

Abstract
Influenza virus neuraminidase (NA) plays a crucial role in facilitating the spread of newly synthesized virus in the host and is an important target for controlling disease progression. The NA crystal structure from the 1918 “Spanish flu” (A/Brevig Mission/1/18 H1N1) and that of its complex with zanamivir (Relenza) at 1.65-Å and 1.45-Å resolutions, respectively, corroborated the successful expression of correctly folded NA tetramers in a baculovirus expression system. An additional cavity adjacent to the substrate-binding site is observed in N1, compared to N2 and N9 NAs, including H5N1. This cavity arises from an open conformation of the 150 loop (Gly147 to Asp151) and appears to be conserved among group 1 NAs (N1, N4, N5, and N8). It closes upon zanamivir binding. Three calcium sites were identified, including a novel site that may be conserved in N1 and N4. Thus, these high-resolution structures, combined with our recombinant expression system, provide new opportunities to augment the limited arsenal of therapeutics against influenza.
Influenza virus is highly contagious and can cause severe respiratory illness and death. Of the three types of influenza virus, type A infects a wide range of avian and mammalian species and can be further classified into subtypes according to the serological reactivity of its surface glycoprotein antigens, hemagglutinin (HA) and neuraminidase (NA). Sixteen serotypes of HA (H1 to H16) and 9 of NA (N1 to N9) circulate in avian and mammalian hosts. Of nine avian NA subtypes, only N1 and N2 have been seen in human viruses responsible for pandemics and recurrent annual epidemics. Type A viruses account for all of the human pandemics of the last century: the 1918 H1N1 “Spanish,” the 1957 H2N2 “Asian,” and the 1968 H3N2 “Hong Kong” influenza viruses.
The 1918 influenza pandemic remains the most devastating single pandemic of any infectious disease in recorded history. The virus pandemic spread globally, infecting 25 to 30% of the world's population and killing at least 20 to 50 million worldwide, including more than half a million people in the United States (13, 19). At the time of the pandemic, the causative agent was not known, and no virus survived intact. However, sequences of all coding regions of the 1918 influenza virus were recently derived from archived autopsy material and from an Alaskan victim buried in permafrost (3, 35-38, 44). Such information has provided the foundation for the resurrection of the 1918 virus and has enabled investigations of its sequence (3, 35-38, 43) and functional characteristics (22, 47-49), as well as structural elucidation of its major HA coat protein (39, 42).
A tetrameric protein, influenza virus NA is a glycohydrolase that catalyzes the cleavage of terminal α-ketosidically linked sialic acids from a large variety of glycoproteins, glycolipids, and oligosaccharides (16, 21). NA is important during the final stages of influenza virus infection, where it removes sialic acid from infected cell surfaces and newly formed virions, thus facilitating progeny virus release and spread of the infection to neighboring cells (5, 34). NA can also promote penetration of the virus through the ciliated epithelium of the human airway by removing the decoy receptors on mucins, cilia, and the cellular glycocalyx (29). One study showed that the 1918 influenza virus, reconstructed by reverse genetics (15), had exceptionally high virulence in mouse models and chicken embryos (47). These data suggested that NA contributes to the enhanced infectivity of the 1918 influenza virus, although the sequence of its gene does not reveal any obvious features to explain these findings.
Inhibition of NA function appears critical in limiting the progression of influenza virus infection in the host, and crystallographic analyses of NAs have provided a platform for structure-based drug design. These structures contributed to the successful development of two potent and selective inhibitors, zanamivir (Relenza) (51) and oseltamivir (Tamiflu) (20). Both drugs were designed to mimic transition state analogues based on crystal structures of N2 and N9 NAs. Notwithstanding, both compounds proved to be highly potent inhibitors of NAs from other strains, including N1, as well as for influenza B viruses. This anticipated success was based on the premise that the active-site residues are highly conserved across all subtypes of type A as well as type B influenza virus (51). However, recent studies have now shown that drug-resistant mutants can be subtype specific, which suggests some variation in the active site and its geometry and in subsequent inhibitor binding modes among the different subtypes (8, 17).
Crystallographic analysis of macromolecules requires relatively large quantities of highly pure protein. Hence, recent structural studies of viral proteins from highly pathogenic influenza virus strains have been greatly facilitated by recombinant protein expression technology. Previously, soluble HA proteins from both the 1918 H1N1 influenza virus (A/South Carolina/1/18) and the 2004 H5N1 (A/Vietnam/1203/04) strains were successfully expressed using a baculovirus expression system (BES) and crystallized for structural analysis (41, 42). In contrast, all current NA crystal structures have so far come from protein isolated from live virus. In this study, we report, for the first time, that recombinant NA from the 1918 influenza virus (1918 NA) can be prepared for structural analysis, thus bypassing the stringent biosafety level 3 requirement for protein isolation directly from live virus. The ectodomain of the NA gene, corresponding to amino acids 82 to 468 (according to N2 numbering) from the 1918 influenza virus A/Brevig Mission/1/1918 (18NA), was cloned and expressed using the BES. The crystal structure of 18NA was determined to 1.65-Å resolution and its complex with the antiviral zanamivir (18NA/Z) to 1.45-Å resolution.
MATERIALS AND METHODS
Cloning.
Based on N2 numbering, cDNA corresponding to residues 83 to 468 of the ectodomain of the NA from A/Brevig Mission/1/1918 (18NA) was cloned into the baculovirus transfer vector pAcGP67A (BD Biosciences) to allow for efficient secretion of recombinant protein. In order to promote tetramerization among monomeric 18NA, a 15-residue repeat tetramerization domain from human vasodilator-stimulated phosphoprotein was appended (24). A construct containing an N-terminal “tetramerization domain” that incorporated a thrombin site between the tetramerization domain and the N terminus of the ectodomain of 18NA was produced. A hexahistidine tag was also introduced at the N terminus of the construct to enable purification by Ni2+ affinity chromatography. The recombinant 18NA protein secreted into the cell culture medium contains three additional plasmid-encoded residues at the N terminus (ADPHHHHHHSSSDYSDLQRVKQELLEEVKKELQKVKEEIIEAFVQELRKRGSLVPRGSPSRS, where the sequence in boldface is the His tag, the tetramerization domain is underlined, and the thrombin cleavage site is in italics).
Expression and purification.
Suspension cultures of insect Sf9 and Hi5 cells were cultured in HyQ SFX-Insect medium (HyClone). The constructed plasmid was cotransfected into Sf9 cells with BaculoGold DNA (BD Biosciences) using Cellfectin reagent (Invitrogen) according to the manufacturer's instructions. After three rounds of viral amplification in Sf9 cells at a multiplicity of infection of 1, the protein was expressed for 3 days in CellSTACK (Corning) culture chambers at 28°C using Hi5 cells (1.5 liters of 7 × 108 Hi5 cells) with a multiplicity of infection of 5. Typical expression preparation was performed on a 9-liter scale. Hi5 cells were removed by centrifugation (6,000 × g; 20 min), and soluble 18NA was recovered from the cell supernatant by metal affinity chromatography using Ni-nitrilotriacetic acid (NTA) resin (Qiagen). The Ni-NTA resin with 18NA absorbed was washed and then exchanged into thrombin digestion buffer (20 mM Tris, pH 8.0, 150 mM NaCl, and 2.5 mM CaCl2). Thrombin digestion was carried out at 4°C overnight using 3 U per mg of protein. The supernatant from the Ni-NTA resin mixture, incubated at 4°C overnight, was collected and concentrated. The cleaved 18NA was further purified from thrombin and any aggregated material by size exclusion chromatography on a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) in 20 mM Tris, pH 8.0, and 150 mM NaCl.
Crystallization and data collection.
Crystallization experiments were set up using the sitting drop vapor diffusion method. Initial crystallization conditions for the native 18NA protein were determined with the EasyXtal μplate G-RB JCSG+ suite (Qiagen). Following optimization, diffraction quality crystals for 18NA were obtained by mixing 0.5 μl of the concentrated protein at 10 mg/ml in 10 mM phosphate, pH 7.30, and 75 mM NaCl with 0.5 μl of the well solution (0.16 M calcium acetate, 0.08 M cacodylate, pH 6.50, 14.4% polyethylene glycol 8000, 20% glycerol) at 22°C. 18NA crystals were harvested directly from the drop. For cocrystallization, the protein sample was prepared in 10 mM Tris, pH 7.30, 100 mM NaCl, and 10 mM CaCl2, to which zanamivir was added in a 10-fold molar excess. Crystals of the 18NA/Z complex were first observed using the Topaz (Fluidigm) nanoscale free interface diffusion system. After optimization, diffraction quality crystals were grown using the sitting drop vapor diffusion method by mixing 0.5 μl of the protein sample with an equal volume of precipitant (20% polyethylene glycol 3000) at 295 K. 18NA/Z crystals were cryoprotected in mother liquor with addition of 20% glycerol before being flash-cooled at 100 K. Diffraction data for the native and the complex crystals were collected at beamlines 9-2 and 11-1, respectively, at the Stanford Synchrotron Radiation Laboratory (SSRL). Data for all crystals were integrated and scaled with Denzo and Scalepack (33). Data collection statistics are outlined in Table 1.
TABLE 1.
Data collection and refinement statistics of the crystal structures of A/Brevig Mission/1/1918 N1 NA and its complex with zanamivir
| Parameter | Resulta for:
|
|
|---|---|---|
| 1918 NA | 1918 NA + zanamivir | |
| Data collection | ||
| Space group | C2221 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 117.73, 138.47, 117.86 | 118.04, 129.25, 118.85 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution range (Å) | 50.00-1.65 (1.68-1.65) | 50.00-1.45 (1.49-1.45) |
| Unique reflections | 113,144 (4,405) | 155,978 (11,401) |
| Completeness (%) | 97.3 (76.4) | 97.6 (86.3) |
| Redundancy | 4.2 (2.8) | 6.8 (5.6) |
| Wilson B value (Å2) | 16.3 | 12.0 |
| Rsymb | 0.09 (0.53) | 0.07 (0.46) |
| <I/σ> | 22.2 (2.0) | 23.9 (2.7) |
| Refinement | ||
| Rcrystc | 0.18 | 0.14 |
| Rfreed | 0.21 | 0.16 |
| Molecules in asymmetric unit | 2 monomers | 2 monomers |
| RMSD from ideal | ||
| Bond length (Å) | 0.013 | 0.013 |
| Bond angle (°) | 1.44 | 1.44 |
| Avg B values (Å2) | ||
| Protein | 16.6 | 10.9 |
| Inhibitor | 8.6/9.2e | |
| Water | 32.0 | 28.3 |
| Ramachandran plot (%) | ||
| Most favored | 84.3 | 84.6 |
| Additionally allowed | 15.1 | 14.8 |
| Generously allowed | 0.6 | 0.6 |
| Disallowed | 0.0 | 0.0 |
Values in parentheses are for the highest-resolution shell.
Rsym = 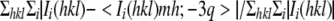 .
.
Rcryst = 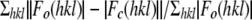 .
.
Rfree is calculated as for Rcryst, but from 5% of the data that were excluded from the refinement.
Values are for the two ligands per asymmetric unit.
Molecular replacement, model building, and refinement.
Both structures were determined by molecular replacement using the program Phaser (31). The unliganded 1918 NA structure was determined using the recent H5N1 NA structure (Protein Data Bank [PDB] code: 2HTY) as a model. The complex structure with zanamivir was subsequently determined using the refined 18NA as an input model. The Matthews coefficients (Vm) (30) for NA (Vm = 2.6 Å3/Dalton) and NA in complex with zanamivir (Vm = 2.5 Å3/Dalton) suggested two copies of monomeric NA per asymmetric unit. The diffraction pattern of alternating sharp and diffuse reflections and a large portion of extraneous electron density overlapping with the dimer model after several rounds of refinement led to the identification of an intrinsic problem associated within the NA crystal that represents a so-called lattice translocation defect (53). The intensity was corrected according to a previously established method (53) with a translocation vector (0, 1/2, 1/2) or (1/2, 0, 1/2) and a lattice defect fraction of 0.226 (54). However, this problem was not observed in the NA complex crystals. The models were initially refined by rigid body refinement in REFMAC5 (32), using all data from 50- to 3-Å resolution. Model building was carried out with the program Coot (14). The last cycles of REFMAC-restrained refinement included data for the highest-resolution shell using isotropic B values. Final statistics for both structures are represented in Table 1. The quality of the structures was analyzed using the JCSG validation suite (www.jcsg.org) including MolProbity (27), WHAT IF (52), Resolve (45), and PROCHECK (25). All figures were generated with PyMol (www.pymol.org).
PDB accession codes.
The atomic coordinates and structure factors of 18NA are available from the RCSB PDB under accession codes 3BEQ for the unliganded 1918 NA and 3B7E for the 1918 NA with zanamivir.
RESULTS
Overall structure.
The 18NA was expressed and purified from the BES and crystallized in space group C2221, with two NA subunits per asymmetric unit for both the native and complex forms. Detailed statistics for the data collection at the SSRL and subsequent processing and model refinement are compiled in Table 1. The native structure was solved by molecular replacement using the H5N1 NA coordinates as the search model (PDB code: 2HTY). Data were refined to crystallographic Rcryst/Rfree values of 0.18/0.21 and 0.14/0.16 for the native and the complex structures, respectively. An atomic model of the inhibitor zanamivir was built into the Fobs − Fcalc difference Fourier map. The refined structures contain the complete globular head region corresponding to residues 82 to 468 from the A/Brevig Mission/1/1918 N1 protein.
The crystal structure of 18NA reveals the standard tetrameric association of identical monomers, which contain a propeller-like arrangement of six four-stranded, antiparallel β-sheets, as described previously for influenza virus A subtypes N1, N2, N4, N8, and N9 (2, 39, 50), as well as for influenza virus B NA (7) (Fig. 1). Comparison with the only other available N1 structure from the H5N1 virus, which shares a sequence identity of 92% in the structurally characterized ectodomains, gave an average root mean square deviation (RMSD) of 0.37 Å for all Cα atoms in both tetrameric NAs. This structural similarity confirms correct folding of the H1N1 NA when recombinantly expressed in the BES. The tetrameric structure has overall dimensions of 94 by 94 by 54 Å3 and has been described as “box shaped” (50). On the membrane-proximal side of the tetramer, a large cavity is formed by all four monomers. The active site is located on the membrane-distal surface, close to the pseudosixfold axis through the center of each monomer, which is tilted approximately 25° away from the fourfold axis of the tetramer, as observed in all known NA structures (7). This orientation of the catalytic site on the NA tetramer, together with a stalk region of approximately 30 to 50 amino acids from the globular head to the membrane, could confer flexibility in reaching potential substrates (4, 38).
FIG. 1.
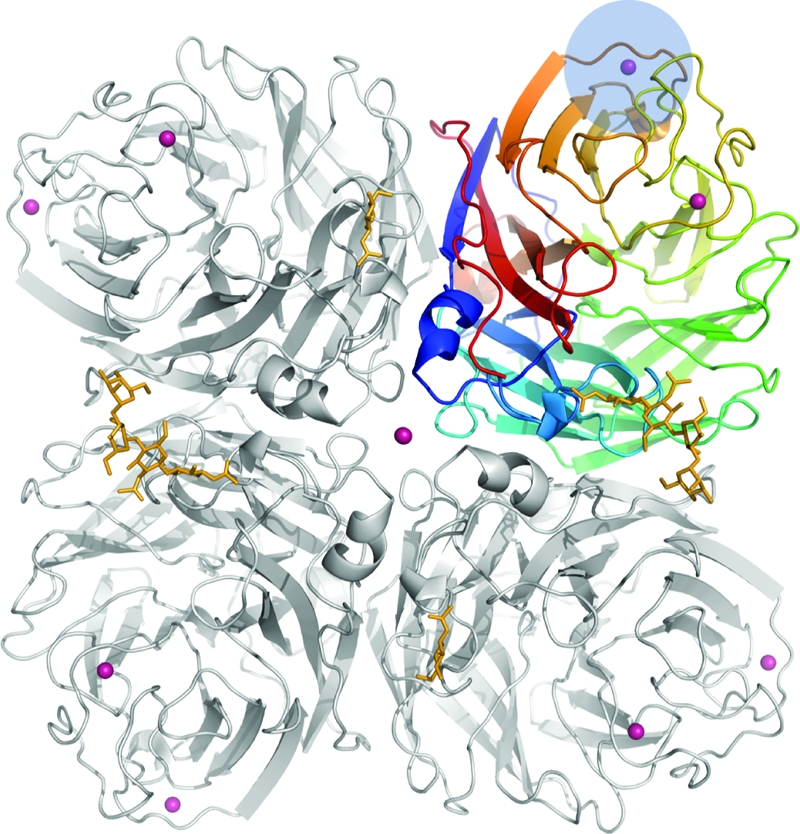
Crystal structure of the 1918 N1 NA tetramer in schematic representation, as viewed from above the viral surface. The tetramer is composed of four identical monomers. One monomer is colored using a rainbow gradient to illustrate the canonical β-propeller arrangement with six four-stranded, antiparallel β-sheets. The active site is located on top of the molecule (membrane distal), close to the local, pseudosixfold symmetry axis. Calcium ions are shown in magenta and glycans in gold. The new calcium ion binding site is highlighted in blue.
The overall structures of NAs are very well conserved among different subtypes of influenza A viruses, as are those from influenza B viruses, despite sequence identities as low as 30% (7). A previous study of NA sequences showed that two phylogenetically distinct groups are found among the nine NA subtypes: N1, N4, N5, and N8 belong to group 1, and N2, N3, N6, N7, and N9 are members of group 2 (39). The major difference between group 1 and group 2 seems to involve the loop region surrounding the active site, including residues 147 to 151 (150 loop), as observed here. Superposition of Cα atoms of the 150 loop between 18NA and N2 NA reveals an RMSD of more than 4 Å. Another feature of group 2 is represented by two highly conserved cysteine residues at positions 175 and 193, which form a disulfide bridge that is absent among group 1 members. Group 1 NAs contain a conserved cysteine at position 161, which was thought to form an interchain disulfide bond between adjacent monomers due to its proximity to the molecular fourfold axis in the N2 structure (50). However, the 18NA structure shows that it exists as a free cysteine and points away from the molecular fourfold axis, with a distance of 16 Å between Cα atoms of these two neighboring Cys residues. Hence, the group 1 subtypes contain one less intrachain disulfide bridge than those in group 2. Another distinct feature of the group 2 subtypes is the N glycosylation site at Asn200. In N2 and N9 NAs, the glycan at Asn200 interacts with an adjacent monomer and could potentially contribute to stabilization of the group 2 NA tetramer. 18NA, as a member of group 1, lacks this glycan, which, therefore, may affect its oligomeric stabilization and may have accounted for some of the difficulty in the past few years of obtaining stable N1 tetramers.
Comparison of the 18NA monomer with other group 1 NA structures reveals a striking similarity. Superimposition of 18NA with N4 (2HTV) and N8 (2HT5) NAs produces RMSDs of 0.57 Å and 0.95 Å (for Cα atoms), respectively. In contrast, superimposition with N2 (PDB code: 1NN2) and N9 (PDB code: 7NN9) from group 2 gives RMSDs of 1.56 Å and 1.55 Å (for Cα), respectively. The NA from influenza virus type B, however, seems to more closely resemble group 1 than group 2, with an RMSD of 0.77 Å to group 1. Compared with group 1 subtypes N4 and N8, N1 has main structural differences located in the loop regions, 328 to 347, 354 to 360, 380 to 392, and 450 to 455 (Fig. 2). The 330 loop, restrained by a disulfide bridge via Cys336, has been previously identified as one of the antigenic sites and contributes to the binding of two antibodies, NC41 and NC10, as shown by analysis using escape mutants and crystal structures (1, 28, 46). NA sequences from both group 1 and group 2 show insertions and deletions in this region. The 380 loop coincides with the putative calcium binding site (discussed below) and adopts very similar conformations in N1 and N4, but not in N8. The 450 loop is located at the bottom of the molecule (membrane proximal) and precedes the C-terminal segment, which runs to the top of the molecule (membrane distal) at the subunit interface. Such variations at the monomer-monomer interface could potentially directly affect the stabilization of the tetramer.
FIG. 2.

Stereo image of superposed monomeric NAs from N1, N4, and N8 in ribbon presentation: N1 in gray, N4 in orange, and N8 in teal. The major variations are located in loop regions: loop A (residues 328 to 347), loop B (354 to 360), loop C (380 to 392), and loop D (450 to 455).
Catalytic site and inhibitor binding.
Residues within the active site are highly conserved among all of the NA subtypes, including eight charged and polar residues (Arg118, Asp151, Arg152, Arg224, Glu276, Arg292, Arg371, and Tyr406) that have direct interaction with the substrate at the catalytic site. The geometry of the catalytic site is structurally stabilized through a network of hydrogen bonds and salt bridges by a constellation of largely conserved framework residues (Glu119, Arg156, Trp178, Ser179, Asp/Asn198, Ile222, Glu227, His274, Glu277, Asn294, and Glu425) (7). In the native 18NA structure, the 150 loop region that borders the active site, including Gly147, Thr148, Val149, Lys150, and Asp151, is noticeably disordered, as reflected in significantly higher average B values (40 Å2) for residues in this loop, compared to that for NA as a whole (16.6 Å2) (Table 1). The 150 loop is displaced from the well-characterized catalytic site in N2 and N9 subtypes, with an RMSD of >4.0 Å, which gives rise to an extra cavity of ∼190 Å3 adjacent to the catalytic site in the N1 structure (Fig. 3). The 150 loop makes a few contacts with the nearby residues in the active site of native 18NA, including hydrogen bonds between Gly147 and Ile437 and between Lys150 and Gln136. The solvent-exposed 150 cavity is defined by about 20 residues (Fig. 4), and only two polar side chains are present in the cavity, Gln136 and Arg156, both of which are highly conserved. Most other residues are hydrophobic and point away from the pocket.
FIG. 3.

Molecular surfaces of the 18NA active site with and without the binding of zanamivir. (A) Native structure showing the large 150 cavity that is unoccupied close to the zanamivir binding site. (B) Inhibitor-bound structure, in which structural changes bring the 150 loop proximal to the zanamivir binding site and, therefore, close the 150 cavity.
FIG. 4.

The 150 cavity of the native 18NA structure. (A) Electrostatic potential of the zanamivir binding site and the 150 cavity. Negatively charged regions are red, positively charged regions are blue, and neutral regions are whitish. (B) Residues bordering the pocket are shown with their side chains. For both images, zanamivir from the complex structure was superimposed onto the native 18NA structure in order to illustrate the extra binding pocket before the conformational changes are induced by inhibitor binding. Both images are viewed in the same orientation with the 150 cavity in the center.
However, upon binding zanamivir, the 150 loop moves closer to the drug-bound pocket, such that it reorganizes to enable more-extensive interactions with the ligand, as well as with other active-site residues in the vicinity (Fig. 5). As a consequence of the conformational changes in the 150 loop, two residues, Asp151 and Arg152, are brought close to allow interactions with the inhibitor. Asp151 is hydrogen bonded via one of the carboxyl oxygens to the secondary guanidinyl nitrogen of zanamivir, and the main-chain carbonyl oxygen interacts with one of the guanidino NH2 groups of the inhibitor. Arg152 also hydrogen bonds to the carbonyl oxygen of the N-acetyl group of zanamivir. Val149 undergoes a Cα translation of about 7.0 Å, as observed in the recent H5N1 NA structure (39). The side chain of Glu119 also adopts a different orientation, to point away from Glu227, interacting with Arg156 instead to accommodate the 4-guanidino group of zanamivir. Other conserved interactions among all other NAs include that of the carboxyl group of zanamivir with three conserved arginine residues, Arg118, Arg292, and Arg371, and that of the two terminal hydroxyl groups on the glycerol side chain on the C-6 position with the carboxylate of Glu276. The active-site conformation of the inhibitor-bound N1 NA is almost identical to that of N2 and N9 NAs. The major conformational changes induced by ligand binding in 18NA were not observed in subtypes N2 and N9, in which the active site remains essentially unchanged, with only minor changes in side chain rotamers (2, 50). Zanamivir shows structurally tight binding to the NA active site, as shown by a network of interactions with the active-site residues (Fig. 5), low B values of between 8 and 9 Å2 (Table 1), and excellent electron density throughout refinement (Fig. 6). This is consistent with a previous study which demonstrated that the current antiviral drugs against influenza virus NA were effective against the 1918 strain (48).
FIG. 5.

Stereo view of the active sites of the superimposed zanamivir-bound (green) and unbound (gray) structures of 18NA. Conserved charged residues, Arg118, Asp151, Arg152, Arg224, Glu276, Arg292, and Arg371, as well as the structurally conserved water molecule involved in the direct interactions with the inhibitor, are shown in ball-and-stick representation. Val 149 in the 150 loop, in which the major conformational changes occur upon inhibitor binding, is also highlighted in stick presentation to illustrate the extent of the loop rearrangement.
FIG. 6.

Active site of 18NA, showing the bound zanamivir with the σA-weighted 2Fobs − Fcalc electron density map contoured at 2.0 σ.
Residues within the active sites of NAs from both the H5N1 and 1918 H1N1 strains are conserved, except for residue 347, which is an Asn in H1N1 and a Tyr in H5N1. Tyr347 hydrogen bonds with the carboxylate group of oseltamivir in the H5N1 NA structure, and therefore, it has been proposed that Tyr347 partially compensates for the loss of interaction with the inhibitor when the Arg292Lys mutation occurs (39). Indeed, it has been suggested that Tyr347 in group 1 NAs accounts for lower resistance to inhibitors in group 1 than in group 2 (39). However, the presence of Asn347 in H1N1 NA and other N1 subtypes (10) suggests that it is not absolutely conserved among group 1. Thus, group 1 NAs that lack Tyr347 may develop resistance to inhibitors similar to that of group 2. Indeed, subtype-specific variations in and around the active site could be exploited in the design of subtype-specific inhibitors and should be taken into account in the design of broad-acting inhibitors.
Metal binding sites.
Calcium ions have been previously shown to be important for the thermostability and enzyme activity of influenza virus NAs (6, 12). Three potential metal binding sites in each monomer of the tetramer were observed in the electron density maps, two of which have been described previously in other NAs, including those from type B influenza virus. The first Ca2+ site is formed by the four backbone carbonyl oxygens from Asp293, Gly297, Gly345, and Asn347, one of the carboxyl oxygens from Asp324, and a water molecule. It was proposed that this calcium site plays an important structural role in stabilizing the architecture and reactive configuration of the catalytic site formed by otherwise flexible loops (40). The second calcium ion is located at the fourfold axis of the NA tetramer, coordinated by five water molecules (Fig. 7A). The four in-plane water molecules are stabilized by the symmetry-related Asp113 in a unidentate fashion, as well as by the main-chain carbonyl oxygen from Lys111 of a neighboring monomer. The fifth water molecule is below the plane and is coordinated by Oγ2 of Asp113. This metal site could be satisfactorily refined with a fully occupied calcium ion in the native structure, whereas in the zanamivir-bound structure it displays strong negative density in an Fobs − Fcalc difference Fourier map. This could be a direct consequence of a high calcium ion concentration in the crystallization solution for the native crystals, which agrees with a previous finding that this site has weak affinity for calcium (7).
FIG. 7.

Calcium-binding sites in 18NA. (A) The calcium (site 2) at the molecular fourfold axis (i.e., crystallographic twofold axis). Only two monomers are shown for clarity, and side chains are labeled (A and B) after residue number to distinguish two monomers in the asymmetric unit. The coordination of the calcium ion is shown by black dashed lines, and hydrogen bonds are shown with green dashed lines. (B) The new calcium binding site 3, highlighting the side chains of Asp379, Asn381, and Asp387; the main-chain carbonyl of Ser389; and two water molecules in direct contact with the calcium ion.
A putative third calcium-binding site, with pentagonal bipyramidal coordination, was observed (Fig. 7B). The seven oxygen ligands involve the main-chain carbonyl oxygen of Ser389, the side chain carbonyl oxygen of Asn381, a carboxyl oxygen of monodentate Asp387, one bidentate Asp379, and two water molecules. The metal/side chain bond lengths are all within 2.25 to 2.50 Å. The identity of the calcium ion is confirmed by the nature of the coordination, the stable refinement of this ion with full occupancy with moderately high-resolution data, and a B value of about 15 Å2, similar to those of the surrounding atoms. Contrary to what was observed with the second calcium site, both the native and the complex structures show very clear electron density, as well as low B values, for this calcium-binding site throughout structural refinement. The residues that make up the new putative calcium binding site are mainly from one loop, including the coordinating Asp379, Asn381, and Asp387 residues. This site is dependent on the local structure of Pro380, Asn381, Gly382, Trp383, and Thr384, which form a tight turn to bring all of the interacting residues into close proximity with the metal ion (Fig. 7B). Other important residues include Gly382 and Ser319. The presence of any side chain at position 382 would distort the coordination geometry of the binding site. Ser319 is hydrogen bonded to Oγ2 of Asp379 in order to optimally position its Oγ1, which is a ligand for the calcium ion. Interestingly, sequence alignment of NAs from different subtypes indicates that residues around this site are largely conserved in N1 and N4 subtypes (Table 2), but not in other subtypes. In particular, Asp379 in 18NA contributes two oxygen ligands for calcium coordination, but the corresponding residues in other subtypes appear to be hydrophobic. This agrees with the observation that the 380 loop shows similar conformations in both N1 and N4 structures, as discussed above. Interestingly, this novel calcium was not observed in the recent N1 and N4 NA structures, although this may be due to either the lower resolution of these structures (39), in comparison to those described here, or the lack of calcium ions in their crystallization conditions. Early studies established the complete dependence of the activity of N1 NAs on calcium ions (9). The contribution of the new Ca binding site to the function of N1 NAs now requires further investigation.
TABLE 2.
Comparison of peptide sequences of influenza A virus NAs of N1 and N4 subtypes in the regions of the putative calcium binding sitea
 |
Residues involved in the direct interaction with the calcium ion are shown in red, and pink indicates residues in which only the backbone carbonyl is involved in the interactions with the calcium ion. Green indicates residues surrounding the site which are structurally important.
Glycosylation.
18NA has seven conserved N-linked glycosylation sites shared by all known avian N1 viral strains. Four are within the stalk region, and the other three sites are in the ectodomain of 18NA: Asn88, Asn146, and Asn234. Asn88 and Asn234 are both located on the undersurface of the molecule, whereas Asn146 is on the membrane-distal face, close to the active site. Modest electron density, contiguous with that of the corresponding carboxamide nitrogens of Asn88, Asn234, and Asn146, was observed for the first N-acetylglucosamine at each site. Conformational flexibility and/or variable occupancy of the carbohydrates could account for weak electron density. Lepidopteran insect cell lines are known to produce high mannose or paucimannose N-glycans (23). Hence, a glycan comprising two N-acetylglucosamines and two mannose moieties was built into the electron density at Asn146 on one of the two monomers present in the asymmetric unit. This terminal mannose was found to be involved in crystal contact and is hydrogen bonded to Lys366, Arg394, and Glu375 on a symmetry-related molecule. The glycan-processing pathways in insect cells differ from those of higher eukaryotes, such that high mannose structures are incorporated at sites where complex N-glycans may otherwise be present. Carbohydrates attached to Asn88, Asn234, and other potential glycosylation sites in the stalk region might be important in protecting the enzyme from proteolytic cleavage around the extended stalk.
DISCUSSION
The tetrameric 18NA structure presented here shows a high degree of structural similarity to NA from the H5N1 strain (39). Previous studies have shown that the primary structural differences that characterize group 1 subtypes (N1, N4, N5, and N8) are associated with the conformation of the 150 loop or 150 cavity, as observed in the avian H5N1 NA structure (39). Since the currently available drugs oseltamivir and zanamivir are effective against all influenza virus strains, it is likely that an induced conformational change occurs upon ligand binding for all group 1 NAs. Development of these inhibitors was based entirely on the available N2 and N9 structures, both of which belong to group 2 and exhibit a rigid substrate binding cavity. Hence, the 150 cavity was previously not considered during drug development. The identification of this additional space, together with the flexible substrate-binding loop, provides new opportunities for subtype-specific drug design. The polar residues Glu136 and Arg156, located at the bottom of the cavity, provide the potential for specific anchoring of inhibitors to this site. Furthermore, Asp151 on the 150 loop is potentially important in stabilizing a water molecule which could act as a proton donor in the catalytic reaction (11). Hence, new drugs could be developed to target the 150 cavity to block the conformational changes required for both optimal substrate-binding and catalysis.
The high-resolution structures of the 1918 N1 NA and its complex with zanamivir have unambiguously revealed conformational changes that occur in 18NA upon inhibitor binding, as observed previously in other group 1 NA structures. Although zanamivir and oseltamivir are effective against different subtypes of influenza viruses, including the human 1918 H1N1 and H5N1 avian strains, a growing concern is that more drug-resistant mutants will emerge under the selection pressure of constant drug use (26). The 1918 NA structure provides improved resolution of the extra N1 NA binding cavity, which should assist in the rational design of inhibitors. This extra pocket is likely a generic feature among group I NAs of avian and mammalian influenza virus, and the residues surrounding this pocket appear to be conserved among these subtypes. This feature could represent a potential new basis for developing subtype-specific inhibitors through either derivatized forms of currently available drugs or via a de novo lead that specifically targets the 150 cavity.
The expression strategy for the production of recombinant NA proteins adopted here represents a practical and highly cost-effective means of generating recombinant antigens for vaccination (18), as well as for producing proteins for high-throughput drug screening processes in vitro. These findings could help reduce the likelihood of drug-resistant forms of NA, at least in group I, although it is not yet known whether mutations in this new pocket that would reduce its effectiveness as a target might subsequently arise.
Acknowledgments
The work was supported by NIH grant AI-058113 (I.A.W.), an Oxford University Clarendon Award, and a UK Overseas Research Student Award. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structure Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
We thank C. Basler (Mount Sinai School of Medicine, New York) for generously providing the full-length clone of A/Brevig Mission/1/1918, P. Horton and P. Carney for technical assistance in optimizing the baculovirus expression system described here, J. C. Paulson (The Scripps Research Institute) for providing zanamivir, X. Dai and D. A. Shore (The Scripps Research Institute) for their assistance in data collection, and D. A. Shore for helpful comments during manuscript preparation.
This is publication 19120-MB from The Scripps Research Institute.
Footnotes
Published ahead of print on 20 August 2008.
REFERENCES
- 1.Air, G. M., W. G. Laver, and R. G. Webster. 1990. Mechanism of antigenic variation in an individual epitope on influenza virus N9 neuraminidase. J. Virol. 645797-5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker, A. T., J. N. Varghese, W. G. Laver, G. M. Air, and P. M. Colman. 1987. Three-dimensional structure of neuraminidase of subtype N9 from an avian influenza virus. Proteins 2111-117. [DOI] [PubMed] [Google Scholar]
- 3.Basler, C. F., A. H. Reid, J. K. Dybing, T. A. Janczewski, T. G. Fanning, H. Zheng, M. Salvatore, M. L. Perdue, D. E. Swayne, A. Garcia-Sastre, P. Palese, and J. K. Taubenberger. 2001. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. USA 982746-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bender, C., H. Hall, J. Huang, A. Klimov, N. Cox, A. Hay, V. Gregory, K. Cameron, W. Lim, and K. Subbarao. 1999. Characterization of the surface proteins of influenza A (H5N1) viruses isolated from humans in 1997-1998. Virology 254115-123. [DOI] [PubMed] [Google Scholar]
- 5.Bucher, D., and P. Palese. 1975. The biologically active proteins of influenza virus: neuraminidase, p. 83-123. In E. D. Kilbourne (ed.), The influenza viruses and influenza. Academic Press, New York, NY.
- 6.Burmeister, W. P., S. Cusack, and R. W. Ruigrok. 1994. Calcium is needed for the thermostability of influenza B virus neuraminidase. J. Gen. Virol. 75381-388. [DOI] [PubMed] [Google Scholar]
- 7.Burmeister, W. P., R. W. Ruigrok, and S. Cusack. 1992. The 2.2 Å resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 1149-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr, J., J. Ives, L. Kelly, R. Lambkin, J. Oxford, D. Mendel, L. Tai, and N. Roberts. 2002. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antivir. Res. 5479-88. [DOI] [PubMed] [Google Scholar]
- 9.Carroll, S. M., and J. C. Paulson. 1982. Complete metal ion requirement of influenza virus N1 neuraminidases. Brief report. Arch. Virol. 71273-277. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J. M., H. C. Ma, J. W. Chen, Y. X. Sun, J. M. Li, and Z. L. Wang. 2007. A preliminary panorama of the diversity of N1 subtype influenza viruses. Virus Genes 3533-40. [DOI] [PubMed] [Google Scholar]
- 11.Chong, A. K., M. S. Pegg, N. R. Taylor, and M. von Itzstein. 1992. Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza virus. Eur. J. Biochem. 207335-343. [DOI] [PubMed] [Google Scholar]
- 12.Chong, A. K., M. S. Pegg, and M. von Itzstein. 1991. Influenza virus sialidase: effect of calcium on steady-state kinetic parameters. Biochim. Biophys. Acta 107765-71. [DOI] [PubMed] [Google Scholar]
- 13.Crosby, A. W. 1989. America's forgotten pandemic: the influenza of 1918. Cambridge University Press, Cambridge, United Kingdom.
- 14.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 602126-2132. [DOI] [PubMed] [Google Scholar]
- 15.Fodor, E., L. Devenish, O. G. Engelhardt, P. Palese, G. G. Brownlee, and A. Garcia-Sastre. 1999. Rescue of influenza A virus from recombinant DNA. J. Virol. 739679-9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottschalk, A. 1957. Neuraminidase: the specific enzyme of influenza virus and Vibrio cholerae. Biochim. Biophys. Acta 23645-646. [DOI] [PubMed] [Google Scholar]
- 17.Ives, J. A., J. A. Carr, D. B. Mendel, C. Y. Tai, R. Lambkin, L. Kelly, J. S. Oxford, F. G. Hayden, and N. A. Roberts. 2002. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antivir. Res. 55307-317. [DOI] [PubMed] [Google Scholar]
- 18.Johansson, B. E., and I. C. Brett. 2007. Changing perspective on immunization against influenza. Vaccine 253062-3065. [DOI] [PubMed] [Google Scholar]
- 19.Johnson, N. P., and J. Mueller. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull. Hist. Med. 76105-115. [DOI] [PubMed] [Google Scholar]
- 20.Kim, C. U., W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver, and R. C. Stevens. 1997. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 119681-690. [DOI] [PubMed] [Google Scholar]
- 21.Klenk, E., H. Faillard, and H. Lempfrid. 1955. Enzymatic effect of the influenza virus. Hoppe Seyler's Z. Physiol. Chem. 301235-246. [PubMed] [Google Scholar]
- 22.Kobasa, D., A. Takada, K. Shinya, M. Hatta, P. Halfmann, S. Theriault, H. Suzuki, H. Nishimura, K. Mitamura, N. Sugaya, T. Usui, T. Murata, Y. Maeda, S. Watanabe, M. Suresh, T. Suzuki, Y. Suzuki, H. Feldmann, and Y. Kawaoka. 2004. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431703-707. [DOI] [PubMed] [Google Scholar]
- 23.Kost, T. A., J. P. Condreay, and D. L. Jarvis. 2005. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 23567-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhnel, K., T. Jarchau, E. Wolf, I. Schlichting, U. Walter, A. Wittinghofer, and S. V. Strelkov. 2004. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. USA 10117027-17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26283-291. [Google Scholar]
- 26.Le, Q. M., M. Kiso, K. Someya, Y. T. Sakai, T. H. Nguyen, K. H. Nguyen, N. D. Pham, H. H. Ngyen, S. Yamada, Y. Muramoto, T. Horimoto, A. Takada, H. Goto, T. Suzuki, Y. Suzuki, and Y. Kawaoka. 2005. Avian flu: isolation of drug-resistant H5N1 virus. Nature 4371108. [DOI] [PubMed] [Google Scholar]
- 27.Lovell, S. C., I. W. Davis, W. B. Arendall III, P. I. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson, and D. C. Richardson. 2003. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50437-450. [DOI] [PubMed] [Google Scholar]
- 28.Malby, R. L., W. R. Tulip, V. R. Harley, J. L. McKimm-Breschkin, W. G. Laver, R. G. Webster, and P. M. Colman. 1994. The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 2733-746. [DOI] [PubMed] [Google Scholar]
- 29.Matrosovich, M. N., T. Y. Matrosovich, T. Gray, N. A. Roberts, and H. D. Klenk. 2004. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 7812665-12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews, B. W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33491-497. [DOI] [PubMed] [Google Scholar]
- 31.McCoy, A. J., R. W. Grosse-Kunstleve, L. C. Storoni, and R. J. Read. 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr. Sect. D Biol. Crystallogr. 61458-464. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 53240-255. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data in oscillation mode. Methods Enzymol. 276307-326. [DOI] [PubMed] [Google Scholar]
- 34.Palese, P., K. Tobita, M. Ueda, and R. W. Compans. 1974. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61397-410. [DOI] [PubMed] [Google Scholar]
- 35.Reid, A. H., T. G. Fanning, J. V. Hultin, and J. K. Taubenberger. 1999. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc. Natl. Acad. Sci. USA 961651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid, A. H., T. G. Fanning, T. A. Janczewski, R. M. Lourens, and J. K. Taubenberger. 2004. Novel origin of the 1918 pandemic influenza virus nucleoprotein gene. J. Virol. 7812462-12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reid, A. H., T. G. Fanning, T. A. Janczewski, S. McCall, and J. K. Taubenberger. 2002. Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J. Virol. 7610717-10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reid, A. H., T. G. Fanning, T. A. Janczewski, and J. K. Taubenberger. 2000. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc. Natl. Acad. Sci. USA 976785-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell, R. J., L. F. Haire, D. J. Stevens, P. J. Collins, Y. P. Lin, G. M. Blackburn, A. J. Hay, S. J. Gamblin, and J. J. Skehel. 2006. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 44345-49. [DOI] [PubMed] [Google Scholar]
- 40.Smith, B. J., T. Huyton, R. P. Joosten, J. L. McKimm-Breschkin, J. G. Zhang, C. S. Luo, M. Z. Lou, N. E. Labrou, and T. P. Garrett. 2006. Structure of a calcium-deficient form of influenza virus neuraminidase: implications for substrate binding. Acta Crystallogr. Sect. D Biol. Crystallogr. 62947-952. [DOI] [PubMed] [Google Scholar]
- 41.Stevens, J., O. Blixt, T. M. Tumpey, J. K. Taubenberger, J. C. Paulson, and I. A. Wilson. 2006. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312404-410. [DOI] [PubMed] [Google Scholar]
- 42.Stevens, J., A. L. Corper, C. F. Basler, J. K. Taubenberger, P. Palese, and I. A. Wilson. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 3031866-1870. [DOI] [PubMed] [Google Scholar]
- 43.Taubenberger, J. K., A. H. Reid, A. E. Krafft, K. E. Bijwaard, and T. G. Fanning. 1997. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science 2751793-1796. [DOI] [PubMed] [Google Scholar]
- 44.Taubenberger, J. K., A. H. Reid, R. M. Lourens, R. Wang, G. Jin, and T. G. Fanning. 2005. Characterization of the 1918 influenza virus polymerase genes. Nature 437889-893. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger, T. C. 2000. Maximum-likelihood density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 56965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tulip, W. R., J. N. Varghese, W. G. Laver, R. G. Webster, and P. M. Colman. 1992. Refined crystal structure of the influenza virus N9 neuraminidase-NC41 Fab complex. J. Mol. Biol. 227122-148. [DOI] [PubMed] [Google Scholar]
- 47.Tumpey, T. M., C. F. Basler, P. V. Aguilar, H. Zeng, A. Solorzano, D. E. Swayne, N. J. Cox, J. M. Katz, J. K. Taubenberger, P. Palese, and A. Garcia-Sastre. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 31077-80. [DOI] [PubMed] [Google Scholar]
- 48.Tumpey, T. M., A. Garcia-Sastre, A. Mikulasova, J. K. Taubenberger, D. E. Swayne, P. Palese, and C. F. Basler. 2002. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA 9913849-13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tumpey, T. M., A. Garcia-Sastre, J. K. Taubenberger, P. Palese, D. E. Swayne, and C. F. Basler. 2004. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA 1013166-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varghese, J. N., W. G. Laver, and P. M. Colman. 1983. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature 30335-40. [DOI] [PubMed] [Google Scholar]
- 51.von Itzstein, M., W. Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. Van Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron, and C. R. Penn. 1993. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363418-423. [DOI] [PubMed] [Google Scholar]
- 52.Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 829, 52-56. [DOI] [PubMed] [Google Scholar]
- 53.Wang, J., S. Kamtekar, A. J. Berman, and T. A. Steitz. 2005. Correction of X-ray intensities from single crystals containing lattice-translocation defects. Acta Crystallogr. Sect. D Biol. Crystallogr. 6167-74. [DOI] [PubMed] [Google Scholar]
- 54.Zhu, X., X. Xu, and I. A. Wilson. 2008. Structure determination of the 1918 H1N1 neuraminidase from a crystal with lattice-translocation defects. Acta Crystallogr. Sect. D Biol. Crystallogr. 64843-850. [DOI] [PMC free article] [PubMed] [Google Scholar]