

Abstract
Iron-sulfur proteins play indispensable roles in a broad range of biochemical processes. The biogenesis of iron-sulfur proteins is a complex process that has become a subject of extensive research. The final step of iron-sulfur protein assembly involves transfer of an iron-sulfur cluster from a cluster-donor to a cluster-acceptor protein. This process is facilitated by a specialized chaperone system, which consists of a molecular chaperone from the Hsc70 family and a co-chaperone of the J-domain family. The 3.0Å crystal structure of a human mitochondrial J-type co-chaperone HscB revealed an L-shaped protein that resembles Escherichia coli HscB. The important difference between the two homologs is the presence of an auxiliary metal-binding domain at the N terminus of human HscB that coordinates a metal via the tetracysteine consensus motif CWXCX9–13FCXXCXXXQ. The domain is found in HscB homologs from animals and plants as well as in magnetotactic bacteria. The metal-binding site of the domain is structurally similar to that of rubredoxin and several zinc finger proteins containing rubredoxin-like knuckles. The normal mode analysis of HscB revealed that this L-shaped protein preferentially undergoes a scissors-like motion that correlates well with the conformational changes of human HscB observed in the crystals.
Iron-sulfur proteins are a ubiquitous class of proteins that play essential roles in a wide range of biochemical processes in the cell (1). The unique characteristic of these proteins is that they harbor a distinctive class of prosthetic groups: the ironsulfur clusters formed from multiple iron and sulfide ions. The most common iron-sulfur clusters are tetranuclear [4Fe-4S]2+,+ and binuclear [2Fe-2S]2+,+. Usually, the clusters are attached to proteins by four cysteine residues, although in several cases other residue types have been as ligands (2). Many iron-sulfur proteins are considered to be evolutionarily ancient because they play important roles in some of the most fundamental biological pathways and processes (1). These processes include electron transfers in respiratory and photosynthetic electron transport chains, nitrogen fixation, and biosynthesis of coenzymes. In addition, iron-sulfur proteins can function as initiators of radical chemistry, as iron and/or iron-sulfur cluster storage proteins, as sensors of cellular iron levels, as sensors of oxygen levels involved in gene regulation of the aerobic and anaerobic respiratory pathways, or as sulfur donors (1, 3, 4).
The functions of iron-sulfur proteins have been studied extensively for decades, but the mechanism and control of their biogenesis has started to be elucidated only recently (5, 6). Three types of systems involved in the assembly of iron-sulfur clusters and maturation of iron-sulfur cluster proteins have been identified so far. These include the nif system of nitrogen fixing bacteria involved in maturation of nitrogenase (7), the suf (sulfur utilization factor) system that plays a significant role during iron and/or sulfur starvation and under oxidative stress conditions (8–10), and the essentially ubiquitous isc (iron-sulfur cluster) system that has a housekeeping role in the cluster assembly and maturation of the iron-sulfur proteins (11). In bacteria, the genes of the isc pathway are organized into an operon that encodes a regulatory protein IscR, a cysteine desulfurase IscS, a scaffold protein for iron-sulfur cluster preassembly IscU, an alternate scaffold and/or iron-binding protein IscA, a J-type co-chaperone HscB, Hsp70-class chaperone HscA, and ferredoxin Fdx (11–13). The majority of proteins from the bacterial isc pathway have been structurally characterized; these include IscS (14), IscU with bound zinc ion (15, 16) and other so far unpublished structures by structural genomics centers (Protein Data Bank codes 1wfz, 1xjs, and 2azh), IscA (20–22), HscB co-chaperone from Escherichia coli (23) the substrate-binding domain of HscA in complex with the IscU recognition peptide (24), and countless ferredoxin structures.
The current model of the biogenesis of iron-sulfur proteins in bacteria proposes two major steps. First, the iron-sulfur cluster is assembled on a scaffold protein (IscU) in a series of reactions facilitated by IscS and an iron donor protein (25–27). In the next step, the preformed iron-sulfur cluster is transferred to the acceptor apo-protein (28). The transfer is facilitated by a specialized molecular chaperone system that consists of a chaperone from the Hsp70 family, HscA, and a J-type co-chaperone, HscB (29). The co-chaperone HscB assists in delivery of IscU to HscA and enhances the intrinsic ATPase activity of the chaperone (30, 31). The chaperone HscA specifically binds a conserved motif in the iron-sulfur scaffold protein IscU (24, 32). Although the molecular mechanism of iron-sulfur cluster transfer has not been fully elucidated, it has been suggested that the chaperones facilitate cluster delivery to an acceptor apoprotein by destabilization of the IscU·[FeS] complex (6). The isc biosynthetic pathway has been also described in fungi and higher eukaryotes. The biosynthesis of iron-sulfur proteins in eukaryotes is, however, less understood than in bacteria (5).
Here we present the x-ray structure of human co-chaperone protein HscB (hHscB)2 (Ref. 33; UniProt ID HSC20_HUMAN) at 3.0 Å resolution. The overall architecture of hHscB resembles that of its E. coli homolog (with 29% sequence identity); however, comparison of the two structures revealed some important differences. Most notably, hHscB possesses a novel metal-binding domain at the N terminus. This domain, which is conserved in HscB homologs from many eukaryotic species, uses a tetracysteine motif situated in the two rubredoxin knuckles to bind a metal ion. Human HscB, which had <30% overall sequence identity to any structure in the Protein Data Bank at the time of its selection and deposition, was chosen as a “sequence-to-structure” target under the National Institutes of Health Protein Structure Initiative.
EXPERIMENTAL PROCEDURES
Expression and Purification of hHscB Constructs—Bioinformatic analysis of the hHscB sequence suggested that residues 1–21 comprise a signal peptide whose presence was expected for a mitochondrial protein. To maximize the chances for success, a series of truncation constructs were prepared from the cDNA encoding hHscB: Δ(1–21)HscB (internal Centre for Eukaryotic Structural Genomics ID go.91294), Δ(1–29)HscB (go.91296), and Δ(1–35)HscB (go.91297). The cDNA encoding these constructs was cloned, and selenomethionine-labeled proteins were expressed and purified following the standard Center for Eukaryotic Structural Genomics pipeline protocols for cloning, protein expression, protein purification, and overall bioinformatics management (34–37). Briefly, truncated cDNAs were amplified by two-step PCR and cloned into pVP16, a custom plasmid derived from pQE80 (Qiagen). Δ(1–26)HscB (go.91295) failed at the PCR stage. All other targets proceeded through successful protein purification. The proteins were expressed in Escherichia coli B834 p(lacI+RARE) cells using 2 liters of auto-induction medium (38). Upon sonication of the harvested cells, the protein in the supernatant was purified by immobilized nickel affinity chromatography, and TEV protease was used to cleave the affinity/solubility tag consisting of His8 maltose-binding protein. After tag capture by subtractive nickel affinity chromatography and a final desalting step, the proteins were concentrated to 10 mg/ml and dialyzed against the protein buffer (50 mm NaCl, 3 mm NaN3, 0.3 mm tris(2-carboxyethyl)phosphine hydrochloride, 5 mm bis-Tris, pH 7.0). Protein aliquots were then drop frozen in liquid nitrogen and stored at 193 K. Protein purifications resulted in ∼30.0 mg of protein with ∼89% selenomethionine incorporation for all constructs. All three purified samples had the same light green color and similar metal analysis results (∼0.3 mol of nickel and ∼0.2 mol of zinc ions/mol of protein).
Crystallization and Data Collection—Protein samples were screened against commercial (Hampton Research, Aliso Viejo, CA) and custom solutions in microplate format and imaged with a CrystalFarm (Bruker AXS, Madison, WI). The best crystals from initial screening were obtained for Δ(1–29)HscB against 25% polyethylene glycol 3350, 200 mm Li2SO4, 100 mm PIPES, pH 6.5 at 293 K. The best optimized crystals of Δ(1–29)HscB were obtained at 293 K by the hanging drop method from a 10 mg ml-1 protein solution in the protein buffer mixed with an equal amount of well solution containing 16% (w/v) polyethylene glycol 3350, 50 mm Li2SO4, 100 PIPES, pH 6.5. Crystals were cryoprotected at 293 K by soaking in four steps in solutions containing 20% (w/v) polyethylene glycol 3350, 50 mm Li2SO4, 100 mm PIPES, pH 6.5, and 0, 7, 14, and 20% (v/v) ethylene glycol, respectively. Cryoprotected crystals were flash frozen in liquid nitrogen. After extensive synchrotron screening of tiny (<50 μm in the largest dimension) and poorly diffracting crystals, the diffraction data were collected from the best specimen with a diffraction limit of about 3 Å. The data set was collected near the selenium K absorption edge (0.97934 Å; 12,660 eV) at the General Medicine and Cancer Institutes Collaborative Access Team 23-ID-D beamline at the Advanced Photon Source at Argonne National Laboratory. The diffraction images were integrated and scaled using HKL2000 (39).
Structure Solution—The selenium substructure of selenomethionine-labeled Δ(1–29)HscB crystals was determined by using HySS (40) and ShelxD (41). The programs identified 10 consensus anomalous sites, which suggested the presence of two molecules in the asymmetric unit. The structure was phased automatically and density-modified by using autoSHARP (42) with the help of auxiliary programs from the CCP4 suite (43). Inspection of the experimental electron density map at this stage revealed several recognizable helices organized into helix bundles. A model of bacterial HscB (23) (Protein Data Bank code 1fpo) was manually placed into the electron density map. Inspection of the crystal packing that resulted from placement of the first molecule confirmed that a second molecule had to be present in the asymmetric unit to bridge the disconnected layers formed by the first molecule and its symmetry mates. However, no additional recognizable features were present in the map to guide placement of the second molecule in the asymmetric unit. Therefore ShelxC, ShelxD, and ShelxE programs (41) were used to attempt an independent phasing. The resulting electron density map showed (marginal) additional features that allowed the second molecule of E. coli HscB to be placed into the asymmetric unit. The angle between the two helical bundle domains of the L-shaped HscB molecule had to be adjusted to obtain a satisfactory fit. The initial model of the asymmetric unit obtained from this procedure was rigid body refined and was used in additional model mask-guided density modification trials as implemented in the density improvement strategy of autoSHARP. This approach resulted in higher quality experimental electron density maps that allowed for more reliable model building. The structure was completed with multiple cycles of iterative manual building in Coot (44), refinement in REFMAC5 (45), and model mask constrained density modification in autoSHARP. All of the refinement steps were monitored using an Rfree value based on 8.1% of the independent reflections. The stereochemical quality of the final model was assessed using MOLPROBITY (46). The figures were prepared using PyMOL (47).
Normal Mode Analysis—Normal mode analysis is a computational technique used to predict the preferred directions of flexibility of protein structures (48–50). The technique is based on the assumption that the native state is the minimum energy configuration and that the potential around it can be approximated by a quadratic function. Given a potential energy model of intramolecular interactions, the second derivative (Hessian) matrix gives the shape of the quadratic energy well around the native state. Diagonalization of the Hessian matrix yields the normal modes of deformation (eigenvectors) and the corresponding harmonic frequencies (square roots of the eigen values). The contribution of each mode to the total flexibility is proportional to the reciprocal of the frequency, because the lowest frequency modes represent the largest, most global deformations. Different models may be used to define the potential, ranging from complicated chemical force fields to simple models based on distances within the protein structure, known as elastic network models (51). In the latter class of models, atoms or residues within a certain distance of each other are connected by linear (Hookean) spring potentials. Recently our group had proposed a residue level model, called the distance network model, that defines interactions between two residues based on distances between atoms from the respective residues (52). In this model, atomic contacts at different distances are given different weights (spring constants) and are then added together to form the total Hessian. In our implementation of the algorithm, the Hessian matrix is calculated using an in-house Perl script, and MATLAB (MathWorks, Natick, MA) is used to diagonalize the Hessian matrix of the system and analyze the normal modes.
Normal modes were computed for chain A of hHscB (Protein Data Bank code 3bvo). The conformational differences between chains A and B of hHscB were compared with the deformations predicted by the lowest frequency normal modes. Dot products were computed between the low frequency modes and the difference vector between the Cα coordinates of monomers A and B. The dot product, defined below, is equivalent to a linear (Spearman) correlation, and its square represents the fraction of variance in the data (conformational change, v) captured by the prediction (normal mode, u).
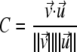 |
(Eq. 1) |
The comparison had to be restricted to a subset of the residues for which coordinates were available in both chains A and B, specifically residues 40–46, 53–143, and 156–234. Because the normal mode vectors were effectively restricted (projected) to a subspace of the total normal mode space, they no longer formed an orthonormal basis set. Although the square dot product with each normal mode still measures the accuracy of prediction, the total sum of the dot products may exceed one. This means that the sum of the square dot products slightly overestimates the fraction of the overall variation of the conformational change represented by those normal modes.
RESULTS
Structure Quality—The mature recombinant human co-chaperone Δ(1–21)HscB did not yield diffraction quality crystals from our crystallization screens. Therefore we cloned and expressed additional N-terminal truncation constructs and tested them in further crystallization trials. The N-terminal truncated version Δ(1–29)HscB (from now on referred to as hHscB) yielded diffraction quality crystals that supported the structure determination. A single-wavelength anomalous diffraction method (53) was used to phase the 3.0 Å diffraction data set collected from a crystal of selenomethionine-labeled hHscB near the selenium K absorption edge. The structure was refined to a crystallographic R factor of 23.6% and free R factor of 28.8%. Details of data collection, phasing, and refinement statistics are summarized in Table 1. hHscB crystallized in space group P21 with two molecules in the asymmetric unit. The final electron density map corresponding to molecule A of the hHscB model, which was of a quality expected for a ∼3 Å map (Fig. 1a), supported the building of a continuous polypeptide chain. However, two segments of molecule A (residues 48–53 and 148–159) had a markedly lower map quality. In fact, residue Asp54 of molecule A could not be modeled satisfactorily and remained an outlier in the Ramachandran plot. The final electron density map corresponding to molecule B of the hHscB model was of much lower quality, and several segments could not be modeled at all. The final model of hHscB therefore consists of residues 39–235 for molecule A and residues 40–46, 53–143, and 153–234 for molecule B. Residues not resolved in the electron density map were not modeled. Medium noncrystallographic symmetry constrains, as defined in the standard refinement protocol of Refmac5, were applied during the model refinement to two segments spanning residues 40–143 and 157–234.
TABLE 1.
Crystal parameters, x-ray data collection, phasing, and refinement statistics
| Peak | |
|---|---|
| Space group | P21 |
| Unit cell parameters (Å, °) | a = 63.6, b = 32.8, c = 114.4, β = 105.2 |
| Data collection statistics | |
| Wavelength (Å) | 0.97934 |
| Energy (eV) | 12,660 |
| Resolution range (Å) | 44.75–3.00 (3.11–3.00) |
| No. of reflections (measured/unique)a | 61747/9770 |
| Completeness (%) | 96.7 (79.8) |
| Rmergeb | 0.086 (0.342) |
| Redundancy | 6.3 (4.3) |
| Mean I/sigma(I) | 10.0 (3.0) |
| Phasing statisticsc | |
| Phasing power (isomorphous/anomalous) | 0.0/1.37 |
| Mean FOM (centric/acentric) | 0.09/0.285 |
| Rcullis (isomorphous/anomalous) | 0.0/0.736 |
| Refinement and model statistics | |
| Resolution range | 44.75–3.00 (2.15–2.10) |
| No. of reflections (total/test) | 9242/753 |
| Rcrystd | 0.236 (0.264) |
| Rfreee | 0.288 (0.386) |
| Root mean square deviation bonds (Å) | 0.008 |
| Root mean square deviation angles (°) | 1.047 |
| ESU from Rfree (Å) | 0.542 |
| TLS groups (residue range) | A39–A70, A71–A147, A148–A158, A159–A235 B40–B70, B71–B143, B158–B234 |
| B factor - Wilson (Å2) | 75.5 |
| Average B factor – molecule A/B (Å2) | 72.4/72.8 |
| No. of protein molecules/all atoms | 2/3103 |
| No. of waters/zinc(II)/sulfates | 0/2/1 |
| Ramachandran Plot by MOLPROBITY (%) | |
| Favored regions | 95.1 |
| Additional allowed regions | 4.6 |
| Outliers | 0.3 (Asp-A54) |
| Protein Data Bank code | 3bvo |
The values in parentheses are for the highest resolution shell
Rmerge = ΣhΣi|Ii(h) – 〈I(h)〉|/ΣhΣiIi(h), where Ii(h) is the intensity of an individual measurement of the reflection, and 〈I(h)〉 is the mean intensity of the reflection
Phasing by SHARP in 36.78–3.00 Å resolution range
Rcryst = Σh|Fobs| – |Fcalc|/Σh|Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively
Rfree was calculated as Rcryst using ∼8.1% of the randomly selected unique reflections that were omitted from structure refinement
FIGURE 1.

Three-dimensional structure of hHscB. a, stereodiagram of a representative portion of the 3.0 Å 2mFo-DFc electron density map (blue mesh) of hHscB and final refined model (sticks). b, stereodiagram of Cα traces of structurally superposed hHscB (red) and E. coli HscB (cyan). The structure of hHscB consists of three domains: the N-domain, which is not found in E. coli HscB, and the J- and C-domains connected by a short linker region (black arrow). The red sphere represents a metal ion coordinated by the N-domain.
Similarities and Differences between E. coli and Human HscB—The phase improvement and subsequent building of the hHscB model was guided by the structure of E. coli HscB (23) (Protein Data Bank code 1fpo; see “Experimental Procedures” for details). The two proteins show 29% sequence identity in the aligned regions and share the same overall fold (Fig. 1b). The most notable difference between the structures is that in addition to domains homologous to the J- and C-domains of E. coli HscB, hHscB possesses a novel N-terminal domain (the N-domain) capable of binding a metal ion (Fig. 1b, metal shown as a red sphere). The second important difference is related to the relative orientations of the J- and C-domain in these molecules. This subject will be treated in detail after the comprehensive description of the structure of hHscB.
The N-Domain of hHsbB Possesses a Tetracysteine Metal-binding Motif—The structure of hHscB can be divided into three topologically distinct domains, the N-domain (residues 39–71), the J-domain (residues 72–145), and the C-domain (residues 156–235; Fig. 1b). The N-domain, which lacks any recognizable secondary structure elements, forms a small globular domain that harbors a metal-binding site. The metal atom is coordinated by a set of four cysteine residues Cys41, Cys44, Cys58, and Cys61 (Fig. 2a, inset, blue sticks) located on the two apposed β-hairpins. The multiple sequence alignment of the N-domain sequences from a range of hHscB homologs (Fig. 2b) revealed a consensus sequence CWXCX9–13FCXXCXXXQ that contains the four cysteines involved in metal coordination. The metal was tentatively modeled as zinc(II), based on the results of a metal analysis performed on the recombinant protein sample used for crystallization. The analysis revealed the presence of a substoichiometric mixture of nickel and zinc ions in the protein sample (0.33 mol of nickel and 0.23 mol of zinc/mol of protein). The observed nickel ions likely represent a contamination resulting from a purification protocol that employed nickel affinity chromatographic steps. However, the true identity of the metal that the N-domain binds in vivo remains an open question.
FIGURE 2.

The N-domain of hHscB. a, a ribbon representation of hHscB is shown in a rainbow-coloring scheme from the N terminus (blue) to the C terminus (red). The helices of the J- and C-domains are labeled with capital letters. The conserved HPD motif of the J-domain (blue helix A and green) and acidic patch of the C-domain (yellow to red) are shown as sticks. The inset shows a stereo diagram of the N-domain in line and ribbon representations. The dashed yellow lines represent possible hydrogen bonds stabilizing the structure. Selected residues referred to in the text are labeled. The red sphere represents a metal ion coordinated by four conserved cysteine residues. b, multiple sequence alignment of the N-terminal domains of the hHscB homologs from a range of species. The sequences of hHscB homologs were selected based on the profile-profile alignment by the FFAS03 server (61) of hHscB against 85% nonredundant data base, and the N-domains were realigned using the Multalin server (58). The alignment suggests a consensus sequence CWXCX9–13FCXXCXXXQ for this domain.
The N-domain Is Similar to C-4 Zinc Finger Domains and Rubredoxin—Based on the Vector Alignment Search Tool server (54), the N-domain is unique and does not closely resemble any previously described proteins. However, several distant structural homologs with 14–18% identity could be identified using the FATCAT server (55). These include solution structures of RanBP2/NZF type zinc finger domains found in nuclear pore complex protein Nup153 (Protein Data Bank codes 2ebr and 2ebq) and protein Hepatitis B Virus-associated factor (Protein Data Bank code 2crc) as well as NZF domain of Npl4 (59) (Protein Data Bank code 1nj3), and C-4 zinc finger domain of HDM2 (60) (Protein Data Bank code 2c6a). Several additional proteins that contain a tetracysteine zinc-binding module distantly similar to the N-domain of hHscB were identified by a profile-profile sequence search using the FFAS03 server (61). These include protein PF0610 from Pyrococcus furiosus (62) (Protein Data Bank code 2gmg), polypeptide 8 of DNA-directed RNA polymerase II (63) (Protein Data Bank code 1i3q, chain I), the RING domain from tripartite motif-containing protein 31 (Protein Data Bank code 2ysl), and finally Sec23a and Sec24a proteins (64) (Protein Data Bank code 2nup).
The metal coordinating loops of all these proteins show local similarity to the “knuckles” of rubredoxin, a well known tetracysteine iron-binding protein involved in a range of redox reactions, including oxidative stress response pathways in microaerophillic/anaerobic bacteria (65). The structural alignment in Fig. 3 depicts the N-domain of hHscB (red), the NZF domain of Npl4 (59) (cyan), and Clostridium pasteurianum rubredoxin (66) (blue). The alignment shows that these proteins share the same overall topology, and their backbones are closely similar around the metal-binding site; however, the structural segments connecting individual rubredoxin knuckles show wide variations.
FIGURE 3.

Structural homologs of the N-domain. A stereo diagram depicts Cα traces of structurally superposed hHscB (blue), zinc-substituted rubredoxin from C. pasteurianum (66) (Protein Data Bank code 1irn, red), and NZF domain of Npl4(59) (Protein Data Bank code 1nj3, cyan). Four cysteine residues located in the apposed rubredoxin knuckles provide coordination for a metal ion (red sphere).
Structural Details of the N-domain—The core of the N-domain of HscB, which is relatively polar, is likely stabilized by hydrogen bonds between the side chain N-atom of Trp48, the side chain amide of Gln65, and the backbone carbonyl of Ala63 (Fig. 2a, inset). Residue Arg55 may form up to three hydrogen bonds to the backbone carbonyls of Gly49, Gly51, and Glu53. Finally, the indole N-atom of Trp42 may form a hydrogen bond to the backbone carbonyl of Leu64. Residues Trp42 and Gln65 are highly conserved in homologs of hHscB and are a crucial part of the consensus motif of the N-domain, CWXCX9–13FCXXCXXXQ, identified from the multiple sequence alignment of these sequences (Fig. 2b). The interface of the N- and J-domains buries surface area of 610 Å2 and is stabilized by multiple residues in van der Waals' contacts, hydrogen bonds, and salt bridges. Specifically, residues forming interdomain hydrogen bonds include Asp72, Ser75, Arg93, Glu113, and His120 of the J-domain and Asp54, Leu64, Gln65, Thr70, and Arg71 from the N-domain. Residues Asp54 and Arg71 of the N-domain are involved in electrostatic interaction with Arg93 of helix B of the J-domain. In addition to the above mentioned residues, the residues involved in significant van der Waals' contacts include Leu76, Leu97, Leu100, Val101, Phe106, Thr112, Phe116, and Leu123 from the J-domain and Trp42, Phe56, Phe57 (conserved in the consensus motif), Ala66, and Pro67 from the N-domain.
Structural Details of the J-domain—The J-domain is arranged into the orthogonal helical bundle with helix hairpins topology (67). The domain consists of five helices connected by short loops (Fig. 2a). Helix A, which follows the N-domain, is connected by an eight-residue linker to the central helix bundle formed by helices B, C, and D. These helices are arranged in an antiparallel direction (B and C versus D) to form a bundle with a helix hairpin topology. The loop connecting helices B and C harbors the signature J-domain motif HPD (residues His102-Pro103-Asp104 of hHscB), which has been implicated in interactions with molecular chaperones of the Hsp70 family (68). Finally, the J-domain is completed by a three-turn long helix E, which packs against helix A, and a linker connecting helices A and B. Helix E is positioned at the base of the central helix bundle and runs at an approximately 80-degree angle away from helix D.
Structural Details of the C-domain—The C-domain is formed by three helices arranged into the three-helical up-and-down bundle with monooxygenase topology (67). The C-domain is connected to the J-domain via a flexible linker (residues 146–157), which was completely disordered in molecule B of hHscB and marginally resolved in the electron density maps for molecule A of hHscB. The J- and C-domains meet at an approximate 90-degree angle to create an L-shaped molecule. The C-terminal domain contains a hydrophobic core formed by the interaction of nonpolar side chains from the three amphipathic helices F, G, and H. Although the three-helix bundle found in the C-domain of hHscB is a common structural motif seen in a wide variety of proteins, the combination of the J- and C-domain of hHscB represents a unique modular arrangement found so far only in E. coli HscB (24) (Protein Data Bank code 1fpo), which was the closest homolog of hHscB in the Protein Data Bank at the time of writing of this manuscript. The C-terminal domain of E. coli HscB has been implicated in binding and targeting IscU to the bacterial chaperone HspA (30). Several acidic residues on the surface of helix F, which are conserved in HcsB homologs from different organisms (residues Glu167, Glu170, and Glu174 in hHscB), have been suggested as sites for interaction with the scaffold protein IscU (23). Complex formation between E. coli IscU and HscB resulted in experimentally observable changes in NMR signals localized to the area comprising these acidic residues (69). In addition, the alanine shaving of the C-domain of Jac1p, a yeast HscB homolog, confirmed that some of these residues contribute to the interaction with Isu1p, a yeast homolog of IscU (70).
Conformational Variability of hHscB—Molecules A and B of hHscB show a somewhat different arrangement of their J- and C-domains. Alignment of J- and N-domains of the two hHscB monomers (root mean square deviation of 0.3 Å) leads to a displacement of the C-terminal domains by as much as 10 Å (Fig. 4a, curved arrow). The J-domain and the C-domain of hHscB are connected by a 12-residue loop (residues 143–157). This loop had poorly defined electron density in molecule A of hHscB and was completely disordered in molecule B, probably because of the high mobility of this region. The corresponding loop in the E. coli HscB has been identified similarly as a highly mobile segment of the protein by B-factor analysis (23) and confirmed by NMR analysis (69). This loop represents the most structurally divergent segment between hHscB and E. coli HscB (Fig. 1b, black arrow). Molecule A of hHscB and molecule B of E. coli HscB show the best superposition, with an all-atom root mean square deviation of 1.2 Å. On the other hand, molecule B of hHscB aligns to molecule B of E. coli HscB with a much lower root mean square deviation (2.8 Å), consistent with the significantly altered arrangement of the J- and C-domains between these proteins. These comparisons suggested that the HscB proteins might be much more conformationally flexible than previously expected (23). To investigate this in a more rigorous way, we analyzed the structure of hHscB using normal mode analysis.
FIGURE 4.

Observed and theoretical conformational variability of hHscB. a, stereodiagram of Cα traces of chain A (red) and chain B (cyan) of hHscB as observed in the crystal lattice. The N- and J-domains of the both chains were structurally superposed to reveal conformational differences in the arrangement of C-domains. The black arrow points to the topologically equivalent residues at the tip of the C-domain that were displaced by ∼10 Å. The red sphere represents a metal ion coordinated by the N-domain. b, the colored rods represent a direction (white to red) and amplitude of motion of the hHscB Cα-atoms along the three lowest frequency nontrivial normal modes (left to right). All of the modes are also depicted using simplifying schematic drawings. The first lowest frequency nontrivial normal mode (left) can be understood as scissors-like opening and closure of the molecule of hHscB. The second lowest frequency nontrivial normal mode (middle) reveals a rotational motion of the N- and J-domains and sideways twisting motion of the C-domain. Finally, the third lowest frequency nontrivial normal mode (right) reveals the rotational motion of the C-domain and sideways twisting motion of the N- and J-domains.
Normal Mode Analysis of hHscB—First, normal modes of the A chain of hHscB were calculated using the Distance Network Model as described under “Experimental Procedures.” Under the harmonic assumptions, the modes with the lowest frequencies contribute the most to the conformational flexibility of the structure. The fraction of the total variation in all of the normal modes contributed to by each of the 10 modes with the lowest frequencies was calculated as a fraction of the total reciprocal frequencies squared. Table 2 shows that the three lowest frequency modes account for over 46% of the variation out of the ensemble of 591 normal modes, illustrating the reduction of complexity accomplished by normal mode analysis. The first lowest nontrivial normal mode (Fig. 4b, left panel) predicts a scissors-like motion with individual blades formed by the N- and J-domains and C-domain, respectively. The second lowest nontrivial mode (Fig. 4b, middle panel) suggests a movement involving a rotation of the N- and J-domains along the axis of the J-domain and sideways twisting of the C-domain. In the third lowest nontrivial mode, the roles of the blades switch, i.e. the rotation of the C-domain along the axis of the C-domain is accompanied by coupled sideways twisting of the N- and J-domains (Fig. 4b, right panel). The complex movements of hHscB corresponding to these modes can be viewed in the supplementary movies accompanying this manuscript (supplemental Movies S1–S3). Next, the deformations predicted by the 10 lowest frequency normal modes were compared with the conformational differences between the A and B chains of hHscB observed in the asymmetric unit of crystal. The comparison was restricted to the residues for which coordinates are available in both chains, because the model for chain B lacks coordinates for 22 residues. Table 2 shows square dot products between the normal modes and the vector of conformational changes between chains A and B. The lowest frequency mode coincides very well with the conformational changes, accounting on its own for 73% of the conformational change. Together, the three lowest frequency modes account for over 90% of the conformational changes between the two chains in the asymmetric unit. Furthermore, we computed the normal modes for E. coli HscB and compared them to those of hHscB (matching residues 1–74 with 71–144 and residues 90–165 with 160–235 of the J- and C-domains of E. coli HscB and hHscB, respectively). The conformational variability content of the E. coli HscB normal modes shows a very similar distribution to those of hHscB (Table 2, compare first and third columns). The correlation between the first lowest frequency nontrivial modes of the two structures was 0.906. The correlation for the corresponding second and third modes were 0.323 and 0.241, respectively. However, comparing mode 2 of hHscB and mode 3 from E. coli HscB resulted in the correlation of 0.857, and comparison of mode 2 from E. coli HscB with mode 3 from hHscB gave the correlation of 0.612. These results thus indicate that the relative importance of these topologically analogous modes flipped for the two structures.
TABLE 2.
Analysis of normal modes of hHscB and E. coli HscB
|
Nontrivial normal
modea
|
hHscB
|
Normalized weight of E. coli
HscBb
|
|
|---|---|---|---|
| Normalized weightb | Fraction of conformational changec | ||
| % | % | % | |
| 1 | 22.28 | 73.07 | 21.31 |
| 2 | 13.04 | 3.12 | 13.38 |
| 3 | 10.99 | 15.51 | 11.04 |
| 4 | 4.48 | 1.02 | 4.48 |
| 5 | 3.62 | 0.01 | 3.66 |
| 6 | 2.61 | 0.26 | 2.62 |
| 7 | 1.92 | 0.04 | 1.92 |
| 8 | 1.83 | 0.06 | 1.82 |
| 9 | 1.43 | 1.58 | 1.44 |
| 10 | 1.22 | 0.01 | 1.22 |
Normal modes not related to protein translation and/or rotation in order of increasing frequency
Fraction of total normal mode variance generated by the mode
Fraction of conformational changes between chains A and B of hHscB predicted by individual normal modes from computed from chain A of hHscB
DISCUSSION
The most important observation provided by the structure of hHscB is that it contains a novel domain capable of binding a metal ion. The PSI-BLAST search revealed that this domain is present in a range of eukaryotic organisms, including higher eukaryotes (mouse, frog, mosquito, and opossum), plants (rice, grape vine, and thale crest), parasites (Plasmodium and Trichomonas vaginalis), and several interesting bacterial species detailed below. The domain coordinates a metal ion via a tetracysteine motif from the consensus sequence CWXCX9–13FCXXCXXXQ, which was identified for this domain from the alignment (Fig. 2b). The recombinant protein sample used for crystallization experiments in this study contained a substoichiometric mixture of nickel and zinc ions. The structure of the metal-binding site of the N-domain of hHscB is reminiscent of “rubredoxin knuckles” found in the iron-binding site of rubredoxin, as well as in a range of related zinc finger proteins. The N-domain could thus act as a zinc finger domain that gives additional functional properties to hHscB compared with E. coli HscB. Hypothetically, the N-terminal domain of hHscB could mediate the interaction with specialized transport proteins and/or facilitate the export of iron-sulfur clusters across the mitochondrial membrane. Another intriguing possibility is that the N-domain of hHscB binds other metals, perhaps an iron ion, and could be involved in one-electron redox reactions. The domain could possibly provide electrons during formation of cubane-like [4Fe-4S] iron-sulfur clusters from linear [2Fe-S] clusters (25, 71). Further studies will be required to identify the physiologically relevant metal bound by hHscB. The structural similarity between iron(II) and zinc(II)-loaded rubredoxin (66) suggests that this issue could not be settled satisfactorily by means of x-ray crystallography alone.
Another fascinating observation about the N-terminal domain also points toward the possible involvement of this domain in redox processes; the domain is found in HscB homologs of only a few bacterial species (Fig. 2b) that possess unusual iron and/or heavy metal-dependent metabolic pathways. These species include magnetotactic bacteria Magnetospirillum magneticum, Magnetospirillum gryphiswaldense, and Magnetococcus, as well as chemolithoautotrophic Leptospirillum ferrooxidans and Anaeromyxobacter dehalogenans. Magnetotactic bacteria make use of a specialized protoorganelle formed by a linear array of cytoplasmic invaginations of the inner membrane filled with an in situ formed magnetic material (17, 18, 72). The material is usually magnetite (a FeO·Fe2O3-based mineral) or less frequently greigite (a Fe(II)Fe(III)2S4-based mineral). L. ferrooxidans is capable of oxidizing iron- or sulfur-containing minerals, such as pyrite, and uses the reducing equivalents obtained this way to fix carbon (19, 56). Finally, Anaeromyxobacter represents a metabolically versatile species that can use iron(III) or other heavy metal ions, including uranium(VI), as terminal electron acceptors (57). These bacteria have been used as bioremediation agents of heavy metal contaminated areas.
An insight into the conformational flexibility of hHscB was achieved by a normal mode analysis. This analysis revealed that hHscB can undergo motions with several major components: a scissors-like closing of the L-shaped molecule and more complex motions that involve simultaneous sideways twisting of one of the “blades” and rotation of the other along its extended axis. The three most prominent normal modes describing these motions accounted for over 46% of the conformational variability expected for all the modes. Importantly, the conformational difference between the hHscB protomers observed in the asymmetric unit correlated well (88%) with the combined displacement predicted by the first and the third normal mode. Robustness of normal mode analysis based on the 3.0 Å structure of hHscB and a potential based on the residue level distance network was further validated by the analysis of a higher resolution E. coli HscB structure (1.8 Å), which shows 29% sequence identity to hHscB in the J- and C-domains. The analysis revealed a strong similarity between the lowest frequency normal modes of the two homologous structures (Table 2). The mode of closest agreement is a scissors-like hinge motion between the J- and C-domains (Fig. 4b, left panel), which appears robust to changes in sequence and to addition of the N-domain. Despite a change in relative frequencies, the second and the third modes are also quite robust, although they show a greater impact of the structural and sequence differences than the first mode. In summary, we observed a very close agreement between the lowest frequency modes of hHscB and E. coli HscB as well as between the normal modes and the experimentally observed conformational changes in the hHscB crystals. This leads us to conjecture that the L-shaped structural motif of hHscB is inherently dynamic, with the normal modes indicating the preferred directions of flexibility. This inherent flexibility may have functional consequences during the iron-sulfur transfer within a complex formed by IscU, HscB, HscA, and the acceptor protein.
Supplementary Material
Acknowledgments
We thank all members of the Centre for Eukaryotic Structural Genomics team involved in this work, especially Dave Aceti, Lai Bergeman, Ronnie Frederick, Katarzyna Gromek, Leigh Grundhoefer, Andrew Larkin, Karl Nichols, Xiaokang Pan, Mike Popelars, John Primm, Kory Seder, Donna Troestler, Frank Vojtik, Brian Volkman, Gary Wesenberg, Russell Wrobel, and Zsolt Zolnai. We thank Anna Füzéry for comments on the manuscript.
The atomic coordinates and structure factors (code 3bvo) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grants P50 GM64598 and U54 GM074901 (to J. L. M.). This work was also supported by the Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-ENG-38, by National Cancer Institute Grant Y1-CO-1020, and by National Institute of General Medical Science Grant Y1-GM-1104. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Movies S1–S3.
Footnotes
The abbreviations used are: hHscB, human HscB; bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; PIPES, piperazine-N,N′-bis(2-ethanesulfonic acid).
References
- 1.Johnson, D. C., Dean, D. R., Smith, A. D., and Johnson, M. K. (2005) Annu. Rev. Biochem. 74 247-281 [DOI] [PubMed] [Google Scholar]
- 2.Meyer, J. (2008) J. Biol. Inorg. Chem. 13 157-170 [DOI] [PubMed] [Google Scholar]
- 3.Rouault, T. A., and Tong, W. H. (2005) Nat. Rev. Mol. Cell. Biol. 6 345-351 [DOI] [PubMed] [Google Scholar]
- 4.Marquet, A., Bui, B. T., Smith, A. G., and Warren, M. J. (2007) Nat. Prod. Rep. 24 1027-1040 [DOI] [PubMed] [Google Scholar]
- 5.Lill, R., and Muhlenhoff, U. (2005) Trends Biochem. Sci. 30 133-141 [DOI] [PubMed] [Google Scholar]
- 6.Vickery, L. E., and Cupp-Vickery, J. R. (2007) Crit. Rev. Biochem. Mol. Biol. 42 95-111 [DOI] [PubMed] [Google Scholar]
- 7.Jacobson, M. R., Cash, V. L., Weiss, M. C., Laird, N. F., Newton, W. E., and Dean, D. R. (1989) Mol. Gen. Genet. 219 49-57 [DOI] [PubMed] [Google Scholar]
- 8.Outten, F. W., Djaman, O., and Storz, G. (2004) Mol. Microbiol. 52 861-872 [DOI] [PubMed] [Google Scholar]
- 9.Patzer, S. I., and Hantke, K. (1999) J. Bacteriol. 181 3307-3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokumoto, U., Kitamura, S., Fukuyama, K., and Takahashi, Y. (2004) J. Biochem. (Tokyo) 136 199-209 [DOI] [PubMed] [Google Scholar]
- 11.Takahashi, Y., and Nakamura, M. (1999) J. Biochem. (Tokyo) 126 917-926 [DOI] [PubMed] [Google Scholar]
- 12.Zheng, L., Cash, V. L., Flint, D. H., and Dean, D. R. (1998) J. Biol. Chem. 273 13264-13272 [DOI] [PubMed] [Google Scholar]
- 13.Ayala-Castro, C., Saini, A., and Outten, F. W. (2008) Microbiol. Mol. Biol. Rev. 72 110-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cupp-Vickery, J. R., Urbina, H., and Vickery, L. E. (2003) J. Mol. Biol. 330 1049-1059 [DOI] [PubMed] [Google Scholar]
- 15.Ramelot, T. A., Cort, J. R., Goldsmith-Fischman, S., Kornhaber, G. J., Xiao, R., Shastry, R., Acton, T. B., Honig, B., Montelione, G. T., and Kennedy, M. A. (2004) J. Mol. Biol. 344 567-583 [DOI] [PubMed] [Google Scholar]
- 16.Liu, J., Oganesyan, N., Shin, D. H., Jancarik, J., Yokota, H., Kim, R., and Kim, S. H. (2005) Proteins 59 875-881 [DOI] [PubMed] [Google Scholar]
- 17.Komeili, A. (2007) Annu. Rev. Biochem. 76 351-366 [DOI] [PubMed] [Google Scholar]
- 18.Komeili, A., Li, Z., Newman, D. K., and Jensen, G. J. (2006) Science 311 242-245 [DOI] [PubMed] [Google Scholar]
- 19.Rawlings, D. E., Tributsch, H., and Hansford, G. S. (1999) Microbiology (Read.) 145 5-13 [DOI] [PubMed] [Google Scholar]
- 20.Bilder, P. W., Ding, H., and Newcomer, M. E. (2004) Biochemistry 43 133-139 [DOI] [PubMed] [Google Scholar]
- 21.Cupp-Vickery, J. R., Silberg, J. J., Ta, D. T., and Vickery, L. E. (2004) J. Mol. Biol. 338 127-137 [DOI] [PubMed] [Google Scholar]
- 22.Morimoto, K., Yamashita, E., Kondou, Y., Lee, S. J., Arisaka, F., Tsukihara, T., and Nakai, M. (2006) J. Mol. Biol. 360 117-132 [DOI] [PubMed] [Google Scholar]
- 23.Cupp-Vickery, J. R., and Vickery, L. E. (2000) J. Mol. Biol. 304 835-845 [DOI] [PubMed] [Google Scholar]
- 24.Cupp-Vickery, J. R., Peterson, J. C., Ta, D. T., and Vickery, L. E. (2004) J. Mol. Biol. 342 1265-1278 [DOI] [PubMed] [Google Scholar]
- 25.Agar, J. N., Krebs, C., Frazzon, J., Huynh, B. H., Dean, D. R., and Johnson, M. K. (2000) Biochemistry 39 7856-7862 [DOI] [PubMed] [Google Scholar]
- 26.Ding, H., Clark, R. J., and Ding, B. (2004) J. Biol. Chem. 279 37499-37504 [DOI] [PubMed] [Google Scholar]
- 27.Urbina, H. D., Silberg, J. J., Hoff, K. G., and Vickery, L. E. (2001) J. Biol. Chem. 276 44521-44526 [DOI] [PubMed] [Google Scholar]
- 28.Muhlenhoff, U., Gerber, J., Richhardt, N., and Lill, R. (2003) EMBO J. 22 4815-4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandramouli, K., and Johnson, M. K. (2006) Biochemistry 45 11087-11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoff, K. G., Silberg, J. J., and Vickery, L. E. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 7790-7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silberg, J. J., Tapley, T. L., Hoff, K. G., and Vickery, L. E. (2004) J. Biol. Chem. 279 53924-53931 [DOI] [PubMed] [Google Scholar]
- 32.Hoff, K. G., Ta, D. T., Tapley, T. L., Silberg, J. J., and Vickery, L. E. (2002) J. Biol. Chem. 277 27353-27359 [DOI] [PubMed] [Google Scholar]
- 33.Sun, G., Gargus, J. J., Ta, D. T., and Vickery, L. E. (2003) J. Hum. Genet. 48 415-419 [DOI] [PubMed] [Google Scholar]
- 34.Jeon, W. B., Aceti, D. J., Bingman, C. A., Vojtik, F. C., Olson, A. C., Ellefson, J. M., McCombs, J. E., Sreenath, H. K., Blommel, P. G., Seder, K. D., Burns, B. T., Geetha, H. V., Harms, A. C., Sabat, G., Sussman, M. R., Fox, B. G., and Phillips, G. N., Jr. (2005) J. Struct. Funct. Genomics 6 143-147 [DOI] [PubMed] [Google Scholar]
- 35.Sreenath, H. K., Bingman, C. A., Buchan, B. W., Seder, K. D., Burns, B. T., Geetha, H. V., Jeon, W. B., Vojtik, F. C., Aceti, D. J., Frederick, R. O., Phillips, G. N., Jr., and Fox, B. G. (2005) Protein Expression Purif. 40 256-267 [DOI] [PubMed] [Google Scholar]
- 36.Thao, S., Zhao, Q., Kimball, T., Steffen, E., Blommel, P. G., Riters, M., Newman, C. S., Fox, B. G., and Wrobel, R. L. (2004) J. Struct. Funct. Genomics 5 267-276 [DOI] [PubMed] [Google Scholar]
- 37.Zolnai, Z., Lee, P. T., Li, J., Chapman, M. R., Newman, C. S., Phillips, G. N., Jr., Rayment, I., Ulrich, E. L., Volkman, B. F., and Markley, J. L. (2003) J. Struct. Funct. Genomics 4 11-23 [DOI] [PubMed] [Google Scholar]
- 38.Blommel, P. G., Becker, K. J., Duvnjak, P., and Fox, B. G. (2007) Biotechnol. Prog. 23 585-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otwinowski, Z., and Minor, W. (1997) Method Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 40.Grosse-Kunstleve, R. W., and Adams, P. D. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59 1966-1973 [DOI] [PubMed] [Google Scholar]
- 41.Sheldrick, G. M. (2008) Acta Crystallogr. Sect. A 64 112-122 [DOI] [PubMed] [Google Scholar]
- 42.de la Fortelle, E., and Bricogne, G. (1997) Method Enzymol. 276 472-494 [DOI] [PubMed] [Google Scholar]
- 43.Collaborative Computational Project Number 4. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 44.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 45.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 46.Lovell, S. C., Davis, I. W., Arendall, W. B., III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 47.DeLano, W. L. (2002) The PYMOL Molecular Graphic System, DeLano Scientific LLC, San Carlos, CA
- 48.Brooks, B., and Karplus, M. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 6571-6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Go, N., Noguti, T., and Nishikawa, T. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 3696-3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tama, F., and Brooks, C. L. (2006) Annu. Rev. Biophys. Biomol. Struct. 35 115-133 [DOI] [PubMed] [Google Scholar]
- 51.Tirion, M. M. (1996) Phys. Rev. Lett. 77 1905-1908 [DOI] [PubMed] [Google Scholar]
- 52.Kondrashov, D. A., Van Wynsberghe, A. W., Bannen, R. M., Cui, Q., and Phillips, G. N., Jr. (2007) Structure 15 169-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rice, L. M., Earnest, T. N., and Brunger, A. T. (2000) Acta Crystallogr. Sect. D Biol. Crystallogr. 56 1413-1420 [DOI] [PubMed] [Google Scholar]
- 54.Madej, T., Gibrat, J. F., and Bryant, S. H. (1995) Proteins 23 356-369 [DOI] [PubMed] [Google Scholar]
- 55.Ye, Y., and Godzik, A. (2003) Bioinformatics 19 (Suppl. 2) 246-255 [DOI] [PubMed] [Google Scholar]
- 56.Rojas-Chapana, J. A., and Tributsch, H. (2004) FEMS Microbiol. Ecol. 47 19-29 [DOI] [PubMed] [Google Scholar]
- 57.Wu, Q., Sanford, R. A., and Loffler, F. E. (2006) Appl. Environ. Microbiol. 72 3608-3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corpet, F. (1988) Nucleic Acids Res. 16 10881-10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, B., Alam, S. L., Meyer, H. H., Payne, M., Stemmler, T. L., Davis, D. R., and Sundquist, W. I. (2003) J. Biol. Chem. 278 20225-20234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu, G. W., Allen, M. D., Andreeva, A., Fersht, A. R., and Bycroft, M. (2006) Protein Sci. 15 384-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaroszewski, L., Rychlewski, L., Li, Z., Li, W., and Godzik, A. (2005) Nucleic Acids Res. 33 284-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, X., Lee, H. S., Sugar, F. J., Jenney, F. E., Jr., Adams, M. W., and Prestegard, J. H. (2007) Biochemistry 46 752-761 [DOI] [PubMed] [Google Scholar]
- 63.Cramer, P., Bushnell, D. A., and Kornberg, R. D. (2001) Science 292 1863-1876 [DOI] [PubMed] [Google Scholar]
- 64.Mancias, J. D., and Goldberg, J. (2007) Mol Cell 26 403-414 [DOI] [PubMed] [Google Scholar]
- 65.Hagelueken, G., Wiehlmann, L., Adams, T. M., Kolmar, H., Heinz, D. W., Tummler, B., and Schubert, W. D. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 12276-12281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dauter, Z., Wilson, K. S., Sieker, L. C., Moulis, J. M., and Meyer, J. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 8836-8840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pearl, F. M., Bennett, C. F., Bray, J. E., Harrison, A. P., Martin, N., Shepherd, A., Sillitoe, I., Thornton, J., and Orengo, C. A. (2003) Nucleic Acids Res. 31 452-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsai, J., and Douglas, M. G. (1996) J. Biol. Chem. 271 9347-9354 [DOI] [PubMed] [Google Scholar]
- 69.Fuźery, A. K., Tonelli, M., Ta, D. T., Cornilescu, G., Vickery, L. E., and Markley, J. L. (2008) Biochemistry 47 9394-9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andrew, A. J., Dutkiewicz, R., Knieszner, H., Craig, E. A., and Marszalek, J. (2006) J. Biol. Chem. 281 14580-14587 [DOI] [PubMed] [Google Scholar]
- 71.Unciuleac, M. C., Chandramouli, K., Naik, S., Mayer, S., Huynh, B. H., Johnson, M. K., and Dean, D. R. (2007) Biochemistry 46 6812-6821 [DOI] [PubMed] [Google Scholar]
- 72.Bazylinski, D. A., and Frankel, R. B. (2004) Nat. Rev. 2 217-230 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.