

Abstract
Non-structural protein 3 (NS3) is a multifunctional enzyme possessing serine protease, NTPase, and RNA unwinding activities that are required for hepatitis C viral (HCV) replication. HCV non-structural protein 4A (NS4A) binds to the N-terminal NS3 protease domain to stimulate NS3 serine protease activity. In addition, the NS3 protease domain enhances the RNA binding, ATPase, and RNA unwinding activities of the C-terminal NS3 helicase domain (NS3hel). To determine whether NS3hel enhances the NS3 serine protease activity, we purified truncated and full-length NS3-4A complexes and examined their serine protease activities under a variety of salt and pH conditions. Our results indicate that the helicase domain enhances serine protease activity, just as the protease domain enhances helicase activity. Thus, the two enzymatic domains of NS3-4A are highly interdependent. This is the first time that such a complete interdependence has been demonstrated for a multifunctional, single chain enzyme. NS3-4A domain interdependence has important implications for function during the viral lifecycle as well as for the design of inhibitor screens that target the NS3-4A protease.
NS3-4A is a multifunctional enzyme with serine protease and helicase activities that are essential for hepatitis C viral (HCV)3 replication (1–3). The NS3 N-terminal domain adopts a chymotrypsin-like fold, and it displays serine protease activity in the presence of the NS4A co-factor protein (4, 5) (Fig. 1A). The genomic HCV RNA is initially translated into a long polyprotein that must be cleaved into the core, envelope, and nonstructural (NS) proteins, which are required for viral replication and packaging. The NS3-4A protease is essential for this process, as it cleaves several sites within the HCV polyprotein (4). It may also enhance HCV replication by cleaving host proteins such as Cardif (also known as IPS-1, MAVS, or VISA), which are involved in innate immunity (6–9). In this way, NS3-4A may down-regulate the cellular antiviral response.
FIGURE 1.

Composition and purification of NS3-4A. The NS3-4A complex organization and construct design are illustrated schematically (A and B). In panel A, pro refers to the serine protease domain and the Roman numerals indicate the respective NS3 helicase subdomains. The regions where ATP, RNA, and the NS4A co-factor bind are indicated as well. The protein construct expressed in E. coli is depicted in panel B. The numbers below the map refer to the HCV polyprotein numbering of the amino acids of NS3-4A. In panel C, His-SUMO-NS3 and His-SUMO-NS4A fusion construct designs are presented. These proteins were purified separately and their His-SUMO tags were removed using the same methods as for NS3-4A. Subsequently, the native NS3 or native NS3 protease domain was reconstituted with native NS4A (see “Experimental Procedures”).
The NS3 C-terminal domain (NS3hel) belongs to the DEXH/D-box subgroup of helicase superfamily 2 (10). It exhibits NTPase activity and, in the presence of the protease domain, it is a reactive RNA helicase (11–13). The NS3 helicase, along with the HCV NS5A phosphoprotein and the HCV NS5B RNA-dependent RNA polymerase, is required for replication of the viral RNA genome (14) and is possibly required for viral packaging as well (15). Thus, NS3-4A must be able to function as a serine protease, an RNA helicase, and a packaging enzyme in an HCV-infected cell in order for the virus to propagate.
Recently, we observed that RNA binding and unwinding by NS3hel are greatly enhanced by the covalently attached NS3 protease domain (13). This suggests that, despite their seemingly unrelated enzymatic functions, the helicase and protease domains have evolved to become interdependent and functionally coupled. It is well established that the NS3 protease domain requires binding of NS4A for reactivity (4, 16–18), and that the protease domain can function in the absence of NS3hel (19–23). However, it has not been determined whether the NS3hel domain enhances the inherent activity of the NS3 protease domain. If the hel domain does influence the protease domain function, this finding will guide the design of NS3 constructs that are used for future functional, structural, and screening studies on the NS3 protease. It would also provide a clear-cut, biochemically characterized example of functional coevolution among viral protein domains. Finally, it would implicate the protein-protein interface between the helicase and protease as a major functional element and potential drug target. We therefore set out to determine whether the NS3 protease is stimulated by the presence of the NS3hel domain.
To address this issue, we coexpressed and purified native, full-length NS3-4A serine protease complexes using a bacterial expression system and we measured NS3-4A protease activities with a fluorescently labeled HCV substrate mimic. Here, we show that the native, full-length NS3-4A complexes are conformationally homogeneous and that they display robust serine protease activity under a variety of salt and pH conditions, including conditions that support efficient helicase activity.
In parallel experiments, we prepared reconstituted NS3-NS4A complexes in which the two proteins were individually expressed and then added together to form the active protease. The reconstituted complexes enabled us to compare the protease activity of full-length NS3 (NS3 + NS4A) with that of the NS3 protease domain in isolation (NS3 protease + NS4A). We observe that the full-length NS3 + 4A construct is a much more reactive protease, indicating that the NS3hel domain enhances activity of the NS3-4A serine protease domain. Taken together with prior work, these findings indicate that the two disparate enzyme domains of the NS3 protein are strongly interdependent: the NS3 protease domain enhances the activity of NS3hel (13) and NS3hel enhances activity of the protease domain.
EXPERIMENTAL PROCEDURES
Materials—RNA oligonucleotides described in this work were obtained from Dharmacon and DNA oligonucleotides were obtained from Invitrogen. All other reagents were obtained from Fisher unless otherwise indicated.
Cloning—All DNA primers used for cloning are described under supplemental Table S1. We cloned NS3+ and NS3-4A+-expressing cDNA from the hepatitis C viral genotype 1a (version H77, kindly donated by Dr. Charlie Rice) into the vector pET15b (Novagen). The DNA oligonucleotides used for PCR of NS3 and NS3-4A were NS3 1a-1 and NS3 1a-2 or NS4A(1a)β BamHI. An internal XhoI site in the NS3(1a)+ gene was removed through a silent base pair change using a QuikChange kit (Stratagene). The DNA oligonucleotides used for the QuikChange reaction were NS3 1a-3 and NS3 1a-4.
The NS3+ and NS3/4A+ genes from hepatitis C viral genotype 1b (version N, kindly provided by Dr. Stan Lemon (24)) were amplified and cloned into pET15b according to the methods of Beran et al. (25). The upstream DNA oligonucleotide used for PCR amplification was NS3α XhoI and the downstream DNA oligonucleotide was NS3β BamHI or NS4Aβ.
NS3+ and NS3–4A+ of both the 1a and 1b genotypes were cloned into pET-SUMO (Invitrogen) using ExTaq PCR (Takara) followed by ligation with linear pET-SUMO according to the manufacturer's protocol. The DNA oligonucleotides used were the same as those described above for pET15b cloning, except that the 5′ PCR oligonucleotide in the genotype 1a case was NS3(1a) SUMO start and in the genotype 1b case was NS3(1b) 5′ SUMO start.
To produce pET-SUMO-NS4A(1a)+, the NS4A(1a)+ gene was PCR amplified using the DNA oligonucleotides NS4A 1a SUMO start and NS4A 1a SUMO end and then cloned into pET-SUMO. To produce the isolated NS3 protease domain, a stop codon was inserted in the NS3(1a)+ gene after amino acid 188 in the pET-SUMO-NS3(1a)+ plasmid (13). Mutagenesis was conducted via QuikChange (Stratagene), using DNA oligonucleotides NS3prot(1a)-1 and NS3prot(1a)-2. To create the NS3/4A polyprotein with an Ala/Ala cleavage site instead of a Thr/Ser cleavage site, the wild-type NS3–4A(1b)+ construct was mutated by QuikChange (Stratagene), using oligonucleotides AA NS3–4A-1 and AA NS3–4A-2. All constructs were verified initially through PCR screening as necessary and subsequently sequenced for accuracy (Keck Facility, Yale University).
Protein Purification—Because there are a multitude of reports describing the purification of various autocleaved and reconstituted forms of the NS3-4A complex (17, 22, 23, 26, 27) and because all of these factors greatly influence NS3 enzymatic function (13), we believe it is necessary to describe our constructs, purification strategies, and reconstitutions in detail. All proteins were purified according to the methods described in Beran et al., (13, 25). Briefly, 4 liters of Escherichia coli culture in LB (supplemented with 35 μg/ml kanamycin) were grown at 37 °C with shaking. Upon reaching the exponential growth phase, the temperature was shifted to 15 °C, the cultures were supplemented with 1 mm isopropyl 1-thio-β-d-galactopyranoside (final concentration), and subsequently incubated overnight at 15 °C with shaking. After lysing the cells using an Emusiflex C5 cell disruptor (Avestin), the cellular extract was centrifuged at 10,000 rpm (14,500 × g) at 4 °C for 10 min before being passed through a nickel column. The His6 and His6-SUMO fusion proteins were purified through a nickel column (Qiagen), treated with 10 units of SUMO protease (Invitrogen) overnight at 4 °C, and subsequently passed through a gel filtration column (HiLoad Superdex 200 16/60) equilibrated with a buffer containing 25 mm Hepes (pH 8.0), 0.3 m NaCl, 10% glycerol, 1 mm dithiothreitol, and 0.2% Triton X-100. Ninety-six 1.2-ml gel filtration fractions were collected at a rate of 0.4 ml/min.
The NS3-4A complex eluted between fractions 45 and 55, presumably in a detergent micelle (17). Fractions containing NS3-4A were identified using the RET-S1 protease assay (the RET-S1 protease assay details are described later in this section) as well as through SDS-PAGE analysis. Moreover, anti-NS3 and anti-NS4A (monoclonal antibodies in both cases) Western blotting were used to confirm the identity of the purified protein complex, which was then used for experimentation (see Fig. 2, B and C, and the Western blotting procedure described later in this section).
FIGURE 2.

NS3-4A purifies as two separable proteins. A, purified proteins were subjected to denaturing electrophoresis on a 4–12% gradient gel. Purified NS3 (lane 2), NS3-4A (lane 3), and NS3/4A polyprotein (lane 4) were subjected to electrophoresis side by side for comparison of mobilities. The band shown in lane 3 represents the NS3 component of a native, fully cleaved NS3-4A preparation. The band shown in lane 4 represents an NS3/4A polyprotein preparation produced by mutating the Thr/Ser cleavage site between NS3 and NS4A to a non-cleavable sequence (AA). In panels B and C, anti-NS3 and anti-NS4A Western blot analysis confirm the identity of our purified proteins (see “Experimental Procedures”). Panel B depicts an anti-NS3 Western blot. In panel B, lane 1 contains purified NS3, lane 2 contains purified, full-length NS3-4A, and lane 3 contains purified NS3/4A polyprotein. Truncated forms of NS3 are visible below the full-length protein in each lane in the anti-NS3 blot. These truncated forms of NS3 are likely produced during bacterial expression as well as during the multiday purification performed in the absence of protease inhibitors. These truncated forms could represent either N- or C-terminal truncations of NS3 as the monoclonal anti-NS3 antibody binds to the central region of the helicase domain. Panel C depicts an anti-NS4A Western blot. In panel C, lane 1 contains purified NS3, lane 2 contains purified, full-length NS3-4A, and lane 3 contains purified NS3/4A polyprotein. Truncated forms of the NS3/4A polyprotein are visible below the full-length polyprotein in the anti-NS4A blot. These truncated forms of NS3/4A polyprotein likely represent N-terminal degraded NS3/4A as the monoclonal anti-NS4A antibody binds the final 11 C-terminal residues of NS4A.
Free NS3 protein eluted from the gel filtration column between fractions 60 and 70. Fractions containing NS3 were identified using RNA unwinding assays (25) and SDS-PAGE analysis. In addition, anti-NS3 Western blotting was used to confirm the identity of the purified protein, which was then used for experimentation (see Fig. 2B and the Western blotting procedure described later in this section).
Because there is a 13-kDa size difference between His-SUMO tagged and untagged protein, the untagged proteins were initially analyzed side by side using SDS-PAGE with uncleaved His-SUMO NS3–4A or uncleaved His-SUMO-NS3 to verify that the purified proteins were, in fact, untagged species (data not shown).
After purification, preparations were divided into 10-μl aliquots and stored at -80 °C. Protein concentrations were determined using a Bradford assay (Bio-Rad). The NS3 and NS3-4A proteins were examined for purity and their sizes compared using SDS-PAGE (Fig. 2A). Protein samples (40 pmol) were subjected to electrophoresis on a NuPAGE 4–12% BisTris gel (Invitrogen) in MES-SDS buffer for 2 h at 200 V. The gel was subsequently stained with Coomassie Blue.
For reconstitution of the protease complex using purified NS3 and purified NS4A proteins (“NS3 + NS4A”), we expressed His-SUMO-NS3 as well as His-SUMO-NS4A in E. coli using the pET-SUMO vector system (Invitrogen). After partial purification using a nickel column, His-SUMO-NS3 and a 4-fold excess of His-SUMO-NS4A were mixed and incubated together at 4 °C overnight in the presence of 10 units of SUMO protease (Invitrogen). Reconstituted, untagged NS3 + 4A was subsequently isolated from untagged NS3 and untagged NS4A (based upon size differences) by passing the protein mixture through a gel filtration column (HiLoad Superdex 200 16/60) using the same methods as described above for autocleaved NS3-4A. The same procedure was followed to reconstitute and purify untagged NS3 protease domain with untagged NS4A (“NS3 protease domain + NS4A”). In this case, NS3 protease domain alone eluted between gel filtration fractions 75 and 80 and NS3 protease domain + NS4A eluted between fractions 70 and 75. The RET-S1 serine protease assay as well as SDS-PAGE analysis was used to identify the fractions containing reconstituted NS3 protease domain + NS4A. In all cases, protein concentration was determined using a Bradford assay (Bio-Rad).
Western Blot Analysis—SDS-PAGE was performed using a Bio-Rad MiniProtean II apparatus according to the manufacturer's protocols. The Western blot transfer was performed using a Bio-Rad transfer apparatus according to the manufacturer's instructions. Anti-NS3 and anti-NS4A monoclonal antibodies were purchased from Virogen (Watertown, MA) and diluted according to the manufacturer's instructions. In these experiments (as shown in Fig. 2, B and C), 25 pmol of each protein was subjected to electrophoresis on a 12% SDS-polyacrylamide gel for 1 h at 200 V. Subsequently, the proteins were transferred to a nitrocellulose membrane by applying 100 V for 1 h at 4 °C. Finally, the blots were incubated with either anti-NS3 or anti-NS4A monoclonal antibody and subsequently developed using a Pierce Supersignal Western blot analysis kit.
Protease Assays—Protease assays were performed at 37 °C
using the Resonance Energy Transfer-S1 substrate (RET-S1) (Anaspec) designed
by Taliani et al. (5).
RET-S1 is an NS4A/NS4B junction mimic that fluoresces upon cleavage. All
assays, unless otherwise indicated, were performed in 60-μl reaction
volumes containing 40 nm NS3-4A and 5 μm RET-S1. The
data shown in Fig. 6 and
Table 1 were collected using
120 nm protein with the indicated amounts of RET-S1 substrate. In
Table 1, the
kcat values were calculated based upon the observation
that 75% of the protein was active in each case
(Fig. 5). The buffer conditions
were the same as those normally used for helicase assays
(13): 25 mm
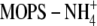 (pH 6.5), 3 mm
MgCl2, 1% glycerol, 2 mm dithiothreitol, 30
mm NaCl, 0.2% (v/v) Triton X-100, unless otherwise indicated. Data
were collected using a Cary Eclipse Spectrophotometer (Varian) with a
temperature-controlled cuvette holder. The excitation wavelength was 350 nm
and the emission wavelength was 440 nm. The initial background value (the
value at time 0, just before substrate addition) was subtracted from all
subsequent time points.
(pH 6.5), 3 mm
MgCl2, 1% glycerol, 2 mm dithiothreitol, 30
mm NaCl, 0.2% (v/v) Triton X-100, unless otherwise indicated. Data
were collected using a Cary Eclipse Spectrophotometer (Varian) with a
temperature-controlled cuvette holder. The excitation wavelength was 350 nm
and the emission wavelength was 440 nm. The initial background value (the
value at time 0, just before substrate addition) was subtracted from all
subsequent time points.
FIGURE 6.

Steady-state proteolysis rates of RET-S1 by NS3-4A, NS3 + NS4A, and NS3 protease domain + NS4A in the presence of varying substrate concentrations. The data were fit to the Michaelis-Menten equation to determine Km, Vmax, and kcat values. The data shown is the average of three experiments.
TABLE 1.
The NS3 helicase domain enhances NS3-4A protease activity The proteolysis data were determined by monitoring RET-S1 cleavage on a fluorescence spectrophotometer (see “Experimental Procedures”). The data shown were determined using proteins of the 1a genotype and is the average of three experiments. The error values represent standard deviation.
| Protein | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|
| μm | pmol RET-S1 cleaved/s | pmol RET-S1 cleaved/s/pmol enzyme | pmol RET-S1 cleaved/s/pmol enzyme/μm substrate | |
| NS3/4A | 10.0 ± 3.0 | 17 ± 2 | 3.15 ± 0.30 | 0.315 |
| NS3 + NS4A | 3.0 ± 1.0 | 0.50 ± 0.05 | 0.09 ± 0.01 | 0.030 |
| NS3 protease domain + NS4A | 4.0 ± 1.2 | 0.11 ± 0.01 | 0.020 ± 0.002 | 0.005 |
FIGURE 5.

Steady-state proteolysis of RET-S1 by reconstituted NS3 + NS4A and NS3 protease domain + NS4A. The steady-state velocity for NS3 + NS4A is 0.005 ± 001 μm/s (pierced circles) and for NS3 protease ± NS4A is 0.001 ± 0.001 μm/s (solid squares). The y intercept of the fitted lines show NS3 + NS4A and NS3 protease domain + NS4A to have 75 ± 10% active fraction and 75 ± 12% active fraction, respectively. The data shown are the average of three experiments and the error values represent standard deviation.
The fraction of active NS3-4A protein was determined for both tagged and untagged preparations by incubating a small amount of enzyme (40 nm) with a large amount of RET-S1 substrate (5 μm). A line was fit to the slope of the proteolysis data during the steady-state phase of reaction. This line was extrapolated to the intersection with the y axis (Sigma Plot, Systat Software), which corresponds to the concentration of active enzyme present in the reaction (28). In all cases throughout this work, the error values shown indicate standard deviation. Importantly, similar values for the fraction of active enzyme (∼75%) were observed when the NS3-4A concentration was raised to 120 nm or when the RET-S1 concentration was raised well above the Km (10 ± 3 μm) (Table 1) to 20 μm (data not shown). Similarly, when reconstituted NS3 protease domain + NS4A and NS3 + NS4A were assayed with RET-S1 concentrations well above the Km values (8–10 μm RET-S1, whereas Km = 4.0 ± 1.2 and 3.0±1.0 μm, respectively) (Table 1), a similar enzymatic active fraction was observed for both complexes (∼75%) (data not shown). When protease activities of NS3-4A(1a) and NS3-4A(1b) were compared using the RET-S1 analyses, results were similar for both genotypes.
RESULTS
The NS3-4A Complex Can Be Purified in a Highly Active Form—To study NS3-4A protease function, we needed to purify NS3 in complex with its NS4A co-factor (i.e. NS3-4A). In previous studies, NS3-4A has been overexpressed in insect cells (27) or in E. coli (12, 17). In all of these cases, additional amino acids were fused to the NS3 N terminus for the purpose of protein purification (12, 17) and lysine residues were added to the NS4A C terminus to increase solubility (17). To our knowledge, a recombinant NS3-4A complex that lacks tags or additional modifying amino acids has never been produced. We therefore designed an approach for generating native, full-length NS3-4A complex (e.g. lacking modifications to the terminal sequences of NS3 or NS4A) using the pET-SUMO expression system in E. coli (Fig. 1B and see “Experimental Procedures”). In this system, the protein of interest is tagged at the N terminus with the SUMO polypeptide, which enhances solubility of the protein and is then readily removed upon incubation with the SUMO protease, thereby resulting in native protein that lacks additional amino acids (29).
We expressed NS3-4A as a His-SUMO-NS3/4A polyprotein in E. coli, designing the construct so that NS3 would be expected to spontaneously cleave NS4A from its C terminus (Fig. 1B) (17, 27), thereby forming an active protease complex (4). After purification on a nickel column, the His-SUMO fusion protein was cleaved by SUMO protease from NS3, generating a native N terminus (Fig. 1B). The protein mixture was subsequently passed through a gel filtration column to separate native protein complex from uncleaved fusion protein (see “Experimental Procedures”). The resultant cleaved, purified NS3-4A complex was ∼95% pure (Fig. 2A).
To determine whether the purified NS3-4A protein had autocatalytically cleaved properly, forming two separate proteins (NS3 and NS4A), we subjected the complex to denaturing electrophoresis side by side with NS3 and an uncleavable NS3/4A polyprotein control. The uncleavable NS3/4A polyprotein was produced by mutating the conserved junction amino acid sequence between NS3 and NS4A from Thr/Ser to Ala/Ala. SDS-PAGE and Western blot analysis show that the NS3 component of the wild-type NS3-4A preparation has the same electrophoretic mobility as NS3 (in Fig. 2A, compare lanes 2 and 3 of the Coomassie-stained gel and in Fig. 2B, compare lanes 1 and 2 of the anti-NS3 Western blot). However, the uncleaved NS3/4A polyprotein migrates more slowly than NS3, as expected due to its larger size (in Fig. 2A, compare lane 4 to lanes 2 and 3 and in Fig. 2B, compare lanes 1 and 2 with lane 3). Coomassie staining does not reveal any NS3/4A polyprotein in our NS3-4A preparation (Fig. 2A, lane 3). However, Western blots are more sensitive than Coomassie staining, and consistent with this, the anti-NS3 Western blots reveal a small fraction of uncleaved NS3/4A polyprotein in the wild-type NS3–4A preparation (Fig. 2B, note the faint upper band in lane 2 that migrates similarly to the NS3/4A polyprotein in lane 3). Taken together, these results demonstrate that the preparation of wild-type NS3-4A complex is composed primarily of fully cleaved NS3 and NS4A molecules. The catalytic function and conformational homogeneity of this preparation was also assessed using enzymological approaches (see below).
The presence of NS4A in the NS3-4A preparation was assessed using denaturing electrophoresis and Western blot analysis with antibodies against NS4A. The anti-NS4A Western blots indicate that the wild-type NS3-4A preparation consists of both free NS4A (which is difficult to detect because it is so small and diffuses rapidly from the gel) and a fraction of uncleaved NS3/4A polyprotein (in Fig. 2C, compare lanes 2 and 3). The free NS4A migrates at the correct position, ∼6 kDa, near the bottom of the gel (Fig. 2C, lane 2). Taken together, the electrophoretic and immunoaffinity methods provide physical evidence that the NS3-4A preparation consists of cleaved NS3 and NS4A components. Nonetheless, it was still essential to demonstrate that the complex was catalytically active and to assess the fraction of active molecules in the preparation.
To quantify the fraction of fully processed, active NS3-4A in the preparation, we analyzed the proteolysis of a depsipeptide substrate known as “RET-S1,” which was originally derived from the NS4A–NS4B cleavage junction (5). As expected (16), both free NS3 and the uncleavable mutant NS3/4A polyprotein are proteolytically inactive, as they fail to react with RET-S1 (see below). However, the wild-type NS3-4A preparation displays efficient protease activity against RET-S1, with a rate constant of 0.020 ± 0.001 μm product/s). This value is significantly faster than values obtained by others for NS3-4A with a non-native N terminus incubated with a 5AB peptide substrate (27). Importantly, burst kinetics experiments reveal that the NS3-4A preparation contains 75 ± 14% active enzyme (Fig. 3), indicating that the majority of the population is properly folded, assembled, and catalytically active. That this value falls short of 100% is consistent with the electrophoretic analyses, which indicated that ∼20% of the wild-type NS3-4A preparation remains uncleaved (Fig. 2). Taken together, the physical and functional information provided by both the electrophoretic and proteolysis assays indicate that we have successfully overexpressed untagged, unmodified NS3-4A in a form that is conformationally homogeneous, appropriately cleaved, and highly reactive. This approach now makes it possible to quantitatively assess the proteolysis and helicase activities of the NS3-4A complex, and to compare these activities with the behavior of other NS3 constructs.
FIGURE 3.

Steady-state proteolysis of RET-S1 by NS3-4A and His-NS3-4A. The y intercept of the line in each case corresponds to the active fraction of protein (see “Experimental Procedures”). For NS3-4A (solid circles), the active fraction was 75 ± 14%. For His-NS3-4A (hatched squares), the active fraction was 25 ± 12%. NS3/4A polyprotein did not display any measurable serine protease activity (solid squares). The data shown are determined using proteins of the 1b genotype. Similar results were observed using NS3-4A(1a) and His-NS3-4A(1a). This data are the average of three experiments and the error values represent standard deviation.
Most previous work on NS3 has utilized N-terminal His-tagged variants of the protein (12, 13, 30, 31). For comparison purposes, we also constructed an NS3-4A complex with a His tag on the N terminus of NS3. Interestingly, kinetic analysis indicates that the His-tagged NS3-4A preparation reacts 2-fold more slowly than the untagged variant (0.010 ± 0.001 μm product/s), and contains only a 25 ± 12% active population (Fig. 3 and “Experimental Procedures”). A His tag at the NS3 N terminus is therefore detrimental for folding and/or assembly of the protease domain, and it also diminishes the ultimate levels of proteolytic activity by the tagged NS3-4A complex. These findings are consistent with the fact that N-terminal NS3 His tags are in close proximity to the protease active site.
Uncleaved NS3/4A Polyprotein Lacks Protease and Helicase Activities—The uncleavable NS3/4A polyprotein construct (containing 2 alanines where the threonine/serine cleavage sequence N-terminal to NS4A would have been) was synthesized originally as a tool that would provide a size standard for determining the presence of any uncleaved NS3/4A polyprotein in wild-type preparations of the complex. However, the mutant NS3/4A construct has also provided valuable additional information about the activities that we should expect for HCV polyproteins. To our knowledge, catalytic activities of uncleaved NS3/4A constructs have never been tested. Here we observed, using the mutant polyprotein, that uncleaved NS3/4A molecules are catalytically inactive (i.e. they lack both serine protease and helicase activity). They fail to display protease activity for substrates provided in trans (i.e. RET-S1) (Fig. 3), and they lack helicase activity (data not shown). Thus, proper assembly of the cleaved, NS3-4A complex is required for all the enzymatic activities of this protein. It is therefore likely that many sections of the polyprotein are not appropriately folded or functional until the individual proteins are cleaved apart.
NS3-4A Exhibits Robust Serine Protease Activity Under a Variety of Salt and pH Conditions—It has previously been reported that NS3-4A only functions as a robust serine protease under conditions of high salt (≥150 mm NaCl) and high pH (7.5–8.0) (17, 27). These same conditions are incompatible with those required for NS3 helicase activity (13, 17, 32), and it therefore has been suggested that the two activities (proteolysis and unwinding) might be mutually exclusive (17). We therefore sought to determine whether protease activity of the wild-type NS3-4A complex is restricted to the narrow range of previously reported conditions. To this end, we measured NS3-4A serine protease activity under a variety of pH (6.5 and 8.0) and salt conditions (30, 75, and 200 mm NaCl) (Fig. 4). We did not observe significant differences in the active fraction of protein (i.e. the y intercept of the velocity plots) under these varying pH and salt conditions, particularly when accounting for error (Fig. 4 legend). The steady-state proteolysis velocities in 30 mm NaCl at pH 6.5 and 8.0 were observed to be 0.006 ± 0.001 and 0.019 ± 0.001 μm/s, respectively. In 200 mm NaCl at pH 6.5 and 8.0, the steady-state proteolysis velocities were observed to be 0.010 ± 0.001 μm/s in both cases. Therefore, native, full-length NS3-4A is a robust serine protease under a broad range of salt and pH conditions, including those that are compatible with helicase activity.
FIGURE 4.

Steady-state velocity curves for NS3-4A proteolysis of RET-S1 under a range of pH and salt conditions. The steady-state rates of proteolysis were 0.006 ± 0.001 μm product/s at pH 6.5, 30 mm NaCl (solid circles), 0.019 ± 0.001μm product/s at pH 8, 30 mm NaCl (pierced circles), 0.019 ± 0.001 μm product/s at pH 6.5, 75 mm NaCl (solid diamonds), 0.020 ± 0.001 μm product/s at pH 8, 75 mm NaCl (pierced diamonds), 0.010 ± 0.001 μm product/second at pH 6.5, 200 mm NaCl (solid squares), and 0.010 ± 0.001 μm product/s at pH 8, 200 mm NaCl (pierced squares). The active fraction in each case, as determined by intersection with the y intercept, was 68 ± 9% for pH 6.5, 30 mm NaCl, 84 ± 16% for pH 8.0, 30 mm NaCl, 84 ± 15% for pH 6.5, 75 mm NaCl, 70 ± 12% for pH 8.0, 75 mm NaCl, 95 ± 5% for pH 6.5, 200 mm NaCl, and 95 ± 5% for pH 8.0, 200 mm NaCl. The data shown were determined using NS3-4A of the 1b genotype and represent the steady-state data points fit to a line. Similar results were observed with NS3-4A (1a). This data are the average of three experiments and the error values represent standard deviation.
NS3hel Enhances NS3-4A Serine Protease Activity—Given that efficient NS3 helicase activity requires the protease domain (13), we asked whether NS3 protease activity is enhanced by the helicase domain. To answer this question, it was necessary to build new types of protein expression constructs because polyprotein precursors containing the isolated protease domain (i.e. a His-SUMO-NS3 protease domain/NS4A polyprotein construct) did not undergo autocatalytic cleavage to form an NS3 protease-4A complex (data not shown). We therefore expressed the NS3 protease domain and full-length NS4A as separate His-SUMO fusion proteins (Fig. 1C), which were each affinity purified on nickel columns. The partially purified His-SUMO-NS3 protease domain and His-SUMO-NS4A proteins were then combined in the presence of SUMO protease and incubated to form an active complex of the native NS3 protease domain and native NS4A (NS3 protease + NS4A, see “Experimental Procedures”). Similarly, we reconstituted full-length NS3 + NS4A as a control for comparative purposes (Fig. 1C) (see “Experimental Procedures”).
Similar to the preparation of co-expressed NS3-4A, 75 ± 10% of the reconstituted NS3 + NS4A preparation and 75 ± 12% of the reconstituted NS3 protease + NS4A preparations were proteolytically active (Fig. 5). Comparison of the kinetic parameters for NS3-4A, NS3 protease domain + NS4A, and NS3 + NS4A proteolysis of RET-S1 revealed that the Km values for cleavage of substrate did not differ significantly (Km = 10.0 ± 3.0 μm for NS3-4A, 4.0 ± 1.2 μm for NS3 protease + NS4A, and 3.0 ± 1.0 μm for NS3 + NS4A) (Fig. 6 and Table 1), indicating that RET-S1 binds similarly to all the constructs.
However, the maximal velocities of proteolysis by the three enzyme constructs were substantially different. The rates of cleavage for NS3-4A, NS3 + 4A, and NS3 protease + 4A were 17.0 ± 2.0, 0.50 ± 0.05, and 0.11 ± 0.01 pmol of RET-S1 cleaved/s, respectively (Fig. 6 and Table 1). The large rate differences between the co-expressed NS3-4A and the reconstituted NS3 + NS4A constructs suggest that the NS4A cofactor does not associate and promote protease folding as effectively when presented in trans (i.e. 25 mm Hepes (pH 8.0), 0.3 m NaCl, 10% glycerol, 1 mm dithiothreitol, 0.2% Triton X-100).
Many conditions were explored to optimize reconstitution with NS4A in trans. Despite varying detergent concentrations (0.1–1%, using Triton X-100 or CHAPS), varying glycerol concentrations from 10 to 50%, and varying NaCl from 30 to 200 mm, increases in proteolytic activity were not observed (data not shown).
Taken together, these data suggest that the protease domain active site is optimally formed only upon co-expression with NS4A. Nonetheless, the reconstituted complexes retain significant levels of activity and provide valuable tools for structure/function studies (5, 22).
Given the inherently higher reactivity of co-expressed NS3-4A, it is clear that proteolytic activity of the isolated protease domain (NS3 protease + 4A) is directly comparable only with the reconstituted complex containing full-length NS3 (NS3 + 4A). When these two constructs are compared, the isolated protease domain is much less reactive than full-length NS3 (Fig. 6B). Indeed, NS3 protease + NS4A is 5- and 6-fold less efficient than NS3 + NS4A when Vmax and kcat/Km values are compared, respectively (Table 1). It is notable that the major effects are on kcat, which suggests that the helicase domain directly influences catalytic function of the protease active site. Therefore, NS3 protease activity is enhanced by the presence of the NS3hel domain, indicating that the two domains have evolved to become completely interdependent.
DISCUSSION
By creating new types of NS3 constructs and monitoring their enzymatic activities, we have evaluated the structural integrity and catalytic potential of the serine protease domain. We have established that NS3 protease activity is strongly influenced by its structural context, and that positive and negative effects on catalysis are induced by the presence of adjacent domains and uncleaved polyprotein moieties, respectively. Taken together, our findings suggest that NS3 protease activity is tuned and regulated by other viral components, which have coevolved to maximize the role of the protease in the HCV viral lifecycle.
The Helicase and the Protease Are Interdependent Enzymes within a Single Protein—Like many viral proteins, NS3 is multifunctional and it contains different enzymatic activities within a single protein chain. It has long been known that the protease and ATPase/helicase activities of NS3 are located on separate domains of NS3, and given the disparate nature of these activities, and their presumably different roles in function of the virus, it was generally assumed that the protease and helicase activities had no relationship to one another. However, structural analysis revealed that the protease domain is located in close proximity to the ATPase and RNA binding sites of NS3hel (33, 34). We therefore wondered whether the two enzymes in NS3 might actually be interdependent, having evolved to function optimally as a unit. Allosteric coactivation would have important ramifications for the function of the HCV replication complex and for the testing of HCV inhibitors, which tends to be studied with the isolated domains.
In our first study of this problem, we showed that RNA binding, ATPase, and RNA unwinding activities of the helicase are all stimulated by the presence of the covalently attached NS3 protease domain (13). Here we demonstrate that the NS3-4A serine protease activity is stimulated by the presence of NS3hel as well. Importantly, the NS3hel domain must be covalently attached to the NS3 protease domain, as adding NS3hel in trans had no effect on NS3 protease domain + NS4A protease activity (data not shown). Thus, the different enzymatic domains of NS3 are fully interdependent, which suggests that they have coevolved and that their presence on a single protein may have provided a selective advantage to the virus. Perhaps most importantly, the results suggest that these seemingly disparate enzymatic activities may somehow be coupled during some aspect of viral function.
The NS3/4A Polyprotein Fails to Catalyze Proteolysis or Unwinding—Just as the helicase domain activates serine protease activity, the presence of an uncleaved NS3/4A junction inhibits serine protease activity. The data presented herein show that polyprotein cleavage must occur before NS3 becomes proteolytically active. Even NS3 helicase activity requires cleavage of the NS3/4A junction sequence before any RNA unwinding activity is observed (data not shown). Taken together, these data indicate that polyprotein processing must necessarily be a very early event, as it is required for catalytic functions of viral constituents.
Proteolysis and Helicase Activities Are Not Mutually Exclusive—Previous work has suggested that NS3-4A protease activity occurs under conditions that are incompatible with helicase activity. This finding implies that helicase and protease activities might be mutually exclusive, or that their viral functions are completely unrelated. Importantly, it had been reported that NS3-4A protease activity is very sensitive to salt concentrations when the protein is studied at low concentrations (<1 nm) and less sensitive to salt concentrations at higher protein concentrations (≥1 nm) (22). We therefore conducted protease assays with relatively high NS3-4A concentrations (40–120 nm) that fall within the range typically employed for RNA helicase assays (12, 13, 25, 31). Our ability to observe helicase and protease activity under the same conditions may also stem from the fact that we utilized untagged NS3-4A, which is more reactive than His-tagged NS3-4A. An N-terminal His tag reduces both the rate of serine protease activity and the active fraction of protein (Fig. 3). Indeed, proteolysis by untagged NS3-4A is not highly salt dependent and the reaction is efficient under diverse conditions (Fig. 4).
Earlier work has demonstrated that NS3 functions as a robust RNA helicase at NaCl concentrations as high as 100 mm and it can unwind RNA at even higher NaCl concentrations, although less efficiently (13). The fact that NS3 can cleave protein or unwind RNA in the same range of salt and pH conditions (including ionic conditions that approach physiological (150 mm)) indicates that NS3-4A is capable of transitioning smoothly between proteolysis and RNA unwinding during various stages of HCV replication. Immediately after NS3-4A autocleaves from the HCV polyprotein, it may unwind HCV RNA or use NS3hel as a motor to translocate along the HCV polyprotein and scan for subsequent peptide cleavage sites. Protein translocation by NS3hel has never been demonstrated, but its potential importance is underscored by the translocase activities of related proteins such as ClpX (35–37).
Reconstituted Forms of NS3-4A Are Significantly Less Reactive Than Autocleaved NS3-4A—To perform this study, it was necessary to create reconstituted forms of the NS3-4A complex, in which the 4A protein was added in trans to the serine protease domain or to the full-length NS3. This provided an opportunity to compare the reactivities of NS3-4A molecules that, despite identical sequences, were produced in different ways (through autocleavage of polyprotein or reconstitution from separate proteins). Here we observe that autoproteolyzed NS3-4A is a much more active serine protease than complexes that were reconstituted from separate NS4A molecules (NS3 + NS4A). Consistent with this, the NS3-4A complex that is disrupted by dilution into low salt buffers (17) cannot be restored to normal levels of the protease activity by the addition of salt (data not shown). Specifically, we observe that NS3-4A has a Vmax for RET-S1 proteolysis that is 34 times faster than NS3 + NS4A (Table 1). Intriguingly, the Km values for NS3-4A and NS3 + NS4A proteolysis of RET-S1 do not vary significantly (Table 1). Taken together, the kinetic analysis of proteolysis by NS3-4A, NS3 + NS4A, and NS3 protease + NS4A suggests that all of these constructs bind substrate with a similar affinity, but they have large differences in efficiency of chemical catalysis.
One possible explanation is that the NS4A co-factor may not properly intercalate with the NS3 protease domain when NS3 and NS4A are simply mixed, potentially resulting in misalignment of active site residues within the NS3 protease domain. Autocleavage between NS3 and NS4A and the concurrent intercalation of NS4A into the NS3 protease domain requires a specific cleavage site sequence that slows the proteolysis reaction (Thr/Ser as opposed to Cys/Ser at other HCV polyprotein sites) (38). Presumably, this slow autocleavage between NS3 and NS4A facilitates the correct intercalation of NS4A into the NS3 protease domain as NS4A is being cleaved from the NS3 C terminus (38). Therefore, whereas our findings suggest that the helicase domain allosterically influences the protease active site, an alternative interpretation of the results is that a coexpressed helicase domain is required for proper folding of the protease domain and/or appropriate docking of the 4A cofactor. In addition, the expression strategy employed in this work may not optimally recapitulate folding of the individual proteins. But regardless of the mechanistic basis for the apparent codependence of the protease and the helicase, the data clearly show that reconstituted NS3 and 4A constructs are significantly impaired, which has major implications for the design of protein constructs used in drug screening and for structure/function efforts on HCV.
Many investigators have aimed to disrupt NS3-4A protease activity as an approach toward antiviral therapeutics and various forms of the complex have been employed in these studies (19–23, 39). Although it has been suggested that NS3hel might enhance NS3-4A protease activity (23), this synergy was not demonstrated until now. Our findings suggest that screens for NS3-4A protease inhibitors should involve full-length NS3 constructs rather than truncated protease domains. Moreover, these screens should utilize constructs in which NS3-4A is produced through autoproteolysis rather than reconstitution. Indeed, a recent study comparing the effects of NS3-4A serine protease inhibitors on full-length NS3-4A produced through autocleavage, NS3 + NS4A peptide, and NS3 protease domain + NS4A peptide demonstrated that BILN 2061 and VX-950 are much better inhibitors of autocleaved NS3-4A serine protease activity than of reconstituted NS3 + NS4A peptide or NS3 protease domain + NS4A (22). The greater protease activity of autocleaved NS3-4A creates the potential for increased sensitivity when measuring protease activity in the presence of new drugs (i.e. more easily measured inhibition of protease activity in screening assays of compound libraries), which will lead to a greater diversity of promising lead compounds. Given the synergies observed among components of the NS3 helicase-protease, it will be interesting to monitor the influence of NS4A on RNA binding, ATPase, and RNA unwinding activities of NS3 in future studies.
Supplementary Material
Acknowledgments
We thank Dr. William Delaney, Dr. Brett D. Lindenbach, Dr. Victor Serebrov, and Steven Ding for critically reading the manuscript and numerous helpful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM60620. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.

Author's Choice—Final version full access.
Footnotes
The abbreviations used are: HCV, hepatitis C virus; NS, nonstructural; BisTris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; MES, 2-(N-morpholino)ethanesulfonic acid.
References
- 1.Lam, A. M., and Frick, D. N. (2006) J. Virol. 80 404-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999) Science 285 110-113 [DOI] [PubMed] [Google Scholar]
- 3.de la Cruz, J., Kressler, D., and Linder, P. (1999) Trends Biochem. Sci. 24 192-198 [DOI] [PubMed] [Google Scholar]
- 4.Failla, C., Tomei, L., and De Francesco, R. (1994) J. Virol. 68 3753-3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taliani, M., Bianchi, E., Narjes, F., Fossatelli, M., Urbani, A., Steinkuhler, C., De Francesco, R., and Pessi, A. (1996) Anal. Biochem. 240 60-67 [DOI] [PubMed] [Google Scholar]
- 6.Xu, L. G., Wang, Y. Y., Han, K. J., Li, L. Y., Zhai, Z., and Shu, H. B. (2005) Mol. Cell 19 727-740 [DOI] [PubMed] [Google Scholar]
- 7.Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., and Tschopp, J. (2005) Nature 437 1167-1172 [DOI] [PubMed] [Google Scholar]
- 8.Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., Ishii, K. J., Takeuchi, O., and Akira, S. (2005) Nat. Immunol. 6 981-988 [DOI] [PubMed] [Google Scholar]
- 9.Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005) Cell 122 669-682 [DOI] [PubMed] [Google Scholar]
- 10.Jankowsky, E., and Fairman, M. E. (2007) Curr. Opin. Struct. Biol. 17 316-324 [DOI] [PubMed] [Google Scholar]
- 11.Suzich, J. A., Tamura, J. K., Palmer-Hill, F., Warrener, P., Grakoui, A., Rice, C. M., Feinstone, S. M., and Collett, M. S. (1993) J. Virol. 67 6152-6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frick, D. N., Rypma, R. S., Lam, A. M., and Gu, B. (2004) J. Biol. Chem. 279 1269-1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beran, R. K., Serebrov, V., and Pyle, A. M. (2007) J. Biol. Chem. 282 34913-34920 [DOI] [PubMed] [Google Scholar]
- 14.Binder, M., Quinkert, D., Bochkarova, O., Klein, R., Kezmic, N., Bartenschlager, R., and Lohmann, V. (2007) J. Virol. 81 5270-5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi, M., Ma, Y., Yates, J., and Lemon, S. M. (2007) J. Virol. 81 629-638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartenschlager, R., Lohmann, V., Wilkinson, T., and Koch, J. O. (1995) J. Virol. 69 7519-7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallinari, P., Paolini, C., Brennan, D., Nardi, C., Steinkuhler, C., and De Francesco, R. (1999) Biochemistry 38 5620-5632 [DOI] [PubMed] [Google Scholar]
- 18.Shimizu, Y., Yamaji, K., Masuho, Y., Yokota, T., Inoue, H., Sudo, K., Satoh, S., and Shimotohno, K. (1996) J. Virol. 70 127-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimasi, N., Martin, F., Volpari, C., Brunetti, M., Biasiol, G., Altamura, S., Cortese, R., De Francesco, R., Steinkuhler, C., and Sollazzo, M. (1997) J. Virol. 71 7461-7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin, C., Lin, K., Luong, Y. P., Rao, B. G., Wei, Y. Y., Brennan, D. L., Fulghum, J. R., Hsiao, H. M., Ma, S., Maxwell, J. P., Cottrell, K. M., Perni, R. B., Gates, C. A., and Kwong, A. D. (2004) J. Biol. Chem. 279 17508-17514 [DOI] [PubMed] [Google Scholar]
- 21.Lamarre, D., Anderson, P. C., Bailey, M., Beaulieu, P., Bolger, G., Bonneau, P., Bos, M., Cameron, D. R., Cartier, M., Cordingley, M. G., Faucher, A. M., Goudreau, N., Kawai, S. H., Kukolj, G., Lagace, L., LaPlante, S. R., Narjes, H., Poupart, M. A., Rancourt, J., Sentjens, R. E., St. George, R., Simoneau, B., Steinmann, G., Thibeault, D., Tsantrizos, Y. S., Weldon, S. M., Yong, C. L., and Llinas-Brunet, M. (2003) Nature 426 186-189 [DOI] [PubMed] [Google Scholar]
- 22.Mao, S. S., Dimuzio, J., McHale, C., Burlein, C., Olsen, D., and Carroll, S. S. (2008) Anal. Biochem. 373 1-8 [DOI] [PubMed] [Google Scholar]
- 23.Thibeault, D., Bousquet, C., Gingras, R., Lagace, L., Maurice, R., White, P. W., and Lamarre, D. (2004) J. Virol. 78 7352-7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda, M., Yi, M., Li, K., and Lemon, S. M. (2002) J. Virol. 76 2997-3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beran, R. K., Bruno, M. M., Bowers, H. A., Jankowsky, E., and Pyle, A. M. (2006) J. Mol. Biol. 358 974-982 [DOI] [PubMed] [Google Scholar]
- 26.Steinkuhler, C., Tomei, L., and De Francesco, R. (1996) J. Biol. Chem. 271 6367-6373 [DOI] [PubMed] [Google Scholar]
- 27.Sali, D. L., Ingram, R., Wendel, M., Gupta, D., McNemar, C., Tsarbopoulos, A., Chen, J. W., Hong, Z., Chase, R., Risano, C., Zhang, R., Yao, N., Kwong, A. D., Ramanathan, L., Le, H. V., and Weber, P. C. (1998) Biochemistry 37 3392-3401 [DOI] [PubMed] [Google Scholar]
- 28.Copeland, R. A. (2005) Evaluation of Enzyme Inhibitors in Drug Discovery, John Wiley & Sons, Inc., Hoboken, NJ
- 29.Li, S. J., and Hochstrasser, M. (1999) Nature 398 246-251 [DOI] [PubMed] [Google Scholar]
- 30.Zhang, C., Cai, Z., Kim, Y. C., Kumar, R., Yuan, F., Shi, P. Y., Kao, C., and Luo, G. (2005) J. Virol. 79 8687-8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang, P. S., Jankowsky, E., Planet, P. J., and Pyle, A. M. (2002) EMBO J. 21 1168-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam, A. M., Rypma, R. S., and Frick, D. N. (2004) Nucleic Acids Res. 32 4060-4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim, J. L., Morgenstern, K. A., Lin, C., Fox, T., Dwyer, M. D., Landro, J. A., Chambers, S. P., Markland, W., Lepre, C. A., O'Malley, E. T., Harbeson, S. L., Rice, C. M., Murcko, M. A., Caron, P. R., and Thomson, J. A. (1996) Cell 87 343-355 [DOI] [PubMed] [Google Scholar]
- 34.Yao, N., Reichert, P., Taremi, S. S., Prosise, W. W., and Weber, P. C. (1999) Structure 7 1353-1363 [DOI] [PubMed] [Google Scholar]
- 35.Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T., and Baker, T. A. (2000) Mol. Cell 5 639-648 [DOI] [PubMed] [Google Scholar]
- 36.Singh, S. K., Grimaud, R., Hoskins, J. R., Wickner, S., and Maurizi, M. R. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 8898-8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortega, J., Singh, S. K., Ishikawa, T., Maurizi, M. R., and Steven, A. C. (2000) Mol. Cell 6 1515-1521 [DOI] [PubMed] [Google Scholar]
- 38.Wang, W., Lahser, F. C., Yi, M., Wright-Minogue, J., Xia, E., Weber, P. C., Lemon, S. M., and Malcolm, B. A. (2004) J. Virol. 78 700-709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dahl, G., Sandstrom, A., Akerblom, E., and Danielson, U. H. (2007) Antiviral Ther. 12 733-740 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.