

Abstract
Retinol dehydrogenase 13 (RDH13) is a recently identified short-chain dehydrogenase/reductase related to microsomal retinoid oxidoreductase RDH11. In this study, we examined the distribution of RDH13 in human tissues, determined its subcellular localization and characterized the substrate and cofactor specificity of purified RDH13 in order to better understand its properties. The results of this study demonstrate that RDH13 exhibits a wide tissue distribution and, by contrast with other members of the RDH11-like group of short-chain dehydrogenases/reductases, is a mitochondrial rather than a microsomal protein. Protease protection assays suggest that RDH13 is localized on the outer side of the inner mitochondrial membrane. Kinetic analysis of the purified protein shows that RDH13 is catalytically active and recognizes retinoids as substrates. Similar to the microsomal RDHs, RDH11, RDH12 and RDH14, RDH13 exhibits a much lower Km value for NADPH than for NADH and has a greater catalytic efficiency in the reductive than in the oxidative direction. The localization of RDH13 at the entrance to the mitochondrial matrix suggests that it may function to protect mitochondria against oxidative stress associated with the highly reactive retinaldehyde produced from dietary β-carotene.
Keywords: dehydrogenase, mitochondria, reductase, retinaldehyde, retinol
Short-chain dehydrogenases/reductases (SDRs) comprise a large family of functionally heterogeneous proteins that participate in the metabolism of steroids, prostaglandins, retinoids, aliphatic alcohols and xenobiotics [reviewed in refs. 1,2]. Members of the SDR superfamily are found in the cytoplasm, mitochondria, nuclei, peroxisomes and endoplasmic reticulum. Many enzymes exhibit the same substrate and cofactor specificity, but different subcellular localization and tissue distribution [reviewed in ref. 3].
To date, about 3000 primary structures from various species have been annotated in sequence databases as members of the SDR superfamily on the basis of SDR signature features, such as the TGX3GXG motif of the nucleotide binding region and the catalytically active tetrad N-S-Y-K, which constitutes the active site [1]. At least 63 SDR genes have been identified in the human genome database [1]. For many of these putative oxidoreductases, the cellular functions are yet to be determined.
Retinol dehydrogenase 13 (RDH13) is a recently identified member of the SDR superfamily of proteins that shares sequence similarity with RDH11 (also known as retinal reductase 1 [4–6]), RDH12 [6,7] and RDH14 (previously known as PAN2 [8]) proteins. RDH11, RDH12 and RDH14 have been characterized and found to be microsomal proteins that recognize retinoids [4–8] and medium-chain aldehydes [7] as substrates, with NADP+/NADPH as the preferred cofactors. However, the substrate and cofactor specificity of RDH13 remains unknown, as it failed to exhibit any enzymatic activity under the conditions of previous assays [6]. Thus, it is not clear whether RDH13 represents a catalytically active member of the SDR superfamily.
This study was undertaken in order to better understand the properties of RDH13 and to identify its potential substrates. We examined the distribution of RDH13 in human tissues, determined its subcellular localization, expressed and purified the recombinant protein, and characterized its substrate and cofactor specificity. The results of this study reveal significant differences between RDH13 and the other members of the RDH11–14 group of proteins, and offer an important insight into the properties of this new member of the SDR superfamily.
Results
Tissue distribution of RDH13
It has been shown that a protein recognized by anti-RDH13 serum is present in the inner segments of rod and cone photoreceptors [6]; however, the distribution of RDH13 in extra-ocular tissues has not yet been determined. Therefore, we examined the expression pattern of RDH13 in eight human tissues using polyclonal antiserum raised against bacterially expressed and purified RDH13. Western blot analysis revealed that anti-RDH13 serum recognized a protein of the expected size (~ 36 kDa) in seven of the eight tissues (Fig. 1). The intensity of immunostaining was strongest in the kidney, heart and lung, but the corresponding protein band was also detectable in the prostate, testis and ovary. These results demonstrate that RDH13 is a relatively widespread protein and that its expression level varies considerably in different tissues.
Fig. 1.
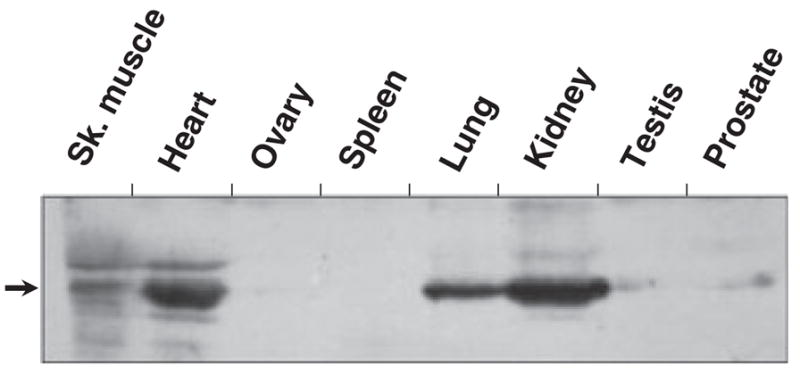
RDH13 expression in human tissues. Samples (100 μg) of tissue homogenates were separated by SDS-PAGE and analyzed by western blotting using anti-RDH13 serum, as described in Experimental procedures. The arrow indicates the position of the RDH13 protein. Sk. muscle, skeletal muscle.
Subcellular localization of RDH13
RDH13 shares the greatest sequence similarity with RDH11, RDH12 and RDH14, which are integral membrane proteins of the endoplasmic reticulum. To determine whether RDH13 is also targeted to the endoplasmic reticulum, we analyzed its subcellular localization in prostate cancer LNCaP cells, which express endogenous RDH13 at high levels. LNCaP cells were homogenized and the subcellular fractions were resolved by discontinuous sucrose density gradient [9]. Equal aliquots of the gradient fractions were subjected to denaturing SDS-PAGE and analyzed by western blotting using anti-RDH13 serum. As shown in Fig. 2, RDH13 was detected in fractions 3–7 of the gradient. To identify the organelles present in these fractions, we used antibodies against organelle-specific marker proteins. Lamin, a nuclear protein, was found only in the bottom two fractions (6 and 7), where nuclei, cell debris and unbroken cells were expected to be present. Golgin exhibited two peaks of distribution, one at the 0.8 M/1.2 M sucrose interface (fractions 3 and 4), as expected, and also in the unbroken cells area (fractions 6 and 7). Calnexin, the marker for the endoplasmic reticulum, appeared to be spread throughout the gradient, whereas porin, an integral protein of the outer mitochondrial membrane, was most abundant in fractions 3–7, similar to RDH13. Thus, the flotation pattern of RDH13 coincided best with that of porin, suggesting that, by contrast with the other members of the RDH11–14 cluster, RDH13 is a mitochondrial and not an endoplasmic reticulum protein.
Fig. 2.
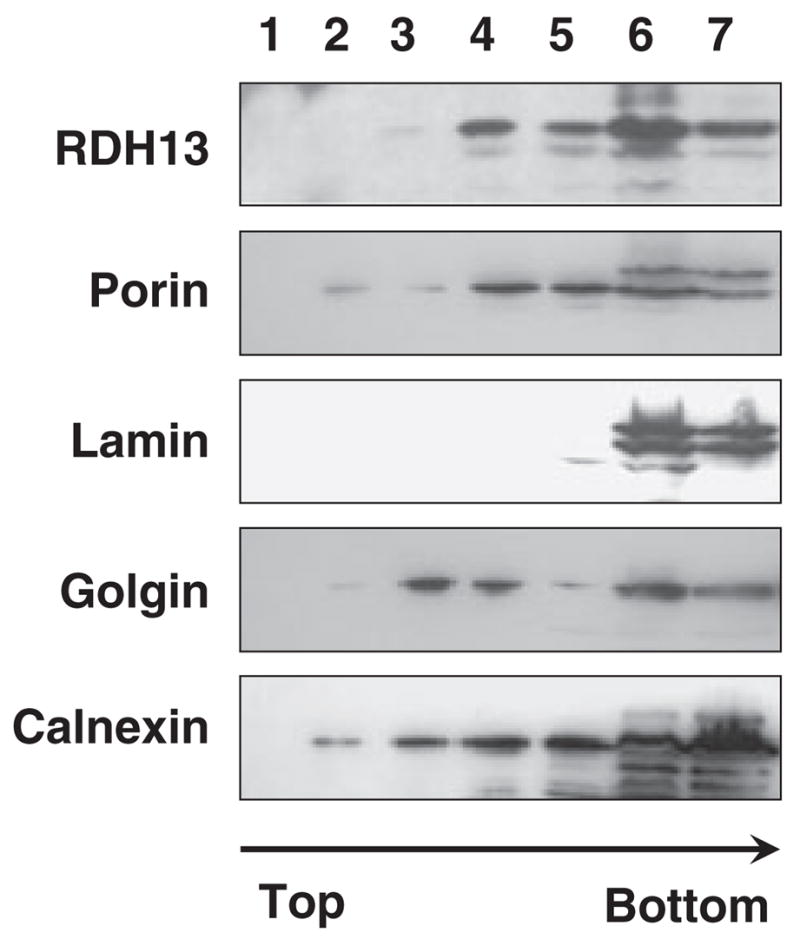
Subcellular localization of RDH13 in LNCaP cells. Subcellular fractions of LNCaP cells were separated by sucrose gradient, as described in Experimental procedures, and analyzed by western blotting using antibodies against RDH13 or specific marker proteins of cellular organelles, as indicated. Fractions are numbered from the top of the gradient.
Mitochondria have a highly compartmentalized structure, which can influence the substrate and cofactor availability for RDH13. To determine the submitochondrial localization of RDH13, freshly isolated mitochondria were fractionated into the intermembrane space, outer membrane, matrix and inner membrane, and the fractions were analyzed by western blotting using anti-RDH13 serum. RDH13 protein was found to be most abundant in the fraction containing the inner mitochondrial membranes (Fig. 3A), suggesting that it is a membrane-bound protein. To determine whether RDH13 is a peripheral or an integral membrane protein, the inner mitochondrial membranes or whole mitoplasts were treated with NaCl/Pi, 1 M NaCl, 100 mM Na2CO3 or 1% Triton X-100, as described in Experimental procedures. The samples were centrifuged and the distribution of RDH13 between the pellet and supernatant was analyzed by western blotting. As shown in Fig. 3B, RDH13 protein remained associated with the membranes after treatment with NaCl/Pi or NaCl, but was completely solubilized by Na2CO3 and Triton X-100 treatments. As integral membrane proteins cannot be extracted by alkaline treatment [10], these results indicate that RDH13 is a peripheral membrane protein.
Fig. 3.
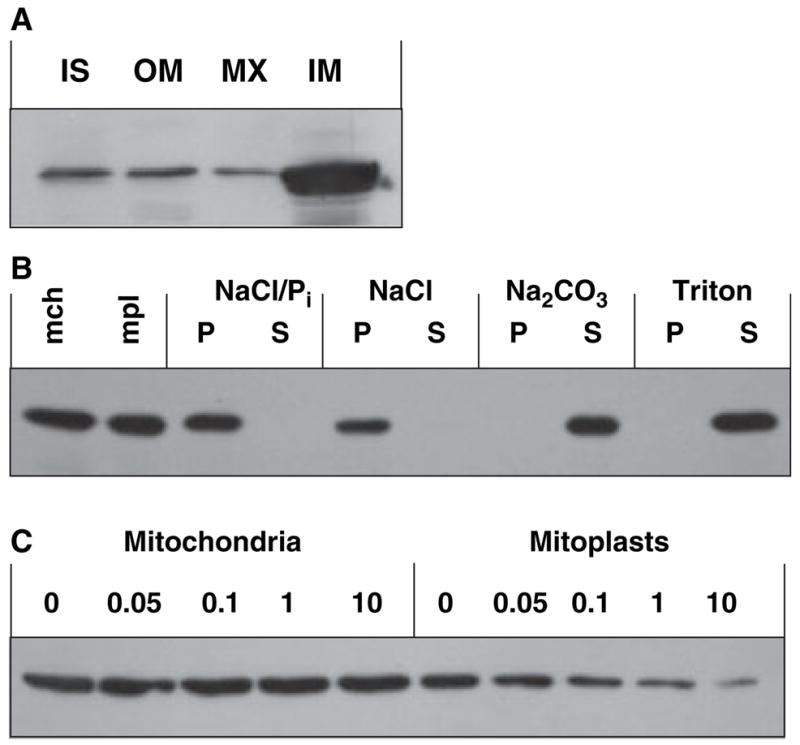
Submitochondrial localization of RDH13. (A) Mitochondria were fractionated into intermembrane space (IS), outer membranes (OM), matrix (MX) and inner membranes (IM). One-fiftieth of each fraction was separated by SDS-PAGE and the distribution of RDH13 was determined by western blotting. (B) Mitoplasts (mpl) were prepared by hypotonic or digitonin treatment of mitochondria (mch) and incubated with NaCl/Pi, NaCl, Na2CO3 or Triton X-100 (Triton). Treated samples were centrifuged and the distribution of RDH13 between soluble and insoluble fractions was analyzed by western blotting. P, pellet; S, supernatant. The results were identical for digitonin- and hypotonically prepared mitoplasts. (C) Mitochondria or mitoplasts were incubated with the indicated amounts of trypsin (μg) for 30 min on ice, followed by the addition of soybean trypsin inhibitor. RDH13 protein stability was monitored by western blotting.
To determine whether RDH13 is localized on the matrix side of the inner membrane or faces the intermembrane space, we carried out protease protection assays. Mitochondria or mitoplasts (lacking the outer membrane) were treated with increasing concentrations of trypsin, and the stability of RDH13 protein was analyzed by western blotting. RDH13 was completely resistant to trypsin digestion in intact mitochondria at all concentrations of trypsin. By contrast, in mitoplasts, there was a progressive loss of RDH13 protein (Fig. 3C). This result indicates that the outer membrane protects RDH13 from trypsin in intact mitochondria, and the removal of the outer membrane exposes RDH13 to trypsin. Thus, RDH13 appears to be localized on the outer side of the inner mitochondrial membrane, facing the intermembrane space.
Finally, we determined whether RDH13 contains a cleavable mitochondrial targeting signal sequence. Analysis of the primary structure of RDH13 using the MitoProt II algorithm [11] suggested a potential cleavage site at amino acid 62, with a probability of export to mitochondria of 0.77. However, RDH13 produced by in vitro translation, using expression construct under the T7 promoter in pCR4.2-TOPO and the TNT Coupled Reticulocyte Lysate Transcription/Translation System (Promega, Madison, WI, USA), had the same size in SDS-PAGE as the fully processed protein in LNCaP cells (data not shown), indicating that RDH13 lacks a cleavable mitochondrial target sequence. This result is consistent with the localization of RDH13 on the outer side of the inner mitochondrial membrane.
Substrate and cofactor specificity of purified RDH13–His6
A previous study has examined RDH13 for activity towards retinaldehyde in whole Sf9 cells [6]. This analysis failed to detect any increase in retinaldehyde reduction by RDH13-expressing cells compared with control cells. We re-examined the catalytic activity of RDH13 by expressing the protein in Sf9 cells as a fusion with the C-terminal His6 tag in order to purify RDH13 to homogeneity and characterize its properties under well-defined conditions. Similar to native RDH13, recombinant RDH13–His6 was detected in the mitochondrial fraction of Sf9 cells and exhibited the same association with the inner mitochondrial membrane as the native protein (data not shown). Interestingly, the expression of RDH13 in Sf9 cells was accompanied by the appearance of a weak retinaldehyde reductase activity in the mitochondrial fraction, suggesting that RDH13 is active towards retinaldehyde (data not shown).
To obtain further evidence to demonstrate that the increase in mitochondrial retinaldehyde reductase activity was associated with RDH13 expression, we purified RDH13–His6 using Ni2+ affinity chromatography. This single-step purification procedure produced an almost homogeneous protein (Fig. 4). Activity assays showed that purified RDH13–His6 was indeed active towards all-trans-retinaldehyde and appeared to prefer NADPH to NADH as a cofactor, because the conversion of 5 μM all-trans-retinaldehyde in the presence of 1 mM NADPH was about 20-fold greater than that in the presence of 1 mM NADH. However, the specific activity of different RDH13–His6 preparations varied from 47 to 130 nmol· min−1·mg−1.
Fig. 4.
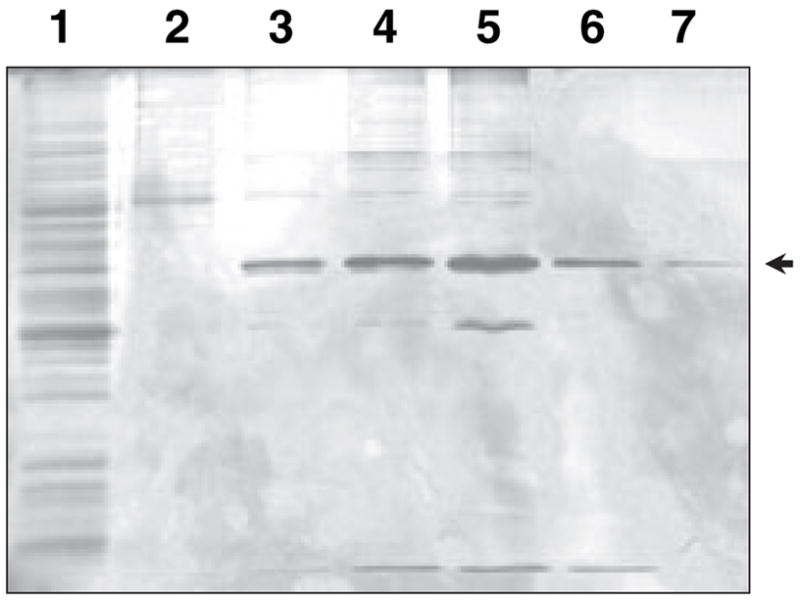
Purification of RDH13–His6 from Sf9 cells. RDH13–His6 was purified by Ni2+ affinity chromatography, and the fractions from various stages of purification were analyzed by SDS-PAGE followed by silver staining. Lane 1, homogenate; lane 2, wash with 10 mM imidazole; lanes 3–7, elution of RDH13–His6 with a stepwise imidazole gradient: 50 mM (3), 100 mM (4), 200 mM (5), 300 mM (6), 400 mM (7). Arrow indicates the position of RDH13–His6.
In this respect, we observed that, if dithiothreitol was omitted from the elution buffer during RDH13–His6 purification, the purified enzyme had a very low activity, but could be reactivated by the addition of dithiothreitol. A comparison of the more active and less active preparations of RDH13 by gel electrophoresis revealed that, in the absence of dithiothreitol, RDH13 appeared as two protein bands, one corresponding to the monomeric form of the protein and the other to the dimeric form (Fig. 5). After the addition of dithiothreitol, the dimer disappeared, shifting to the faster moving monomeric form of RDH13–His6. Glutathione (5 μM), which is the dominant low-molecular-weight thiol in the cell, had the same activating effect on RDH13 as dithiothreitol (data not shown). These results indicate that reducing conditions are essential for the maintenance of the active state of RDH13, and that nonreducing conditions promote the formation of inactive RDH13 dimers. In this respect, RDH13 appears to be similar to another member of the SDR superfamily, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) [12]. Like RDH13, 11β-HSD2 formed inactive dimers in the absence of 2-mercaptoethanol or dithiothreitol. The authors proposed that the inactive dimers could represent a latent form of the enzyme, and dimerization could serve as a mechanism for modulating the enzyme’s activity [12]. RDH13 activity was also affected by the nature of the detergent: the substitution of 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) for Tween-20 resulted in complete inactivation of the enzyme. In addition, RDH13 was sensitive to temperature, becoming partially inactivated after 20 min of incubation in the reaction buffer at 37 °C.
Fig. 5.
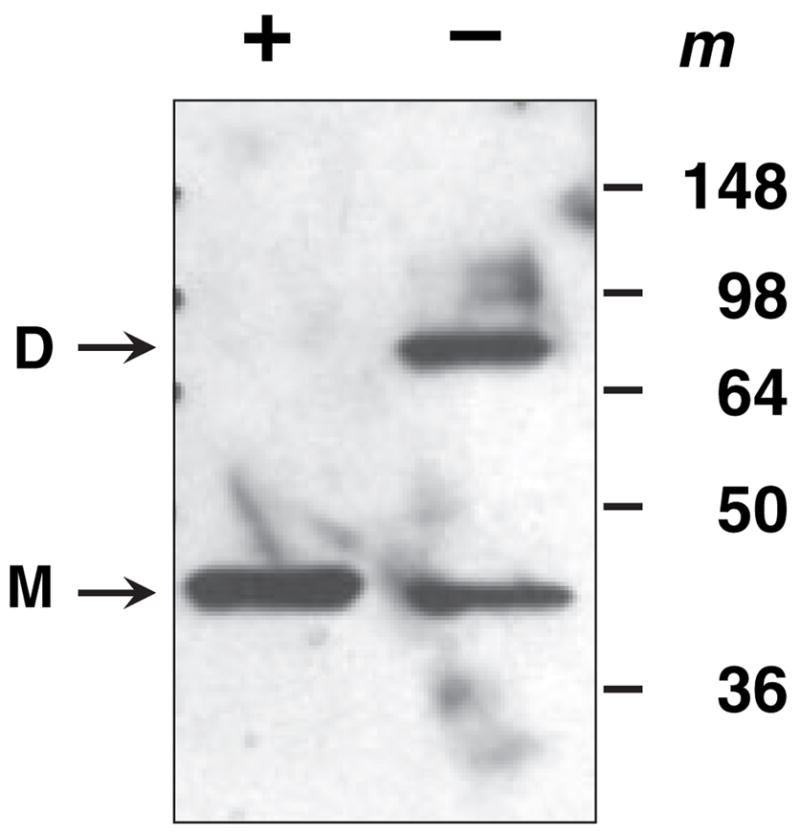
Effect of dithiothreitol on oligomeric state of RDH13–His6. RDH13 was purified and stored at −80 °C in the absence of reducing agents. Samples of this preparation were denatured in a boiling water bath for 5 min using gel loading buffer with (+) or without (−) dithiothreitol and analyzed by SDS-PAGE. The positions of the monomeric (M) and dimeric (D) forms of the protein are indicated on the left. m, molecular mass markers.
To determine the catalytic efficiency of RDH13, we carried out kinetic characterization of the purified enzyme (Table 1). This analysis showed that RDH13 reduced all-trans-retinaldehyde with an apparent Km value of 3.2 ± 0.7 μM and V max value of 230 ± 24 nmol·min−1·mg−1. The apparent Km value for all-trans-retinol (~ 3μM) appeared to be similar to that for retinaldehyde; however, the rate of retinol oxidation by RDH13 was extremely low (~ 5 nmol· min−1·mg−1), which precluded an accurate determination of the kinetic constants. The apparent Km value of RDH13–His6 for NADPH (1.5 ± 0.1μM) was three orders of magnitude lower than that for NADH (~ 6000 μM), consistent with its preference for NADPH as a cofactor. Thus, kinetic analysis reveals that RDH13 exhibits substrate and cofactor specificity very similar to that of RDH11, RDH12 and RDH14.
Table 1.
Kinetic constants of purified RDH13.
| Substrate/cofactor | Km (μM) | Vmax (nmol·min−1·mg−1) |
|---|---|---|
| All-trans-retinaldehyde | 3.2 ± 0.7 | 230 ± 24 |
| All-trans-retinol | ~3a | ~ 5 |
| NADPH | 1.5 ± 0.1 | 230 ± 24 |
| NADH | ~ 6000a | ~ 25a |
The determination accuracy of kinetic constants for the oxidation of retinol or the reduction of retinaldehyde in the presence of NADH as cofactor was limited by the low reaction rates.
RDH13–His6 was also tested for activity towards 17β-, 3α- and 11β-hydroxysteroids, and corresponding ketosteroids, as described for other SDRs [13–15]; however, no significant conversion was observed. Other compounds were examined as potential substrates by evaluating their ability to inhibit the RDH13-catalyzed reduction of all-trans-retinaldehyde. These compounds included short-chain aldehydes, such as nonanal, 6-cis-nonenal and 2-trans-nonenal, because they have been shown to be good substrates for RDH12 [7]. Glyceraldehyde and acetoacetyl-coenzyme A were tested because they have been found to be metabolized by another mitochondrial SDR, 17β-HSD10 [16]. In addition, we tested several commercially available derivatives of cholesterol, such as taurocholic acid, 25-hydroxycholesterol and 25-nor-5-cholesten-3-ol-25β-one, as some steps of cholesterol metabolism are catalyzed by cytochrome P450 enzymes associated with the inner membrane of mitochondria. No compound was inhibitory at a concentration of 50 μM, suggesting that they could not compete with retinaldehyde and, most probably, were not substrates for RDH13.
Thus, we have established that RDH13 is principally different from related RDH11, RDH12 and RDH14 in that it is targeted to the mitochondria, and is not an integral but a peripheral membrane protein associated with the inner mitochondrial membrane. Furthermore, RDH13 is much more labile than RDH11 and related microsomal proteins, and requires reducing conditions to stay active. At the same time, RDH13 is very similar to the members of the RDH11–14 cluster of SDRs in terms of its substrate and cofactor preferences.
Discussion
This study presents the first characterization of the tissue distribution, subcellular localization and catalytic activity of the recently discovered member of the SDR superfamily, RDH13. Western blot analysis of RDH13 distribution in human tissues carried out in this study shows that RDH13 is a widespread protein, being expressed at some level in seven of the eight human tissues examined. This protein expression pattern is in agreement with the presence of RDH13 transcripts in at least 32 adult tissues, as well as in embryonic and cancer tissues, as reported in the Expressed Sequence Tag GenBank database. Human RDH13 shares 83% protein sequence identity with mouse RDH13 and 72% identity with frog RDH13, and the corresponding genes have similar genomic organization [17], indicating that RDH13 is conserved across species. The high degree of protein conservation and the ubiquitous expression pattern suggest that RDH13 plays an important metabolic role. However, until recently, no enzymatic activity for RDH13 had been demonstrated.
RDH13 is most closely related to the NADP+-dependent microsomal enzymes RDH11, RDH12 and RDH14, which exhibit the highest activity as retinaldehyde reductases [4–8]. In this study, we have shown, for the first time, that purified RDH13 exhibits an oxidoreductive activity towards retinoids, strongly prefers NADPH over NADH as a cofactor, and has a much greater catalytic efficiency as a reductase than as a dehydrogenase. The catalytic efficiency of RDH13 as a retinaldehyde reductase is significantly lower than that of a related protein RDH11, primarily because of the much higher Km value for retinaldehyde (3 μM versus 0.12 μM for RDH11 [5]). However, the kcat value of RDH13 for retinaldehyde reduction (8.2 min−1) is comparable with that of RDH11 (18 min−1), and the Km values of the two enzymes for NADPH are also very similar (1.5 and 0.47 μM for RDH13 and RDH11, respectively [5]). Thus, consistent with its sequence similarity to RDH11, RDH12 and RDH14, RDH13 acts as an NADP+-dependent retinaldehyde reductase.
The surprising finding of this study is that RDH13 is localized in the mitochondria rather than in the endoplasmic reticulum, where the other members of RDH11–14 group are localized. This finding is supported by the immunolocalization of RDH13 in whole cells, as reported by Keller and Adamski [18] whilst this manuscript was in preparation. It is possible that mitochondrial RDH13 arose from the mistargeting of microsomal RDH enzymes during evolution, as has been suggested for mitochondrial P450s [19]. The exact sequence targeting RDH13 to the mitochondria remains to be established.
The analysis of the submitochondrial localization of RDH13 carried out here shows that RDH13 is associated with the inner mitochondrial membrane. The primary structure of RDH13 contains two hydrophobic segments, 2–21 and 242–261, which are suffi-ciently long to serve as transmembrane segments; however, as shown in the present study, alkaline extraction completely removes the protein from the membrane, indicating that RDH13 is a peripheral membrane protein [10]. The peripheral association of RDH13 with the membrane further distinguishes this protein from the microsomal retinaldehyde reductases, which are integral membrane proteins that appear to be anchored in the membrane via their N-terminal hydrophobic segments [5].
The results of the protease protection assays carried out in this study suggest that RDH13 is localized on the outer side of the inner mitochondrial membrane, facing the intermembrane space. This submitochondrial localization of RDH13 is consistent with the lack of a cleavable N-terminal mitochondrial targeting pre-sequence in the primary structure of RDH13, as shown by the lack of size difference between the in vitro translated and fully processed native RDH13 protein. It is well established that the mitochondrial targeting sequence is cleaved by matrix proteases on transfer of the protein across the inner mitochondrial membrane, and that all proteins of the mitochondrial outer membrane and some proteins of the intermembrane space and the inner membrane are devoid of such signals [20].
The association of RDH13 with the outer side of the inner mitochondrial membrane suggests that it is likely to be exposed to the cytosolic pool of substrates and cofactors [21], because the outer mitochondrial membrane is highly permeable. This is consistent with the function of RDH13 as a retinaldehyde reductase, as both retinaldehyde and NADPH can diffuse through the outer mitochondrial membrane. It should be noted that, with the exception of one study, which suggests that mitochondria contain cellular retinoic acid binding protein [22], mitochondria have not been previously considered to play a role in retinoid metabolism. However, recently, retinaldehyde has been implicated in the impairment of mitochondrial function resulting from increased consumption of β-carotene [23]. The anti-oxidant properties of β-carotene have been explored in smokers as part of intervention trials [23]. However, under the conditions of severe oxidative stress existing in smokers’ lungs, β-carotene appears to act as a pro-oxidant, causing a higher incidence of cancer. The primary product of the oxidative cleavage of β-carotene is the highly reactive retinaldehyde, which is formed in tissues by the widely expressed β-carotene monooxygenase [24]. Numerous studies have demonstrated that retinaldehyde is toxic for mitochondria. For example, retinaldehyde has been shown to inhibit adenine nucleotide translocase in a concentration-dependent manner [23], uncouple oxidative phosphorylation [25] and inhibit Na+/K+-ATPase activity more strongly than the endogenous major lipid peroxidation product 4-hydroxynonenal [26]. The incubation of mitochondria with retinaldehyde causes a dramatic decrease in the mitochondrial content of glutathione and protein-SH and increases the formation of highly toxic malonic dialdehyde, promoting oxidative stress in the mitochondria [27]. However, by contrast with retinaldehyde, retinol has been found to be protective against oxidative damage [23]. It can be speculated that the localization of detoxifying RDH13 retinaldehyde reductase at the entrance to the mitochondrial matrix may serve as a barrier protecting the mitochondria against the highly reactive retinaldehyde. Retinaldehyde reducing enzymes have been identified previously in the cytoplasm [28], endoplasmic reticulum [4–8] and peroxisomes [29]. This study expands the list of organelles containing retinaldehyde reductases to include mitochondria, suggesting that protection against retinaldehyde is universally required.
The mitochondrial localization might imply that RDH13 has other substrates in addition to retinaldehyde. However, none of the nonretinoid compounds tested in this study have been demonstrated to be utilized by RDH13. Nevertheless, the basic finding that RDH13 has a catalytic activity that can be tested using retinaldehyde provides a new opportunity for screening multiple candidate compounds as competitive inhibitors and potential substrates for RDH13. Additional studies are necessary to explore other potential functions for RDH13 in mitochondria in addition to the reduction of retinaldehyde.
Experimental procedures
DNA expression vectors
A full-length cDNA coding for RDH13 was obtained from the American Type Culture Collection (Manassas, VA, USA, IMAGE: 3687808 clone, ATCC No. 6111051). To prepare RDH13 tagged with the C-terminal His6, RDH13 cDNA was cloned into the pET28a vector (Novagen, Madison, WI, USA) between the NcoI and HindIII restriction sites. Because RDH13 contains an endogenous NcoI site, the coding sequence of RDH13 was PCR amplified starting with the second codon using the forward primer 5′-AG-CCGCTACCTGCTGCCGCT-3′ and the reverse primer 5′-CCAGAAGCTTTCTGGGGAGGGGCTGCTCCCT-3′ containing the HindIII restriction site (site in italic). The first codon (ATG) for RDH13 was provided by pET28a treated as follows. pET28a DNA was digested with NcoI restriction endonuclease, blunt ended using T4 DNA polymerase (New England Biolabs, Inc., Beverly, MA, USA), which created the ATG codon, and then digested with HindIII to provide a sticky end for RDH13 ligation. The PCR-amplified RDH13 lacking the ATG codon was gel purified, digested with HindIII restriction endonuclease and ligated in frame with the ATG codon supplied by the pET28a vector via blunt end/sticky end ligation.
To create a construct encoding RDH13–His6 for expression in Sf9 cells, the RDH13/pET28a vector was digested with XbaI and NotI endonucleases to excise a fragment containing the RDH13 coding sequence and a short portion of the pET28a polylinker. This fragment was ligated in frame with the His6 tag provided by the modified pVL1393 described previously [5]. Recombinant baculovirus was produced by cotransfection of Sf9 cells with the transfer vector and the linearized Sapphire™ Baculovirus DNA (Orbigen Inc., San Diego, CA, USA), according to the manufacturer’s instructions.
RDH13 expression construct in pCR4.2-TOPO was obtained from P. Nelson (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) and used for in vitro transcription/translation assay to determine the size of unmodified protein, as described previously [15].
Preparation of antibodies and western blot analysis
RDH13–His6 in pET28a vector was expressed in Escherichia coli BL21(DE3) strain and purified using Ni2+-nitrilotri-acetic acid metal affinity resin (Qiagen Inc., Valencia, CA, USA), according to the manufacturer’s protocol. The protein appeared to be inactive, but was obtained in quantities sufficient for antiserum production. Rabbit polyclonal antiserum against purified RDH13–His6 was raised at Alpha Diagnostics International Inc. (San Antonio, TX, USA).
For western blot analysis of RDH13 expression, samples of human tissue obtained from the Anatomical Gift Foundation (Laurel, MD, USA) were homogenized in 50 mM Hepes, pH 6.8, 2 mM dithiothreitol, 1 mM benzamidine and 1 mM EDTA, as described previously [14]. Proteins were separated by 12% SDS-PAGE, and transferred to Hybond™-P membrane (Amersham Biosciences, Piscataway, NJ, USA). The membrane was blocked with a 5% solution of BSA in Tris-buffered saline with 0.1% Tween-20, rinsed and incubated with RDH13 antiserum in the same buffer at a 1 : 4000 dilution.
Fractionation of LNCaP cells
Cells were harvested, washed with 10 mM Tris–HCl, pH 7.4, 0.25 M sucrose with protease inhibitors, and disrupted using a Dounce homogenizer. The homogenate was adjusted to 1.4 M sucrose by the addition of 2 M sucrose in 10 mM Tris–HCl. The sample was layered over 2 mL of 1.6 M sucrose in a centrifuge tube, and sequentially overlaid with 3 mL of 1.2 M, 1.5 mL of 0.8 M and 1 mL of 0.25 M sucrose. The gradient was centrifuged for 3 h at 207 000 g. in an SW41Ti Beckman rotor. One and half milliliter fractions were harvested, starting from the top of the gradient [9], and analyzed by western blotting using antiserum against RDH13 and antibodies against porin, golgin (Molecular Probes, Inc., Eugene, OR, USA), lamin (BD Biosciences, Palo Alto, CA, USA) and calnexin (Stressgen Biotechnologies, Victoria, BC, Canada), used at a 1 : 2000 dilution. The detection was performed using an enhanced chemiluminescence western blotting analysis system (Amersham Biosciences), according to the manufacturer’s recommendations.
Isolation of mitochondria and submitochondrial fractionation
LNCaP or Sf9 cells were collected, washed with NaCl/Pi and resuspended in mitochondria isolation buffer (15 mM Tris–HCl pH 7.4, 0.33 M sucrose, 0.025 mM EDTA) with protease inhibitors. Cells were homogenized using a glass–Teflon homogenizer. Unbroken cells, cell debris and nuclei were removed by centrifugation at 1000 g for 10 min. The supernatant was collected and centrifuged at 10 000 g for 10 min. Pellet representing the mitochondrial fraction was resuspended in H medium (70 mM sucrose, 210 mM mannitol, 2 mM Hepes pH 7.4) with protease inhibitors. EDTA was added to a final concentration of 1 mM.
Mitoplasts were prepared using French press, digitonin or hypotonic treatment as indicated. The results obtained with mitoplasts prepared by the three different methods were essentially identical. French press treatment of mitochondria was carried out as described previously [30,31]. Mitoplasts were separated from the outer membranes and intermembrane space proteins by differential centrifugation (10 min, 12 000 g). The 12 000 g pellet containing mitoplasts was resuspended in one-half of the supernatant volume and recentrifuged (10 min, 12 000 g). The 12 000 g supernatants were combined and further fractionated into the outer mitochondrial membranes and intermembrane space proteins by centrifugation for 90 min at 144 000 g. Purified mitoplasts were subjected to three cycles of freezing and thawing and then centrifuged at 144 000 g for 90 min to separate the matrix proteins from inner membrane proteins [32]. The inner membrane fraction was washed three times with NaCl/Pi to remove residual soluble proteins. The volumes of each mitochondrial fraction were recorded, and one-fiftieth of each fraction was analyzed by western blotting using anti-RDH13 serum.
The preparation of mitoplasts using digitonin was carried out by the addition of digitonin to mitochondria to a final concentration of 0.1% at a ratio of 0.125 mg·(mg protein)−1 [33]. Samples were incubated on ice for 15 min, diluted with H medium to 1 mL and centrifuged for 10 min at 10 000 g. Pellets were washed with 1 mL of H medium, centrifuged again for 10 min at 10 000 g and resuspended in the same medium. For hypo-osmotic preparation of mitoplasts, a mitochondrial suspension was diluted 20-fold with 2 mM Hepes, pH 7.4, incubated on ice for 15 min and centrifuged for 10 min at 10 000 g [34]. Pelleted mitoplasts were washed and resuspended in H medium.
Alkaline and detergent extractions
Inner mitochondrial membranes or mitoplasts were treated with 100 μL of one of the following buffers: NaCl/Pi; 1 M NaCl in 20 mM Tris–HCl, pH 7.4; 100 mM Na2CO3, pH 11.5; or 1% Triton X-100 in NaCl/Pi, pH 7.4. The samples were incubated for 30 min on ice, loaded onto 100 μL cushions of 0.5 M sucrose prepared in the respective treatment buffers and centrifuged for 1 h at 200 000 g. Pellets and supernatants were processed as described previously [15], and analyzed by western blotting using anti-RDH13 serum.
Purification of RDH13–His6 fusion protein from Sf9 cells
The expression of RDH13–His6 in insect Sf9 cells was carried out as described previously for RalR1/RDH11 and other microsomal SDRs [13–15]. Briefly, Sf9 cells were infected with the recombinant virus at a virus to cell ratio of 10 : 1 and incubated at 28 °C for 3–4 days. The mitochondrial fraction was isolated as described above, and then solubilized with 15 mM DHPC (Avanti Polar Lipids, Alabaster, AL, USA) in a buffer containing 100 mM potassium phosphate, pH 7.4, 150 mM potassium chloride, 0.1 mM EDTA, 20% glycerol, 5 mM 2-mercaptoethanol, 5 mM imidazole and protease inhibitors. Solubilization was carried out for 30 min on ice with continuous vortexing. To purify RDH13–His6, the extract was incubated with Ni2+-nitrilotriacetic acid resin (Qiagen Inc.) in a batch mode for 30 min on ice. The resin was washed with 120–150 bed volumes of buffer containing 40 mM potassium phosphate, 300 mM potassium chloride, 20% glycerol, 10 mM imidazole, 1 mM DHPC, 5 mM 2-mercaptoethanol and protease inhibitors. RDH13–His6 was eluted with a stepwise gradient of 50–500 mM imidazole in the same buffer, except that the concentration of potassium chloride was 150 mM. Fractions were analyzed by 12% SDS-PAGE. Purified RDH13–His6 preparations were stored at −80 °C. Some loss of enzymatic activity was observed after several months of storage.
HPLC analysis of RDH13 activity
The catalytic activity of RDH13–His6 and the RDH13-containing mitochondrial fraction was assayed as described previously [7]. Retinoids were extracted twice with 2 mL of hexane, separated in a hexane–tert-butyl-methyl ether (96 : 4) mobile phase at a flow rate of 2 mL·min−1 and analyzed using a Waters 2996 Photodiode Array Detector (Waters Corp., Milford, MA, USA). The stationary phase was a Waters Spherisorb S3W column (4.6 mm × 100 mm). On a typical chromatogram, the elution times were as follows: 3.17 min for 9-cis-retinal, 4.38 min for all-trans-retinal, 14.41 min for 9-cis-retinol and 15.59 min for all-trans-retinol. Retinoids were quantified by comparing their peak areas with a calibration curve constructed from the peak areas of a series of standards.
Determination of kinetic constants
The apparent Km values for the reduction of retinaldehyde were determined at 1 mM NADPH and five concentrations of all-trans-retinaldehyde (0.4–6.4 μM). The apparent Km values for the oxidation of retinol were determined at 1 mM NADP+ and six concentrations of all-trans-retinol (0.4–12.8 μM). The apparent Km values for reductive cofactors were determined at 5 μM all-trans-retinaldehyde and five concentrations of NADPH (0.4–6.4 μM) or NADH (0.4–6.4 mM). The reaction volume was varied between 0.5 and 1 mL and the reactions were incubated for 15 min. The concentration of purified RDH13–His6 in the reaction mixture was varied between 0.2 and 0.5 μg·mL−1, so that the amount of product did not exceed 10% of the initial substrate amount. The background value without cofactor was determined for each concentration of substrate and was subtracted from each data point. Reaction rates were determined on the basis of the percentage substrate conversion, as described previously [7]. Initial velocities (nanomole of product formed per minute per milligram of protein) were obtained by nonlinear regression analysis. Kinetic constants were calculated using GRAFIT (Erithacus Software Ltd, Horley, UK) and expressed as the mean ± standard deviation. The results shown are representative of three to four experiments.
The inhibitory effects of various compounds (at 50 μM) on the retinal reductase activity of RDH13 were investigated by adding the compounds to the reaction mixtures with 5 μM retinaldehyde as a substrate. Nonanal, 6-cis-non-enal, 2-trans-nonenal, 25-hydroxycholesterol (Sigma, St Louis, MO, USA) and 25-nor-5-cholesten-3-ol-25β-one (Steraloids, New Port, RI, USA) were added to the reaction mixtures from ethanol stocks; glyceraldehyde, taurocholic acid and acetoacetyl-coenzyme A were added from aqueous stocks.
Acknowledgments
We are grateful to Dr Peter Nelson (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) for providing RDH13 cDNA in pCR4.2-TOPO plasmid. This work was supported by the National Institute on Alcohol Abuse and Alcoholism (Grant AA12153).
Abbreviations
- DHPC
1,2-diheptanoyl-sn-glycero-3-phosphocholine
- HSD
hydroxysteroid dehydrogenase
- RDH
retinol dehydrogenase
- SDR
short-chain dehydrogenase/reductase
References
- 1.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, et al. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact. 2003:143–144. 247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 2.Jörnvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 3.Labrie F, Luu-The V, Lin SX, Labrie C, Simard J, Breton R, Bélanger A. The key role of 17 beta-hydroxysteroid dehydrogenases in sex steroid biology. Steroids. 1997;62:148–158. doi: 10.1016/s0039-128x(96)00174-2. [DOI] [PubMed] [Google Scholar]
- 4.Kedishvili NY, Chumakova OV, Chetyrkin SV, Belyaeva OV, Lapshina EA, Lin DW, Matsumura M, Nelson PS. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1) J Biol Chem. 2002;277:28909–28915. doi: 10.1074/jbc.M202588200. [DOI] [PubMed] [Google Scholar]
- 5.Belyaeva OV, Stetsenko AV, Nelson P, Kedishvili NY. Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry. 2003;42:14838–14845. doi: 10.1021/bi035288u. [DOI] [PubMed] [Google Scholar]
- 6.Haeseleer F, Jang G-F, Imanishi Y, Driessen CAGG, Matsumura M, Nelson PS, Palczewski K. Dual-substrate specificity of short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem. 2002;277:45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry. 2005;44:7035–7047. doi: 10.1021/bi050226k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belyaeva OV, Kedishvili NY. Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Lett. 2002;531:489–493. doi: 10.1016/s0014-5793(02)03588-3. [DOI] [PubMed] [Google Scholar]
- 9.Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM. Chapter 3: subcellular fractionation and isolation of organelles. John Wiley & Sons, Inc.; Hoboken, NJ: 2007. Current Protocols in Cell Biology. [Google Scholar]
- 10.Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Sanchez EP, Ganjam V, Chen YJ, Liu Y, Clark SA, Gomez-Sanchez CE. The 11beta hydroxysteroid dehydrogenase 2 exists as an inactive dimer. Steroids. 2001;66:845–848. doi: 10.1016/s0039-128x(01)00119-2. [DOI] [PubMed] [Google Scholar]
- 13.Gough WH, VanOoteghem S, Sint T, Kedishvili NY. cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3alpha-hydroxysteroids. J Biol Chem. 1998;273:19778–19785. doi: 10.1074/jbc.273.31.19778. [DOI] [PubMed] [Google Scholar]
- 14.Chetyrkin SV, Hu J, Gough WH, Dumaual N, Kedishvili NY. Further characterization of human microsomal 3α-hydroxysteroid dehydrogenase. Arch Biochem Biophys. 2001;386:1–10. doi: 10.1006/abbi.2000.2203. [DOI] [PubMed] [Google Scholar]
- 15.Chetyrkin SV, Belyaeva OV, Gough WH, Kedishvili NY. Characterization of a novel type of human microsomal 3α-hydroxysteroid dehydrogenase: unique tissue distribution and catalytic properties. J Biol Chem. 2001;276:22278–22286. doi: 10.1074/jbc.M102076200. [DOI] [PubMed] [Google Scholar]
- 16.He XY, Merz G, Yang YZ, Mehta P, Schulz H, Yang SY. Characterization and localization of human type10 17beta-hydroxysteroid dehydrogenase. Eur J Biochem. 2001;268:4899–4907. doi: 10.1046/j.0014-2956.2001.02421.2421.x. [DOI] [PubMed] [Google Scholar]
- 17.Kedishvili NY. Retinoid-active short-chain dehy-drogenases/reductases. In: Weiner H, Maser E, Lindahl R, Plapp B, editors. Enzymology and Molecular Biology of Carbonyl Metabolism – 13. Purdue University Press; West Lafayette, IN: 2007. pp. 217–223. [Google Scholar]
- 18.Keller B, Adamski J. RDH12, a retinol dehydrogenase causing Leber’s congenital amaurosis, is also involved in steroid metabolism. J Steroid Biochem Mol Biol. 2007;104:190–194. doi: 10.1016/j.jsbmb.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Werck-Reichhart D, Feyereisen R. Cytochromes P450: a success story. Genome Biol. 2000;1:reviews3003.1–reviews3003.9. doi: 10.1186/gb-2000-1-6-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diekert K, Kispal G, Guiard B, Lill R. An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc Natl Acad Sci USA. 1999;96:11752–11757. doi: 10.1073/pnas.96.21.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon DM, Dancis A, Pain D. Mechanisms of mitochondrial protein import. Essays Biochem. 2000;36:61–73. doi: 10.1042/bse0360061. [DOI] [PubMed] [Google Scholar]
- 22.Ruff SJ, Ong DE. Cellular retinoic acid binding protein is associated with mitochondria. FEBS Lett. 2000;487:282–286. doi: 10.1016/s0014-5793(00)02366-8. [DOI] [PubMed] [Google Scholar]
- 23.Siems W, Wiswedel I, Salerno C, Crifò C, Augustin W, Schild L, Langhans CD, Sommerburg O. Beta-carotene breakdown products may impair mitochondrial functions – potential side effects of high-dose beta-carotene supplementation. J Nutr Biochem. 2005;16:385–397. doi: 10.1016/j.jnutbio.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Lindqvist A, Andersson S. Biochemical properties of purified recombinant human beta-carotene 15, 15′-monooxygenase. J Biol Chem. 2002;277:23942–23948. doi: 10.1074/jbc.M202756200. [DOI] [PubMed] [Google Scholar]
- 25.Stillwell W, Nahmias S. Effect of retinol and retinoic acid on P/O ratios of coupled mitochondria. Biochem Int. 1983;6:385–392. [PubMed] [Google Scholar]
- 26.Siems WG, Sommerburg O, Hurst JS, van Kuijk FJ. Carotenoid oxidative degradation products inhibit Na+-K+-ATPase. Free Radic Res. 2000;33:427–435. doi: 10.1080/10715760000300961. [DOI] [PubMed] [Google Scholar]
- 27.Siems W, Sommerburg O, Schild L, Augustin W, Langhans CD, Wiswedel I. Beta-carotene cleavage products induce oxidative stress in vitro by impairing mitochondrial respiration. FASEB J. 2002;16:1289–1291. doi: 10.1096/fj.01-0765fje. [DOI] [PubMed] [Google Scholar]
- 28.Crosas B, Hyndman DJ, Gallego O, Martras S, Parés X, Flynn TG, Farrés J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J. 2003;373:973–979. doi: 10.1042/BJ20021818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei Z, Chen W, Zhang M, Napoli JL. Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: induction by the PPAR alpha ligand clofibrate. Biochemistry. 2003;42:4190–4196. doi: 10.1021/bi026948i. [DOI] [PubMed] [Google Scholar]
- 30.Decker GL, Greenawalt JW. Ultrastructural and biochemical studies of mitoplasts and outer membranes derived from French-pressed mitochondria. J Ultrastr Res. 1977;59:44–56. doi: 10.1016/s0022-5320(77)80027-0. [DOI] [PubMed] [Google Scholar]
- 31.Hoppel CL, Kerner J, Turkaly P, Turkaly J, Tandler B. The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J Biol Chem. 1998;273:23495–23503. doi: 10.1074/jbc.273.36.23495. [DOI] [PubMed] [Google Scholar]
- 32.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 33.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glick BS. Pathways and energetics of mitochondrial protein import in Saccharomyces cerevisiae. Methods Enzymol. 1995;260:224–231. doi: 10.1016/0076-6879(95)60140-6. [DOI] [PubMed] [Google Scholar]