

Abstract
The dynamics of G protein-mediated signal transduction depend on the two-dimensional diffusion of membrane-bound G proteins and receptors, which has been suggested to be rate-limiting for vertebrate phototransduction, a highly amplified G protein-coupled signaling pathway. Using fluorescence recovery after photobleaching (FRAP), we measured the diffusion of the G protein transducin α-subunit (Gαt) and the G protein-coupled receptor rhodopsin on disk membranes of living rod photoreceptors from transgenic Xenopus laevis. Treatment with either methyl-β-cyclodextrin or filipin III to disrupt cholesterol-containing lipid microdomains dramatically accelerated diffusion of Gαt in its GTP-bound state and of the rhodopsin-Gαβγt complex but not of rhodopsin or inactive GDP-bound Gαβγ. These results imply an activity-dependent sequestration of G proteins into cholesterol-dependent lipid microdomains, which limits diffusion and exclude the majority of free rhodopsin and the free G protein heterotrimer. Our data offer a novel demonstration of lipid microdomains in the internal membranes of living sensory neurons.
Two-dimensional diffusion of membrane proteins is central to G protein-mediated signal transduction. Phototransduction, the G protein cascade of vision, begins with diffusion of photoexcited rhodopsin (metarhodopsin II; R*)2 on the photoreceptor disk membranes to collide with and activate the peripherally membrane-bound G protein transducin, Gαβγt-GDP, at a rate of several hundred per second. The activated GTP-bound α-subunit of transducin, Gαt-GTP, dissociates from R* and Gβγt and diffuses on the membrane surface to bind and activate the effector, a membrane-bound cyclic GMP phosphodiesterase, PDE6. The recovery of a photoresponse is similarly composed of a series of diffusional encounters between phototransduction proteins on the membrane. It has been suggested that such diffusion is rate-limiting for both the activation and recovery phases of phototransduction based on results with genetically modified mice (1). However, the nature, regulation, and functional consequences of membrane-delimited diffusion in G protein signaling have yet to be characterized. Roles for lipid microdomains, such as rafts, in the regulation of membrane protein dynamics have been implicated in G protein signaling and other signaling pathways (2), but current evidence is not definitive, and it is unknown whether lipid microdomains play a role in phototransduction.
EXPERIMENTAL PROCEDURES
Buffers—Standard buffers contained (in mm): buffer A (Ringer's solution), HEPES 5, pH 7.7, NaCl 110, CaCl2 2.0, KCl 2.5, MgCl2 1.2, saturated with 95% O2, 5% CO2; buffer B, HEPES 10, pH 7.2, MgCl2 3.0, arginine 105, glutamic acid 105, EGTA 1.0, DTT 1.0; buffer C (Marc's modified Ringer's solution), HEPES 5, pH 7.5, NaCl 100, CaCl2 2.0, KCl 2.0, MgCl2 1.0; buffer D, sucrose 250, KCl 75, spermidine trihydrochloride 0.5, spermine tetrahydrochloride 0.2, pH 7.4; buffer E, Tris 5.0, pH 7.2, MgCl2 0.5, DTT 2; buffer F, MOPS 20, pH 7.5, NaCl 50, MgCl2 2.0, EDTA 0.1, DTT 2; buffer G, HEPES 50, pH 7.5, EDTA 1.0, EGTA 3.0, MgCl2 5.0, DTT 1, GDP 0.1, phenylmethylsulfonyl fluoride; buffer H, Tris 20, pH 7.5, MgCl2 5, EDTA 0.1, DTT 2, GDP 0.1, and phenylmethylsulfonyl fluoride ∼20 mg/liter; buffer I, MOPS 20, pH 7.5, NaCl 50, MgCl2 2, EDTA 0.1, DTT 2; buffer J, Tris 25, glycine 192, 0.1% (w/v) SDS, pH 8.3.
DNA Constructs—DNA encoding bovine transducin Gαt with enhanced green fluorescent protein (EGFP) fused into a loop in the helical domain (Gαt-EGFP), as shown in Fig. 1, was inserted into a baculovirus vector for expression in Sf9 cells or into a plasmid containing the Xenopus opsin promoter (illustrated in Fig. 1) for transgenesis. The rhodopsin-EGFP-1D4 transgene plasmid (3) was a gift from Dr. Daniel Oprian of Brandeis University. The pXOP-EGFP-Gαt and pVL1392-EGFP-Gαt expression plasmids were constructed using standard PCR mutagenesis and subcloning techniques. In both cases, the coding sequence (GenBank™ accession number AY292281) encodes the first 115 amino acids of bovine Gαt followed by EGFP, connected with a 6-residue linker sequence (SGGGGS) at each end, followed by the remainder of the bovine Gαt sequence. The transgene construct was made by inserting the EGFP-Gαt sequence in place of EGFP in the pXOP-EGFP vector described previously (4), so that the Xenopus rhodopsin promoter (5) is upstream of the translation start site, and the SV40 poly(A) site is downstream of the translation termination site. For transgenesis, pXOP-EGFP-Gαt was cut by ApaLI and MluI, and the 4.1-kb fragment having the Xenopus rhodopsin promoter, EGFP-Gαt and SV40 poly(A) was gel-purified (Qiaex II, Qiagen) with final elution in water. To produce transgenic Xenopus expressing EGFP only, pXOP-EGFP was digested by RsrII and ApaLI, and a 4.0-kb fragment having a rhodopsin promoter, EGFP coding sequence, and SV40 poly(A) was gel-purified and eluted in water. The plasmid for baculovirus production was prepared by inserting a fragment encoding EGFP-Gαt into NotI- and SmaI-digested pVL1392 vector (Pharmingen).
FIGURE 1.

Functional EGFP transducin was expressed in transgenic tadpole rod outer segments. A, map of transgene construct pXOP-EGFP-Gαt. On the map line, the 1.4-kilobase Xenopus rhodopsin promoter sequence is shown in purple, the EGFP coding sequence is shown in green, the bovine transducin cDNA sequence is shown in red, and the SV40 poly(A) sequence is shown in blue. The restriction enzyme sites are: A, ApaLI; X, XhoI; B, BspEI; Ba, BamHI; and M, MluI. B, immunoblotting with Gαt-specific antibodies. Lane 1, wild-type (wt) bovine Gαt; lane 2, insect cell purified EGFP-Gαt; lanes 3 and 4, eye homogenates from two different transgenic tadpoles (lane 3 is the strongest transgene signal observed; lane 4 is typical); lane 5, wild-type tadpole eye homogenate. C, kinetics of nucleotide exchange. The amounts of GTPγ35S bound to recombinant wild-type bovine transducin (open circles) or EGFP-transducin (filled circles) catalyzed by light-activated rhodopsin were measured at different time points by a filtration assay. The curve is a nonlinear least squares fit of experimental data to first order uptake with a rate constant of 8.2 min-1. D, photoreceptor cells of a transgenic tadpole expressing EGFP-Gαt. Fluorescence localization of EGFP-Gαt in a transgenic tadpole retina frozen section is shown in E–G. E, EGFP-Gαt is shown in green. F, tadpole rods (rhodopsin immunostaining) are shown in red. G shows digitally summed images of E and F. Scale bars represent 20 μm.
Expression and Purification of Proteins—Recombinant baculoviruses were isolated following co-transfection of the linearized BaculoGold viral DNA (Pharmingen) and the transfer vector into Sf9 cells following the manufacturer's instructions. Sf9 (from Spodoptera frugiperda, ATCC CRL-1711) cells were cultured to a density of ∼2 × 106/ml and infected with wild-type bovine Gαt and γ1/β1.2 or with EGFP-Gαt and γ1/β1.2 recombinant baculoviruses. The infected cells were incubated at 28 °C for 48 h. Cells were harvested by centrifugation, and the cell pellet was resuspended in 40 ml of buffer G and then sonicated on ice. The lysate was centrifuged at 100,000 × g for 40 min at 4 °C. The pellet was resuspended in buffer H supplemented with 4% sodium cholate (Sigma) or 0.5% polyoxyethylene-10-laurylether (Sigma) and then homogenized using 16–21-gauge needles. The homogenate was centrifuged at 100,000 × g for 40 min at 4 °C after incubation at 4 °C for 3 h. The pellet was extracted with 4% sodium cholate or 0.5% polyoxyethylene-10-laurylether in buffer H again. The supernatants from two extractions were combined and diluted 4-fold using buffer H. The detergent extract was loaded onto a 100-ml DEAE-Sepharose Fast Flow (Sigma) column under gravity at 4 °C. After washing the column with 500 ml of buffer H, EGFP-Gt was eluted with a 200-ml gradient of NaCl from 0 to 1 m in buffer H. Fractions containing EGFP-Gt were pooled and dialyzed with buffer I.
Characterization of Purified Gαt-EGFP—Gαt-EGFP expressed in Sf9 cells was purified by anion exchange chromatography, reconstituted with Gβγt purified from bovine rod outer segments, and assayed along with purified bovine Gαβγt for [35S]GTPγS uptake in the presence of photoisomerized rhodopsin in urea-washed rod disk membranes. Purified EGFP-Gαβγt or wild-type bovine Gαβγt was mixed with urea-washed ROS (6) on ice under a dim red light in buffer F in a 300-μl assay volume. After incubation at room temperature for 5 min, the assay was started by exposure to room light and the addition of [35S]GTPγS solution to a final concentration of 2.5 μm GTPγS and 10 nm rhodopsin. At various time points, 20-μl portions of the reaction mixture were filtered through 0.45-μm nitrocellulose filters, which were washed three times with buffer F and air-dried before liquid scintillation counting. The very low amounts of radioactivity binding to the filters without Gαβγt were subtracted. No detectable binding of [35S]GTPγS was observed without the addition of ROS.
Preparation of Bovine ROS Lipid Vesicles—For reconstitution of purified EGFP-Gαβγt, small unilamellar vesicles were prepared by extrusion from extracted bovine rod outer segment lipids as described (7). Bovine ROS were prepared using standard procedures, and membrane pellets were obtained by centrifugation at 100,000 × g for 30 min at 4 °C. The pellets were resuspended in buffer E and then mixed with an equal volume of chloroform/methanol/12 n HCl (100:100:1, v/v/v), vortexed, and incubated at 23 °C for 20 min. After centrifugation at 100,000 × g at 4 °C for 20 min, the aqueous layer was collected and extracted again with an equal volume of chloroform/methanol/12 n HCl (100:100:1, v/v/v). The organic phases containing ROS lipid were pooled and dried under a stream of argon. The dried ROS lipids were resuspended in buffer F and subjected to at least five freeze/thaw cycles in liquid N2. The lipid suspension was then extruded 10 times through 0.1-μm polycarbonate filters using a liposome extruder (Lipex Biomembranes). The concentration of lipid was determined by the assay of inorganic phosphorous.
For reconstitution of purified EGFP-Gαβγt, a polylysine-coated coverslip was covered with the suspension of small unilamellar vesicles to form supported bilayers. Purified Gαt-EGFP bound to GDP and Gβγt was added to the supported bilayers and washed briefly with buffer to remove unbound protein. The reconstituted bilayers were then used for fluorescence recovery after photobleaching (FRAP) measurements.
Transgenesis—Procedures with animals were carried out according to an approved protocol in accordance with guidelines of the Institutional Animal Care and Use Committee of Baylor College of Medicine. Transgenesis was carried out essentially as described previously (8) by intracytoplasmic injection. About 400,000 snap-frozen sperm nuclei in 4 μl were added to 250–500 ng of transgene DNA in 2.5 μl, incubated for 15 min at room temperature, and then diluted 410-fold with buffer D for injection. Xenopus laevis eggs were injected in 0.4× Marc's modified Ringer's solution containing 6% (w/v) Ficoll (GE Healthcare). Properly gastrulating embryos were selected, raised in 0.1× Marc's modified Ringer's solution until approximately stage 42, and then transferred to dechlorinated water. Tadpoles were anesthetized in 0.01% 3-aminobenzoic acid ethyl ester (Sigma) and monitored for EGFP expression using a fluorescence dissecting microscope (Leica MZFL III). Developmental stages of embryos were determined according to Nieuwkoop and Faber (4).
Immunoblotting and Densitometry—Eyes were collected from four transgenic and four wild-type tadpoles and homogenized in 100 μl of buffer J. 20 μl of each sample (about 10 μg of total protein) was applied to SDS-PAGE alongside purified bovine Gt and insect cell-purified EGFP-Gαt. Immunoblotting was processed according to a standard protocol (9) on proteins separated by SDS-PAGE. Electrophoretic transfer to supported nitrocellulose (NitroPure, Osmonics, Inc.) was carried out in buffer J supplemented with 20% (v/v) methanol for 60 min at 350 mA at 4 °C. The membrane was blocked by 5% nonfat dry milk-TBS/T solution (20 mm Tris-HCl, pH 7.2, 150 mm NaCl, 0.1% Tween 20) for 1 h followed by incubation with primary antibody, Gαt1 (K-20, Santa Cruz Biotechnology), at a 1:600 dilution for 4 h at room temperature. The secondary antibody used was horseradish peroxidase-conjugated (Promega) anti-rabbit IgG with detection by chemiluminescence using the ECL system (Amersham Biosciences). For densitometry, x-ray films were scanned and bands quantified by UN-SCAN-IT (Silk Scientific Inc.) software.
Immunofluorescence Staining—Transgenic tadpoles at stage 45 were euthanized, and heads were fixed in 4% paraformaldehyde-phosphate-buffered saline (PBS, pH 7.2) for 10–16 h at 4 °C and then placed in 30% sucrose-PBS for 1 h at 4 °C. Then they were embedded in OCT compound (Tissue-Tek), and frozen sections were cut at 12 μm. All post-fixation and staining steps were performed at room temperature. Tissue sections were post-fixed in 1:1 methanol:acetone (v/v) for 10 min and rehydrated twice in PBS for 10 min. Sections were blocked for 1 h with 10% goat serum (Sigma) in PBS. Then the sections were incubated overnight with primary antibody to rhodopsin, 1D4, at a concentration of 1 μg/ml in 10% goat serum in PBS. After being washed three times for 5 min in PBS, sections were incubated with secondary antibody, Cy5™-conjugated goat anti-mouse immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) at a 1:300 dilution (5 μg/ml) in 10% goat serum-PBS for 1 h. Sections were washed twice for 10 min in PBS, mounted in aqueous mounting medium (Gel/Mount; Biomeda, Foster City, CA), and examined using a Zeiss 510 LSM confocal microscope.
Preparation of Samples for FRAP Measurements—To measure the diffusion coefficients of EGFP-rhodopsin and EGFP-Gαt in rod photoreceptor cells, Xenopus tadpoles at about stage 45 were anesthetized in 0.01% 3-aminobenzoic acid ethyl ester and sacrificed, and eyes were dissected in control Ringer's solution. To isolate single photoreceptor cells, dissected tadpole eyes were put on a slide and squashed gently by a coverslip coated with rhodopsin antibody, B6-30N (10), used to immobilize the rod cells. To measure the diffusion of Gαt in different states, tadpoles eyes were incubated in oxygenated Ringer's solution supplemented with 10 mm glucose and periodically (once/min) perfused with fresh oxygenated Ringer's with 10 mm glucose; incubated in buffer B with 1 mm ATP and 1 mm GTPγS (supplemented with 50 μm/ml α-toxin; for forming constitutively active Gαt); incubated in Ringer's solution with 6 mm 2-deoxyglucose (2-DG; Sigma) and 10 mm sodium azide (Sigma; nucleotide depletion for accumulating R*-Gαβγt); or incubated in Ringer's solution with 10 mm hydroxylamine (for accumulating Gαβγt). Raft-disrupting drugs used were 10 mm methyl-β-cyclodextrin (MβCD, Sigma; for solubilizing and removing cholesterol) or 2 μg/ml filipin III (Cayman Chemical; for binding cholesterol to form aggregates). To render photoreceptor cells permeable to externally added nucleotides and raft-disrupting drugs, tadpole eyes were incubated with buffer B supplemented with 50 μg/ml α-toxin (List Biological Laboratories, Inc.) and combined with one or two reagents as described above. The cholesterol-loaded MβCD (1.2 mm cholesterol (Sigma) in 10 mm MβCD) was prepared in degassed buffer B supplemented with 50 μm diethylenetriamine pentaacetic acid, sealed in argon, and shaken at room temperature overnight. Previous studies found that treatment of rod disk membranes with these concentrations of cholesterol and MβCD increases disk membrane content of cholesterol from 15 to 38 mol % (11). The above media were all supplemented with 10 mm glucose except in the nucleotide depletion treatments.
To measure the diffusion coefficient of recombinant EGFP-Gαβγt on a supported bilayer of reconstituted lipids from bovine ROS, a poly-d-lysine (Roche Applied Science) precoated coverslip was dipped into bovine ROS lipid vesicles (see below) and washed twice with 5 ml of buffer F. Then 50 μl of purified EGFP-Gαβγt was added to the coverslip, which was then washed and placed on a slide for FRAP measurements.
FRAP Measurements by Confocal Microscopy—Photobleaching experiments were carried out using a Zeiss LSM 510 confocal microscope with Zeiss LSM software, version 2.0 or version 2.8. Retinal samples were imaged using Plan-Neofluar 40×/1.3 oil lens, and supported lipid bilayers were imaged using Plan-Neofluar 40×/1.3 oil lens. The 488 nm line of a 15-milliwatt argon laser was used to bleach fluorescence of samples. Typical settings for prebleach and recovery image scans were 0.5–5% of maximum laser power. A single photoreceptor cell with EGFP fluorescence was selected and located to the center of a field, which was usually 29.6 × 29.6 μm or 256 × 256 pixels; 256 gray level images were collected in the xy plane. Maximum scan speed was selected to take images. The fastest scan time used was 0.098 s, although most images were taken between 0.148 and 0.395 s, depending on the size of the image field. For lateral diffusion (perpendicular to the long axis of the rod), no delay was set between images. For longitudinal diffusion (parallel to the long axis of the rod), data were collected at least once every second. Usually three to five pictures were taken before bleaching. Bleaching was at maximum laser power, and a narrow rectangular area was selected using the bleach control program. For lateral diffusion, the long axis of the selected area was parallel to the long axis of rod outer segments, and the width was usually set to 7–13 pixels (∼1 μm). For longitudinal diffusion, the length was set to ∼1 μm along the long axis of rod outer segments encompassing about 38 of the nearly 2000 disks in a rod cell (as shown in Fig. 3A), and the width was set to cover the entire width of the cell (6–8 μm). For diffusion of EGFP-Gαβγt on a reconstituted ROS lipid layer, a circle with a diameter of ∼9 μm was selected for bleaching. The minimum bleach time was 5 ms, although typical time was 50 ms. For recovery, a time series of up to 90 recovery images was collected. For slow diffusion, one or more images were taken at a long time point to determine to what extent the recovery approached completion. For every image at each time point, the average integrated intensities of three areas were recorded: the area selected for bleaching, a non-bleached area on the photoreceptor cell, and a non-bleached area of background.
FIGURE 3.

An example of measurement of lateral diffusion by FRAP. A, images of a photobleached transgenic rod outer segment at different time points as indicated. The scale bar represents 2.0 μm. B, fitting of corrected recovery data to Equation 1 (see “Experimental Procedures”). C, shows intensity integrated along the y axis of each pixel position along the x axis (inset: labeled 0–90) of the selected region of each image in panel A. D, intensity of the bleached stripe (red; see inset), background (black), and non-bleached part (green) of the rod outer segments at each time point of the FRAP measurement is shown. The blue curve (crt) shows the intensity of the bleached stripe after subtracting background and correcting for fading of fluorescence due to photobleaching during image acquisition.
Usually data were captured as a time series of images in a single focal plane. The objectives used provided sufficient depth of field to ensure fairly uniform bleaching along the z axis, as verified by collection of z-stacks across the entire cell.
FRAP Data Analysis—Data were analyzed using programs implemented in MATLAB®. The recorded intensities from the bleached and non-bleached areas were corrected for background by subtracting from the value for each pixel the average intensity of the selected background area to give Iraw and Inon, respectively. To correct for fading of fluorescence due to taking images, the intensity of the non-bleached area on photoreceptor cells over time was fitted using the first order decay Equation 1. In the equation, Inon(t) is the recorded average integrated intensity of the selected area at time t, and Inon(0) the intensity recorded in the first image collected.
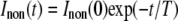 |
(Eq. 1) |
After determination of the parameter T by nonlinear least squares fitting, the intensities in image were corrected by Equation 2, in which I(t) is the corrected intensity of the bleached area.
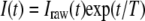 |
(Eq. 2) |
The pixel values in the images were integrated along the y axis, from the top to the bottom of the bleach stripe, to generate one-dimensional (x) integrated intensity profiles for each time point. For an initially uniform distribution across a rectangular membrane of width l, with average intensity I(0), the evolution of the integrated intensity Istripe within a central region of -w/2 ≤ x ≤ w/2 following an initial photobleaching with a Gaussian profile of full-width w, centered at x = 0, is given by the following equation (12),
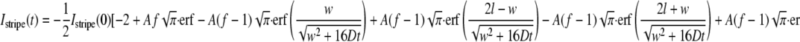 |
(Eq. 3) |
where A is the depth of bleach at x = 0 and t = 0, D is the diffusion coefficient, and f is the immobile fraction. For l, which varies along the z axis, as is the case for disks of circular profile, the root-mean-square value of 0.82l, where l is the cell diameter, can be substituted. The value of D extracted is insensitive to small changes in the value of l. The time course of integrated intensity across the pixels selected for bleaching (-w/2 to w/2 in length units) was fit to this function, with best estimates for f, D, A, and w selected by standard least-squares criteria. Initial estimates for A and w were extracted from the first image collected after photobleaching by fitting to Equation 4 where I(x, 0) is the corrected intensity of each pixel at t = 0,
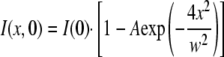 |
(Eq. 4) |
Fits were accepted only if the values for A and w obtained after least-squares fitting of the entire time course were within ±8% of the values estimated from the first image. Final w values agreed within one pixel or less.
The values of D were not corrected for the effects of invaginations of the disk membrane known as incisures; previous experiments by Wey et al. (13) provide an estimate of a factor of 2.7 that may be applied to the D values in Table 1 to obtain that D values would be expected to be observed in the absence of these barriers.
TABLE 1.
Lateral diffusion coefficients of EGFP, EGFP-Gαt, and rhodopsin-EGFP
| Sample | Treatment | Da | n |
|---|---|---|---|
| μm2/s | |||
| ROS with EGFP-Gαt (f < 1%) | Gα-GTP (glucose only) | 0.19 ± 0.02 | 15 |
| Gα-GTP (α-toxin only) | 0.18 ± 0.02 | 8 | |
| Gα-GTPγS (α-toxin/ATP/GTPγS) | 0.21 ± 0.01 | 10 | |
| R*-Gαβγ (2-DG/sodium azide) | 0.047 ± 0.005 | 10 | |
| R*-Gαβγ/no rafts (α-toxin/MβCD/2-DG/NaN3) | 0.15 ± 0.01 | 7 | |
| Gαβγ-GDP (hydroxylamine) | 0.37 ± 0.06 | 7 | |
| Gα-GTP/no rafts (α-toxin/MβCD) | 0.32 ± 0.02 | 11 | |
| Gα-GTP/no rafts (α-toxin/filipin III) | 0.29 ± 0.02 | 9 | |
| Gα-GTP/+ chol. (α-toxin/MβCD/cholesterol) | 0.14 ± 0.01 | 9 | |
| Gαβγ-GDP/no rafts (α-toxin/MβCD/hydroxylamine) | 0.32 ± 0.03 | 8 | |
| Purified EGFP-Gαt (f = 1%) | Free Gt-EGFP (reconstituted on lipid bilayers) | 42.8 ± 4.9 | 3 |
| ROS with EGFP (f = <1%) | Free EGFP | 22 | Est.b |
| ROS with rhodopsin-EGFP (f = <1%) | R* (glucose only) | 0.14 ± 0.02 | 10 |
| R*/no rafts (MβCD) | 0.17 ± 0.02 | 5 | |
| R*/no rafts (α-toxin/MβCD) | 0.18 ± 0.04 | 3 | |
| R*/no rafts (α-toxin/filipin III) | 0.17 ± 0.01 | 3 |
Mean ± S.E.
Estimated
Theoretical Analysis of Diffusion of Membrane Proteins—Diffusion of membrane components can be predicted by the Saffman-Delbrück equation assuming homogeneous viscosity of the membrane,
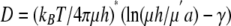 |
(Eq. 5) |
where kB is the Boltzmann constant (1.3807 × 10-23 J/K), T is the absolute temperature (293 K), μ denotes the viscosity of the fluid representing the membrane (∼7 poise), μ′ denotes the viscosity of the surrounding aqueous phase (∼0.02 poise), h denotes the thickness of a disk membrane spanned (8 nm for rhodopsin, 4 nm for lipid tails of transducin), γ is Euler's constant (0.5772), and a denotes the transmembrane radius of a molecule. The radius of one lipid tail is taken to be 0.25 nm, and the radius of the seven-transmembrane domain of rhodopsin is 1.6 nm. Values of 0.35 and 2.0 nm, respectively, were used for two lipid tails on the Gαβγ and rhodopsin dimer and 2.3 nm for the R* dimer bound to Gαβγ.
Photoexcitation of Rhodopsin—The intense laser illumination used for fluorescence excitation necessarily bleached a large fraction of rhodopsin in each cell imaged. At a typical setting of 1.5% of maximal laser power, i.e. 0.225 milliwatt, a 0.148-s scan of a 29.6 × 29.6 μm or 8.76 × 10-6 cm2 field delivers a dose of 8.61 × 1015 photons s-1cm-2 × 0.148 s or 1.27 × 1015 photons cm-2. The probability of any one rhodopsin being excited is
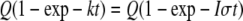 |
(Eq. 6) |
where Q is the quantum yield (0.65), k is Iσ (with I in units of photons/area/s, the absorbance cross-section, and σ in units of area), and t is the duration of the excitation. The absorbance cross-section, σ, of rhodopsin at 488 nm is 1.46 × 10-16 cm2 (calculated from the molar extinction coefficient, ε, according to σ = 3.82 × 10-21 ε cm2 m cm), so the probability of photoisomerization of each rhodopsin during a single image scan is 0.65(1-exp(-0.186)) = 0.11. Thus, a single image scan under these conditions excites sufficient rhodopsins to bind all transducins. In practice, several scans were always needed prior to the beginning of the FRAP experiments to locate the cell, find the correct focus position, and center it in the image field.
RESULTS
Characterization of Purified Gαt-EGFP—To examine the dynamic properties of transducin in intact photoreceptor cells, we produced a fusion of Gαt with EGFP for measurements by FRAP. Previous studies revealed an essentially nonperturbing site for insertion of EGFP within the helical domains of Gα subunits (14, 15). We made the analogous fusion with bovine Gαt (Fig. 1A), with EGFP inserted between Met115 and Pro116, in a loop between twoα-helices. For biochemical characterization, EGFP-Gαt was expressed using a baculovirus vector and purified from insect cells (Fig. 1B). The fusion protein bound photoreceptor Gβγ, bound to GTPγS, and exhibited nucleotide exchange kinetics (Fig. 1C) in the presence of photoexcited rhodopsin (metarhodopsin II, R*) similar to those of wild-type protein. On supported lipid bilayers it displayed very rapid redistribution kinetics (D = 42.8 ± 4.9 μm2/s), consistent with the diffusional properties of a soluble protein or of a peripheral protein to which the membrane provides very little viscous drag (Table 1).
Expression of Gαt-EGFP in Transgenic Rods—To examine G protein dynamics in intact photoreceptor cells, we produced transgenic X. laevis expressing the fusion protein under control of the Xenopus opsin promoter (5, 17), which directs expression to rod photoreceptor cells. The fusion protein is largely localized to the rod outer segments (Fig. 1, D–G) as is the endogenous Gαt, with additional signal visible in the inner segments, likely as a result of the well known light-induced migration of Gαt (18–21). The expression level varied from cell to cell, as observed for other transgenes with the same promoter (22). Immunoblotting was used to estimate the amount of transgene product relative to endogenous Gαt to ensure that results would not be skewed by concentrations much higher than physiological. Results obtained from densitometry studies showed that the amounts of transgene product were less than 8% (Fig. 1B) of those for the endogenous gene, indicating that even in those cells with the brightest fluorescence, EGFP-Gαt protein concentration was unlikely to exceed that of the endogenous Gαt. We observed no dependence of diffusion kinetics on total fluorescence intensity but report here the photobleaching recovery data only from cells with moderate to low intensity levels.
FRAP Measurements—Fluorescence recovery after photobleaching was used to provide an estimate of the diffusion coefficient D and immobile fraction f of rod G protein diffusion under various conditions (Tables 1 and 2). Diffusion of G proteins in rod outer segment membranes can occur either in two dimensions, along the plane of the disk membranes (lateral), or in one dimension, along the long axis of the outer segment across the stack of disks (longitudinal). Their different time scales allow these processes to be separated experimentally. The photobleaching geometries are illustrated schematically in Fig. 2, A and B. We recorded a time series of confocal fluorescence images of cells after introduction of photobleached stripes (Figs. 3A and 4A).
TABLE 2.
Longitudinal diffusion coefficients of EGFP, EGFP-Gαt and rhodopsin-EGFP
| Sample | Treatment | Da | n |
|---|---|---|---|
| μm2/s | |||
| ROS with EGFP-Gαt (f < 1%) | Gα-GTP (glucose only) | 0.016 ± 0.005 | 8 |
| Gα-GTP (α-toxin only) | 0.017 ± 0.005 | 3 | |
| Gα-GTPγS (α-toxin/ATP/GTPγS) | 0.019 ± 0.001 | 4 | |
| R*-Gαβγ (2-DG/sodium azide) | 0.007 ± 0.001 | 8 | |
| Gαβγ-GDP (hydroxylamine) | 0.039 ± 0.005 | 10 | |
| Gα-GTP/no rafts (α-toxin/MβCD) | 0.056 ± 0.005 | 5 | |
| Gα-GTP/+cholesterol (α-toxin/MβCD/cholesterol) | 0.013 ± 0.003 | 3 | |
| Gαβγ-GDP/no rafts (α-toxin/MβCD/hydroxylamine) | 0.037 ± 0.007 | 3 | |
| ROS with EGFP (f < 1%) | Free EGFP | 3.3 ± 0.6 | 6 |
Mean ± S.E.
FIGURE 2.

Lateral (A) or longitudinal (B) diffusion is monitored when a stripe is photobleached at different orientation at rod outer segments. The stripes are shown in gray. Top views of disk within the bleached areas are shown beside the schematically drawn rod outer segments within which incisures are illustrated. Open arrows indicate the directions of diffusion primarily monitored in each geometry.
FIGURE 4.

Example of a FRAP measurement of longitudinal diffusion. A, images of a photobleached transgenic rod outer segments at different time points as indicated. The scale bar represents 2.0 μm. B, fitting of corrected recovery data to Equation 1 (see “Experimental Procedures”). C, shows intensity integrated along the x axis of each pixel position along the y axis (inset: labeled 0–67) of the selected region of each image in panel A. D, intensity of bleached stripe (red; see inset), background (green), and non-bleached part (blue) of the rod outer segments at each time point of the FRAP measurement is shown. The black curve (crt) shows the time-dependent intensity of the bleached stripe after subtracting background and correcting for fading of fluorescence due to photobleaching during image acquisition.
The FRAP measurements using EGFP fusion proteins were first validated by using a rhodopsin-EGFP fusion in transgenic rods, because rhodopsin is the only photoreceptor protein with a documented diffusion coefficient. We used a protein described previously, with a C-terminal EGFP fusion followed by the eight C-terminal amino acids of rhodopsin (3), shown to have specific activity and rod outer segment localization indistinguishable from those of wild-type rhodopsin. The functional properties of this fusion protein have also been extensively characterized (3, 24), and it was found to be indistinguishable from wild-type rhodopsin in its light-dependent activation of transducin and phosphorylation by rhodopsin kinase.
We measured the apparent D value for the rhodopsin-EGFP in transgenic rods to be 0.14 ± 0.02 μm2/s and corrected it for the underestimation of the D in disk membranes caused by incisures (invaginations at the periphery of disks as shown in Fig. 2) as described previously (13) to obtain a value of D = 0.4 μm2/s, in good agreement with diffusion coefficients obtained using other methods (D = 0.2–0.6 μm2/s) (13, 25–27). As reported previously for rhodopsin-EGFP expressed in X. laevis (22), persistent stripes of higher and lower fluorescence intensity corresponding to times of higher and lower transgene expression during disk biogenesis were observed along the disk stacks, confirming that rhodopsin-EGFP cannot diffuse along the long axis of the outer segments because of its confinement on the disk into which it is initially inserted.
The immobile fractions calculated for lateral diffusion (i.e. parallel to the disk membrane surface) were very low (1% or less) for rhodopsin and for Gαt under all conditions tested. This finding indicates that R*, R*-associated Gαβγt, GTP-bound Gαt, and Gαβγt-GDP are able to move freely on a time scale of a few seconds. The very low immobile fraction for R* and its relatively fast diffusion constant (consistent with its geometry; see “Discussion” below) would seem to argue against a significant fraction of rhodopsin or R* being confined to large arrays containing hundreds of rhodopsins in quasi-crystalline aggregates, as suggested by atomic force microscopy of mouse disks (28).
Effects of Activation State of Gαt on Diffusion Kinetics—The values of the diffusion coefficients varied significantly depending on the activation states of Gαt (Table 1). In the presence of GTP, R* is known to dissociate from the Gαt subunit. In the absence of GTP, the G protein is stoichiometrically bound to photoexcited rhodopsin, metarhodopsin II or R*, which is in great excess over G protein in our experiments because of the bright excitation light and the 10-fold molar excess of rhodopsin over Gt in rod cells. GTP depletion by metabolic inhibitors slowed diffusion as a result of accumulation of R*-bound Gαβγt (D = 0.047 ± 0.005 μm2 s-1) relative to the values observed with ample GTP or GTPγS to form active Gαt (D = 0.19–0.21 μm2 s-1). It is not surprising that binding of a monomeric protein with a single lipid tail (Gαt) to a protein complex with 14 transmembrane helices (R* dimer; see “Discussion” below on expected effects of monomers versus dimers of rhodopsin), in addition to the Gβγ dimer with its farnesyl group, would dramatically slow its diffusion. What is very surprising is that addition of the two lipid tails of Gαβγt to an R* dimer would slow the diffusion coefficient for R* by almost a factor of 3.
To observe the diffusion of GDP-bound Gαβγt free of R*, we inactivated R* by using hydroxylamine to remove the agonist, all-trans retinal. Surprisingly, this treatment resulted in Gαt diffusion that was much faster (D = 0.37 ± 0.06 μm2 s-1) than that for Gαt in its active and presumed monomeric state (D = 0.19–0.21 μm2 s-1). These results suggest that both the active GTP form, Gαt, and the R*-bound Gαβγt complex are either involved in very large multimeric complexes or slowed down by a lipid microenvironment distinct from that experienced by Gαβγt, e.g. lipid microdomains or rafts.
To investigate the possibility that EGFP moiety attached to Gαt might give rise to artifactual interactions leading to complex formation and slowed diffusion, we examined the diffusion kinetics of EGFP itself in rod outer segments. EGFP diffuses so rapidly in rod cells that in the 50-ms minimum time for photobleaching, it completely redistributes itself laterally, so that no gradient of fluorescence can be observed. Longitudinal diffusion can be observed, with an apparent diffusion coefficient of 3.3 ± 0.6 μm2/s. When corrected for the tortuosity factor, 0.15 (see below), it yields a lateral diffusion coefficient value of D = 22 μm2/s, 2 orders of magnitude higher than that of the EGFP-Gαt fusion in rod cells, consistent with previous measurements of intracellular EGFP diffusion (29). Thus, it is very unlikely that the hindered diffusion of the EGFP-Gαt fusion is an artifact due to interactions between EGFP and other molecules in the outer segment, and the measurements revealed that the cytoplasmic viscosity experienced by a soluble protein is only about twice that of water.
Effects of Raft-disrupting Drugs—Transducin has been reported to co-purify with “detergent-resistant membranes” after photoactivation of rhodopsin (30, 31). Although these lipid aggregates cannot be reliably identified with lipid rafts within cellular membranes (see “Discussion”), the biochemical results suggest that light-dependent forms of transducin may have enhanced affinity for cholesterol-enriched membranes. To test for a possible role of cholesterol in restricting the diffusion of transducin, we used two standard lipid raft-disrupting reagents, mβCD and filipin III, each combined with α-toxin to ensure that these drugs could enter the cells and interact with disk membranes; similar results were observed without α-toxin. We detected much faster recovery of activated EGFP-Gαt-GTP in both treatments with oxygenated medium containing glucose (Table 1); Gαt diffuses laterally with D of 0.32 ± 0.02 μm2/s after treatment with mβCD and 0.29 ± 0.02 μm2/s with filipin III, similar to D of its diffusion in the hydroxylamine-treated Gαβγt state. In contrast, those treatments had no significant effect on R* and Gαβγ diffusion.
Comparison between Experimental Results and Theoretical Predictions—To interpret the differential effects of raft-disrupting drugs, we compared the observed D values for Gαt and rhodopsin (Table 1) with the theoretical values predicted by the Saffman-Delbrück equation (Equation 5) for proteins embedded in the membrane (32). We used for all complexes the value of membrane viscosity required to obtain the observed D value for EGFP-R*, assuming that rhodopsin is predominantly in dimeric form (33). We note that there is some controversy over whether rhodopsin is a monomer or dimer in ROS membranes (34–37). If we substitute our estimate of the parameter a for monomeric rhodopsin in place of the value used to generate Fig. 5A, in Equation 5 the predicted value of D increases by less than 10%. Diffusion of a membrane protein is dominated by the cross-sectional radius of the transmembrane segment because of the much higher viscosity of the membrane as compared with that of the cytoplasm. By using the transmembrane segment radii and lengths of farnesyl and fatty acid modifications on Gγ and Gα in different states as well as the radius of the R* dimers (note that R* is in ∼10 fold molar excess over Gαt), the diffusion coefficients of Gαβγt and the R* dimer were in good agreement with theoretical predictions with or without lipid microdomain disrupting treatment, whereas those of Gαt and R*2-Gαβγt were much lower than the predicted values. These discrepancies disappeared after lipid raft-disrupting treatments (Fig. 5A). To ensure that the effects of MβCD were due to cholesterol depletion, we measured FRAP under conditions in which MβCD was saturated with added cholesterol and thus was expected to increase the amount of cholesterol in the membranes. In every case, the effects of MβCD were blocked, and diffusion slowed even more than without MβCD treatment (Table 1). Taken together, those results suggest that inactive Gαβγt and both free activated rhodopsin (R*) and rhodopsin are largely excluded from cholesterol-dependent lipid microdomains, whereas Gαβγt-R* and activated Gα-GTP are sequestered into those microdomains or induce the formation of those domains around them.
FIGURE 5.

Deviation of observed lateral diffusion from predictions for membrane homogeneity and derived model of membrane heterogeneity. A, observed diffusion coefficients versus predictions for homogeneous lipid. The bar graph show the ratio of diffusion coefficients between our observation and the theoretical prediction based on homogenous lipid membrane calculated as described under “Experimental Procedures.” The treatments (additions to oxygenated Ringer's solution with 10 mm glucose, except as noted) for each sample were: Gαt, cholesterol added (α-toxin/MβCD/cholesterol); Gαt (no additions or α-toxin/ATP/GTPγS); Rho2-Gαβγt (2-DG/sodium azide, no glucose); Gαβγt (hydroxylamine); Rho2 (rhodospin-EGFP transgenics; no additions); Gαt, cholesterol depleted (α-toxin/MβCD); Rho2-Gαβγt, cholesterol depleted (α-toxin/MβCD/2-DG/sodium azide, no glucose); Gαβγt, cholesterol depleted (α-toxin/MβCD/hydroxylamine); and Rho2, cholesterol depleted (rhodospin-EGFP transgenics, α-toxin/MβCD). Unless otherwise indicated, all measurements used Gαt-EGFP transgenic tadpoles. B, proposed model of disk membrane heterogeneity and diffusion. Based on our findings, the following sequence of events is proposed. In the dark, and immediately after photoexcitation of rhodopsin, both rhodopsin and GDP-Gαβγt diffuse freely in a bulk lipid phase with relatively low effective viscosity. Upon binding of GDP-bound Gαβγt to light-activated rhodopsin (R*), the complex moves from bulk lipid into microdomains (or microdomains assemble around the complex), which may facilitate the conformational changes necessary for GDP-GTP exchange and dissociation of activated GTP-Gαt from R*. GTP-Gαt remains associated with microdomains while in the active state.
Longitudinal Diffusion—Redistribution of EGFP-Gαt along the long axis of the rod outer segment (Fig. 4) was much slower than lateral diffusion. As illustrated in Fig. 2B, diffusion in this direction requires transfer from one disk to another, and is subject to a “tortuosity” factor associated with travel from one side of a disk membrane to another. The ratios of longitudinal to lateral Gαt diffusion coefficients in the outer segment ranged from 0.08 in the GTP-bound active state to 0.11 in the inactive Gαβγ-GDP state. These ratios are somewhat smaller than the tortuosity factor, estimated to be ∼0.15 for cyclic GMP, a soluble molecule that does not require membrane dissociation and reassociation (38). This result suggests that diffusion along the axis of the rod is limited both by the geometry of disk stacks and by the requirement for membrane dissociation. The highest factor for Gαt, 0.18, was observed after cholesterol depletion, perhaps reflecting faster membrane dissociation in the absence of cholesterol.
For longitudinal diffusion, the fraction of activated EGFP-Gαt that appears as immobile is very low (<1%, Table 1) as expected for a peripheral membrane protein, and the apparent diffusion coefficient for the mobile fraction of 0.016–0.019 μm2/s indicates that the root-mean-square displacement along the long axis over the duration of a 4-s dim flash response is nearly 400 nm, a distance corresponding to about 15 disks. Thus inter-disk transfer does occur but is much slower than intra-disk redistribution. Our measurements of longitudinal diffusion make it possible to conclude that passive diffusion is kinetically sufficient to explain the dramatic redistribution of Gt and other phototransduction proteins upon exposure to strong illumination (18–21). These results are consistent with those of others implicating passive diffusion as the most likely mechanism for light-dependent redistribution (39–41).
DISCUSSION
Activated Gαt-GTP is not expected to be entirely in monomeric form, as it is known to associate with both PDE6 and the complex of RGS9-1, Gβ5L, and R9AP (42, 43). Based on a 10:1 molar ratio of rhodopsin to transducin (44) and a 270:1 molar ratio of rhodopsin to PDE6 (45) in frog rods, if all transducin is activated and two transducins bind to one PDE6, ∼7% of activated Gαt should be associated, possibly transiently, with PDE6, which has two additional lipid tails attached. Because of its relatively low concentration in rods, ∼1 RGS9-1/1000 rhodopsins (46), the RGS9-1·Gβ5L·R9AP complex can bind at most 1% of the total Gαt. It is unlikely that we could detect the presence of these small subpopulations by our fitting procedures, even if their diffusion constants were 2-fold higher than that of (presumably) monomeric Gαt-GTP. If their diffusion constants were lower by 1 order of magnitude, they would likely be reflected in our fitting procedures as apparent immobile fractions, f, of 1% or greater. In some fits f was found to be close to 1%, but in none was it found to be greater than 1%.
There have been several reports of detergent-resistant membranes extracted from rod outer segments in which these membranes are identified as lipid rafts (30, 31, 47–51). Unfortunately, these fractions isolated on sucrose gradients after detergent extraction cannot provide a reliable description of lipid microdomains present in intact cells at physiological temperatures prior to detergent addition or of their protein composition under physiological conditions. Isolation of such fractions requires the use of specific concentrations of specific detergents at low temperatures. Use of different detergents, different concentrations of them, or physiological temperatures results in drastically altered molecular compositions of the fractions. A study of the ability of Triton X-100 to induce lipid phase separations concluded, “Hence detergent-resistant membranes should not be assumed to resemble biological rafts in size, structure, composition, or even existence” (52), and similar conclusions were presented in a comprehensive review (53).
Indeed, the picture that emerges from comparing the results of detergent-resistant membrane fractionation of rod outer segment extracts from different groups with one another and with our results is not very clear; the protein and lipid composition appear to differ considerably according to which group prepared them and the conditions used. Thus, in contrast to the FRAP measurements, these detergent fractionation studies do not provide an accurate picture of the distribution of proteins among distinct lipid environments in intact living cells.
The results reported here provide unequivocal evidence that the lipid environment of the rod disk membrane is not homogeneous. A recent description of rod outer segment structure based on cryoelectron tomography (54) revealed structural heterogeneity in disk membranes, with high density patches representing about 70% of the disk volume estimated to contain more than 90% of total rhodopsin. This structural heterogeneity may have its functional correlate in the distinct lipid environments in which rhodopsin and Gt diffuse in different functional states. Our results suggest the following scheme (Fig. 5B): in the dark and immediately after photoexcitation of rhodopsin, both rhodopsin and GDP-Gαβγt diffuse freely in a bulk lipid phase with relatively low effective viscosity (it should be noted that the effective viscosity is a function both of the intrinsic viscosity contributed by the local lipid composition and the effects of membrane “crowding” due to high concentrations of rhodopsin (1, 55)). Upon binding of GDP-bound Gαβγt to light-activated rhodopsin (R*), the complex moves from bulk lipid into microdomains or, alternatively, triggers the assembly of microdomains around it, which may facilitate the conformational changes necessary for GDP-GTP exchange and dissociation of activated GTP-Gαt from R*. GTP-Gαt remains associated with microdomains while in the active state. It will be very interesting in future studies to determine whether these lipid microdomains also contain enriched concentrations of both PDE6 and the GTPase-accelerating protein complex of RGS9-1, Gβ5L, and R9AP, as suggested by the results from biochemical fractionation (30, 48). Such co-sequestration could represent a functional advantage for concentration of activated G protein into an environment with restricted diffusion, whereas R* remains free to search a more mobile environment for inactive G protein heterotrimers. There are many previous reports of membrane proteins, both peripheral and integral, altering the phase behavior of lipids (see for example Refs. 56–59), including induction of lateral phase separation. We have previously reported that GDP-bound Gαβγt, but not GTPγS-bound Gαt, can induce the conversion of artificial lipid bilayers to a tubular structure (16, 23). It remains to be determined what influence the activation state of Gαt may have, if any, on the organization and stability of lipid microdomains in disks.
Acknowledgments
We thank Dr. Thomas Sakmar of Rockefeller University for the gift of recombinant baculovirus.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-EY07981 and R01-EY12505 from NEI. This work was also supported by Welch Foundation Grant Q0035. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: R*, photoexcited rhodopsin (metarhodopsin II); FRAP, fluorescence recovery after photobleaching; MβCD, methyl-β-cyclodextrin; Rho2, rhodopsin dimers; ROS, rod outer segments; 2-DG, 2-deoxyglucose; DTT, dithiothreitol; MOPS, 4-morpholinepropanesulfonic acid; EGFP, enhanced green fluorescent protein; PBS, phosphate-buffered saline; GTPγS, guanosine 5′-3-O-(γthio)triphosphate.
References
- 1.Calvert, P. D., Govardovskii, V. I., Krasnoperova, N., Anderson, R. E., Lem, J., and Makino, C. L. (2001) Nature 411 90-94 [DOI] [PubMed] [Google Scholar]
- 2.Chini, B., and Parenti, M. (2004) J. Mol. Endocrinol. 32 325-338 [DOI] [PubMed] [Google Scholar]
- 3.Jin, S., McKee, T. D., and Oprian, D. D. (2003) FEBS Lett. 542 142-146 [DOI] [PubMed] [Google Scholar]
- 4.Nieuwkoop, P. D., and Faber, J. (1967) Normal Table of Xenopus laevis (Daudin), 2d Ed., Elsevier/North Holland, Amsterdam
- 5.Mani, S. S., Batni, S., Whitaker, L., Chen, S., Engbretson, G., and Knox, B. E. (2001) J. Biol. Chem. 276 36557-36565 [DOI] [PubMed] [Google Scholar]
- 6.Yamanaka, G., Eckstein, F., and Stryer, L. (1985) Biochemistry 24 8094-8101 [DOI] [PubMed] [Google Scholar]
- 7.Wensel, T. G., He, F., and Malinski, J. A. (2005) Methods Mol. Biol. 307 289-313 [DOI] [PubMed] [Google Scholar]
- 8.He, W., Lu, L., Zhang, X., El-Hodiri, H. M., Chen, C. K., Slep, K. C., Simon, M. I., Jamrich, M., and Wensel, T. G. (2000) J. Biol. Chem. 275 37093-37100 [DOI] [PubMed] [Google Scholar]
- 9.Harlow, E., and Lane, D. (1988) Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY
- 10.Adamus, G., Zam, Z. S., Arendt, A., Palczewski, K., McDowell, J. H., and Hargrave, P. A. (1991) Vision Res. 31 17-31 [DOI] [PubMed] [Google Scholar]
- 11.Niu, S. L., Mitchell, D. C., and Litman, B. J. (2002) J. Biol. Chem. 277 20139-20145 [DOI] [PubMed] [Google Scholar]
- 12.Crank, J. (1975) The Mathematics of Difffusion, 2d Ed., Oxford University Press, Oxford, UK
- 13.Wey, C. L., Cone, R. A., and Edidin, M. A. (1981) Biophys. J. 33 225-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes, T. E., Zhang, H., Logothetis, D. E., and Berlot, C. H. (2001) J. Biol. Chem. 276 4227-4235 [DOI] [PubMed] [Google Scholar]
- 15.Janetopoulos, C., Jin, T., and Devreotes, P. (2001) Science 291 2408-2411 [DOI] [PubMed] [Google Scholar]
- 16.Melia, T. J., Sowa, M. E., Schutze, L., and Wensel, T. G. (1999) J. Struct. Biol. 128 119-130 [DOI] [PubMed] [Google Scholar]
- 17.Perkins, B. D., Kainz, P. M., O'Malley, D. M., and Dowling, J. E. (2002) Vis. Neurosci. 19 257-264 [DOI] [PubMed] [Google Scholar]
- 18.Brann, M. R., and Cohen, L. V. (1987) Science 235 585-587 [DOI] [PubMed] [Google Scholar]
- 19.Whelan, J. P., and McGinnis, J. F. (1988) J. Neurosci. Res. 20 263-270 [DOI] [PubMed] [Google Scholar]
- 20.Elias, R. V., Sezate, S. S., Cao, W., and McGinnis, J. F. (2004) Mol. Vis. 10 672-681 [PubMed] [Google Scholar]
- 21.Sokolov, M., Lyubarsky, A. L., Strissel, K. J., Savchenko, A. B., Govardovskii, V. I., Pugh, E. N., Jr., and Arshavsky, V. Y. (2002) Neuron 34 95-106 [DOI] [PubMed] [Google Scholar]
- 22.Moritz, O. L., Tam, B. M., Papermaster, D. S., and Nakayama, T. (2001) J. Biol. Chem. 276 28242-28251 [DOI] [PubMed] [Google Scholar]
- 23.Zhang, Z., Melia, T. J., He, F., Yuan, C., McGough, A., Schmid, M. F., and Wensel, T. G. (2004) J. Biol. Chem. 279 33937-33945 [DOI] [PubMed] [Google Scholar]
- 24.Jin, S., Cornwall, M. C., and Oprian, D. D. (2003) Nat. Neurosci. 6 731-735 [DOI] [PubMed] [Google Scholar]
- 25.Gupta, B. D., and Williams, T. P. (1990) J. Physiol. 430 483-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebman, P. A., and Entine, G. (1974) Science 185 457-459 [DOI] [PubMed] [Google Scholar]
- 27.Poo, M., and Cone, R. A. (1974) Nature 247 438-441 [DOI] [PubMed] [Google Scholar]
- 28.Fotiadis, D., Liang, Y., Filipek, S., Saperstein, D. A., Engel, A., and Palczewski, K. (2003) Nature 421 127-128 [DOI] [PubMed] [Google Scholar]
- 29.Swaminathan, R., Hoang, C. P., and Verkman, A. S. (1997) Biophys. J. 72 1900-1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair, K. S., Balasubramanian, N., and Slepak, V. Z. (2002) Curr. Biol. 12 421-425 [DOI] [PubMed] [Google Scholar]
- 31.Seno, K., Kishimoto, M., Abe, M., Higuchi, Y., Mieda, M., Owada, Y., Yoshiyama, W., Liu, H., and Hayashi, F. (2001) J. Biol. Chem. 276 20813-20816 [DOI] [PubMed] [Google Scholar]
- 32.Saffman, P. G., and Delbruck, M. (1975) Proc. Natl. Acad. Sci. U. S. A. 72 3111-3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fotiadis, D., Liang, Y., Filipek, S., Saperstein, D. A., Engel, A., and Palczewski, K. (2004) FEBS Lett. 564 281-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chabre, M., and le Maire, M. (2005) Biochemistry 44 9395-9403 [DOI] [PubMed] [Google Scholar]
- 35.Chabre, M., Cone, R., and Saibil, H. (2003) Nature 426 30-31 [DOI] [PubMed] [Google Scholar]
- 36.Fotiadis, D., Jastrzebska, B., Philippsen, A., Muller, D. J., Palczewski, K., and Engel, A. (2006) Curr. Opin. Struct. Biol. 16 252-259 [DOI] [PubMed] [Google Scholar]
- 37.Edrington, T. C., Bennett, M. P., and Albert, A. D. (2008) Biophys. J. 95 2859-2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koutalos, Y., Nakatani, K., and Yau, K. W. (1995) Biophys. J. 68 373-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenzweig, D. H., Nair, K. S., Wei, J., Wang, Q., Garwin, G., Saari, J. C., Chen, C. K., Smrcka, A. V., Swaroop, A., Lem, J., Hurley, J. B., and Slepak, V. Z. (2007) J. Neurosci. 27 5484-5494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nair, K. S., Hanson, S. M., Mendez, A., Gurevich, E. V., Kennedy, M. J., Shestopalov, V. I., Vishnivetskiy, S. A., Chen, J., Hurley, J. B., Gurevich, V. V., and Slepak, V. Z. (2005) Neuron 46 555-567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lobanova, E. S., Finkelstein, S., Song, H., Tsang, S. H., Chen, C. K., Sokolov, M., Skiba, N. P., and Arshavsky, V. Y. (2007) J. Neurosci. 27 1151-1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malinski, J. A., Zera, E. M., Angleson, J. K., and Wensel, T. G. (1996) J. Biol. Chem. 271 12919-12924 [DOI] [PubMed] [Google Scholar]
- 43.Hu, G., and Wensel, T. G. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 9755-9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamm, H. E., and Bownds, D. M. (1986) Biochemistry 25 4512-4523 [DOI] [PubMed] [Google Scholar]
- 45.Dumke, C. L., Arshavsky, V. Y., Calvert, P. D., Bownds, M. D., and Pugh, E. N., Jr. (1994) J. Gen. Physiol. 103 1071-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowan, C. W., Fariss, R. N., Sokal, I., Palczewski, K., and Wensel, T. G. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 5351-5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senin, I. I., Hoppner-Heitmann, D., Polkovnikova, O. O., Churumova, V. A., Tikhomirova, N. K., Philippov, P. P., and Koch, K. W. (2004) J. Biol. Chem. 279 48647-48653 [DOI] [PubMed] [Google Scholar]
- 48.Liu, H., Seno, K., and Hayashi, F. (2003) Biochem. Biophys. Res. Commun. 303 19-23 [DOI] [PubMed] [Google Scholar]
- 49.Boesze-Battaglia, K., Dispoto, J., and Kahoe, M. A. (2002) J. Biol. Chem. 277 41843-41849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajala, R. V., Elliott, M. H., McClellan, M. E., and Anderson, R. E. (2006) Adv. Exp. Med. Biol. 572 491-497 [DOI] [PubMed] [Google Scholar]
- 51.Martin, R. E., Elliott, M. H., Brush, R. S., and Anderson, R. E. (2005) Investig. Ophthalmol. Vis. Sci. 46 1147-1154 [DOI] [PubMed] [Google Scholar]
- 52.Heerklotz, H. (2002) Biophys. J. 83 2693-2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munro, S. (2003) Cell 115 377-388 [DOI] [PubMed] [Google Scholar]
- 54.Nickell, S., Park, P. S., Baumeister, W., and Palczewski, K. (2007) J. Cell Biol. 177 917-925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peters, R., and Cherry, R. J. (1982) Proc. Natl. Acad. Sci. U. S. A. 79 4317-4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poveda, J. A., Encinar, J. A., Fernandez, A. M., Mateo, C. R., Ferragut, J. A., and Gonzalez-Ros, J. M. (2002) Biochemistry 41 12253-12262 [DOI] [PubMed] [Google Scholar]
- 57.Mbamala, E. C., Ben-Shaul, A., and May, S. (2005) Biophys. J. 88 1702-1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Birrell, G. B., and Griffith, O. H. (1976) Biochemistry 15 2925-2929 [DOI] [PubMed] [Google Scholar]
- 59.Rietveld, A., van Kemenade, T. J., Hak, T., Verkleij, A. J., and de Kruijff, B. (1987) Eur. J. Biochem. 164 137-140 [DOI] [PubMed] [Google Scholar]