Google Scholar]