

Abstract
ADARs (adenosine deaminases that act on double-stranded RNA) are RNA editing enzymes that catalyze a change from adenosine to inosine, which is then recognized as guanosine by translational machinery. We demonstrate here that overexpression of ADARs but not of an ADAR mutant lacking editing activity could upregulate human immunodeficiency virus type 1 (HIV-1) structural protein expression and viral production. Knockdown of ADAR1 by RNA silencing inhibited HIV-1 production. Viral RNA harvested from transfected ADAR1-knocked-down cells showed a decrease in the level of unspliced RNA transcripts. Overexpression of ADAR1 induced editing at a specific site in the env gene, and a mutant with the edited sequence was expressed more efficiently than the wild-type viral genome. These data suggested the role of ADAR in modulation of HIV-1 replication. Our data demonstrate a novel mechanism in which HIV-1 employs host RNA modification machinery for posttranscriptional regulation of viral protein expression.
Human immunodeficiency virus (HIV) uses many host cellular machineries for its replication. Posttranscriptional modifications of the HIV type 1 (HIV-1) RNA, i.e., alternative splicing, capping, and poly(A) synthesis of the viral pre-mRNA, have been well described (15). However, the process of RNA editing, another subtle type of pre-mRNA modification, is still not well defined, and its role in HIV-1 replication is in doubt.
RNA editing was found to be an important process in regulating gene expression in many eukaryotes and viruses. In hepatitis D virus, the process of RNA editing has been well studied (reviewed in reference 3). A-to-I editing converts a stop codon into a Trp codon, providing envelope protein for virion assembly (4, 25). In the hepatitis C replicon, ADAR1 was shown to abolish replication through editing (33). Hypermutation by RNA editing was found in measles virus mRNA. It is speculated that this process leads to persistence of the virus (5). The editing has also been found in other viruses, such as parainfluenza virus 3 (9), mumps virus (22), and Ebola virus (28).
Evidence of RNA editing in HIV-1 has also been demonstrated. Edited adenosine in the (transactivating response region) (TAR) when the virus is injected into Xenopus oocytes has been reported (31), though the functional consequences of this conversion are not known. It is hypothesized that the phenomenon caused a change in the secondary structure of TAR, which may affect transcription and translation of the viral RNA. Base modification of A to G and C to U has also been shown in the HIV-1 transcript (1). The modification was proposed to play a role in modulation of viral gene expression.
There are many data suggesting that RNA editing might be involved in HIV-1 gene expression. ADARs are the RNA editing enzymes of interested that may be involved in modulation of HIV-1 expression. It was demonstrated that mouse ADAR1 but not ADAR2 is upregulated in lymphocytes in response to inflammation (34). In this study, we demonstrated that ADARs could enhance HIV-1 expression.
MATERIALS AND METHODS
Cell lines, T-lymphocyte isolation, and activation.
293T (adenovirus-transformed human embryonic kidney cell line), COS-7 (adenovirus-transformed African monkey kidney cell line), SupT1 (human T-lymphoblastic leukemia cell line), and H9 (human T-cell line) were subcultured 2 days before RNA isolation.
Twenty milliliters of blood was drawn from healthy volunteers, and peripheral blood mononuclear cells (PBMC) were isolated by centrifugation on Ficoll density medium. Half of the isolated PBMC were sorted for CD4+ T lymphocytes by staining with CD3-fluorescein isothiocyanate and CD4-phycoerythrin (Becton Dickinson) using a FACS Vantage cell sorter (Becton Dickinson). The rest were activated in RPMI-1640 culture medium supplemented with 10% fetal calf serum and 5 μg/ml of phytohemagglutinin (PHA). After 2 days of activation, the cells were harvested and sorted for CD4+ T lymphocytes. The purity of the sorted cells was more than 92%.
RNA extraction and RT-PCR and sequencing.
One milliliter of Trizol reagent (Invitrogen) was added to the collected cells for whole-cell extraction of RNA. RNA was purified according to the manufacturer's protocol. Cytoplasmic and nuclear RNAs were harvested by using the Concert cytoplasmic RNA reagent (Invitrogen) and Trizol reagent (Invitrogen), respectively, as described previously (24). The extracted RNA was treated with RNase-free DNase (Promega). For real-time reverse transcription-PCR (RT-PCR), cDNA was generated by avian myeloblastosis virus reverse transcriptase (Promega) and a random hexamer; the amplification was then performed in a Sybr green dye detection format (LightCycler; Roche). The amplification reactions contained 1× LightCycler Fast Start DNA Master Sybr Green dye I (LightCycler; Roche) and 0.5 mM of each forward and reverse primer. Melting-curve analyses were performed from 65°C to 95°C. RNA extracted from HIV-1-infected PBMC was serially diluted and used for a standard curve setting and the relative quantitative analysis of the viral RNA. Conventional RT-PCR was performed on 35 ng of the RNA using One-Step RT-PCR (Qiagen). The reaction was carried out in a 25-μl volume containing Qiagen One-Step RT-PCR buffer, 0.4 mM deoxynucleoside triphosphate, 0.6 μM of each primer, 1 μl of One-Step enzyme mix (Qiagen), and 4 U of RNase inhibitor (Promega). To control for DNA contamination, RNA samples were subjected to PCR amplification with Taq polymerase in parallel to the RT-PCR. ADAR1 RNA was detected using the primers ADAR (+) 1900 (17) and ADAR1 (−) (AGCGTAGGATTTTGTCACTAC) for 30 PCR cycles (50°C for 30 min, 95°C for 15 min, and cycling at 94°C for 30 s, 51°C for 30 s, and 72°C for 30 s); ADAR2 RNA detection using primers ADAR2 (+) (GATGGTCAGTTCTTTGAAGG) and ADAR2 (−) (GGTCAGGTCACCAAACTTAC) for 35 cycles (50°C for 30 min, 95°C for 15 min, and cycling at 94°C for 30 s, 55°C for 45 s, and 72°C for 30 s); and MLN51 using primers (+)MLN51 (TACTTTTCTGCTCCAGGCGT) and (−)MLN51 (TTACAACCTCAGGTGGAGGG) for 35 cycles (50°C for 30 min, 95°C for 15 min, and cycling at 94°C for 30 s, 55°C for 45 s, and 72°C for 30 s). The following primers were used to detect the expression of specific genes: MxA forward and reverse (13); BSS and GAGA (27) for unspliced HIV-1 RNA; BSS and SJ47A (27) for multiply spliced HIV-1 RNA; E14134 and E14135 (21) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA; and E14134 and GADPH-intron (24) for GAPDH pre-mRNA.
For monitoring of RNA editing, cDNA was amplified with primers pNL4-3(454-472) and pNL4-3(903-937) for the sequences in and around TAR and pNL4-3(7239-7266) and pNL4-3(8480-8459) for the sequences in and around RRE (Rev response element). PCR products were cloned into pGEM-T Easy (Promega), and multiple clones were sequenced in pools of two clones at Macrogen Inc.
DNA constructs.
Plasmids pSS43, pSS151, and pSKW005 were kindly provided by D. Lazinski. pSS151 and pSKW005 contain human ADAR1 and ADAR2 open reading frames, respectively, and pSS43 is a backbone vector of pSS151 and pSKW005 without insert (29). Editing-deficient ADAR1 was generated by mutating pSS151 from HAE to QAA at positions 910 to 912 as previously described (12). The infectious molecular HIV-1 clone pNL4-3 was obtained through the AIDS Research and Reference Reagent Program (4, 7). The pNL4-3 mutant plasmid is the plasmid in which A was replaced with G at positions 8164 and 8166. pLTR-LacZ contains the β-galactosidase gene under the control of the viral promoter long terminal repeat (LTR) from pHxB2. pMJ4, carrying an infectious molecular clone of HIV-1 subtype C from Botswana, was previously described (20). pTat was generated by PCR amplification of Tat exon 1 from pHXB2 and transferred into pCMVβ (Clontech), replacing the β-galactosidase gene.
Transfection, p24 assay, Western blot, green fluorescent protein (GFP) expression detection, and β-galactosidase assay.
pNL4-3 (1.5 μg) was cotransfected with 0.5 μg of either pSS43, pSS151, or pSKW005 into the appropriate cell line (2 × 105 cells of COS-7 or 293T) by using a cationic lipid reagent-mediated transfection technique (DMRIE-C reagent; Invitrogen). Briefly, the plasmids were diluted in 500 μl of serum-free Dulbecco's modified Eagle medium (DMEM). In a separate tube, 8 μl of DMRIE-C reagent was diluted in 500 μl of the same medium. The solutions were then combined, left at room temperature for 20 min, and then added to the cells replacing culture medium. The cells were then incubated at 37°C for 4 h. Transfection solution was then replaced with growth medium. For the ADAR experiment, 1.5 μg of pNL4-3 was cotransfected with the above plasmids in various amounts. Supernatants were collected at 72 h after transfection. The supernatants were assayed for p24 by using an enzyme-linked immunosorbent assay (ELISA) kit (BioMérieux bv). For Western blot analyses, cell lysates and supernatants were subjected to electrophoresis in 12% polyacrylamide gel, blotted on nitrocellulose membranes, and probed with pooled HIV-1-seropositive human sera, rabbit polyclonal anti-ADAR1 (kindly provided by D. Lazinski), or mouse monoclonal antiactin (Santa Cruz).
Cotransfection of the HIV-1- and ADAR-expressing plasmids together with pEGFP-N1 (Clontech) was done by the same method using 0.5 μg of each plasmid. The cells were harvested at 24 and 72 h posttransfection, GFP expression was detected by flow cytometry (FACScan; Becton Dickinson), and supernatants were harvested for the p24 assay at the same time. Cotransfection of ADAR-containing plasmids, pTat, and pLTR-LacZ was performed using 15 μl of the DMRIE-C reagent, with transfection into 7 × 105 cells of COS-7. The transfected cells were collected at 72 h after transfection, and β-galactosidase activity was measured using a colorimetric assay with the substrate o-nitrophenyl-β-d-galactoside.
Infectivity assay in GHOST-CXCR4 cells.
GHOST-CXCR4 cells were obtained through the AIDS Research and Reference Reagent Program (6). The cells were maintained in DMEM supplemented with 10% fetal bovine serum, G418 (500 μg/ml), puromycin (1 μg/ml), and hygromycin (50 μg/ml). Supernatants from transfected cells containing an equal amount of p24 antigen were filtered and treated with 2 U of Turbo DNase (Ambion) for 30 min at room temperature in 1× Turbo DNase buffer to reduce the amount of contaminated DNA. GHOST-CXCR4 cells were infected with the treated supernatant for 12 h and then washed with phosphate-buffered saline three times. The inoculated cells were monitored for green fluorescent infected cells by flow cytometry at days 1 and 3 after fixation in 4% paraformaldehyde. The level of infection in GHOST-CXCR4 cells was confirmed by real-time PCR for Gag HIV DNA using the primers SK38 and SK39 (19) in the Sybr green dye detection format (LightCycler; Roche).
Silencing of gene expression.
293T cells (5 × 104) were plated 1 day before transfection. On the first day of the transfection experiment, 200 pmol of small interfering RNA (siRNA), either siControl, the negative control siRNA (Qiagen), or siADAR1 (14) (Qiagen), was diluted in 180 μl of Opti-MEM (Invitrogen). In another tube, 4 μl of Oligofectamine (Invitrogen) was diluted in 11 μl of Opti-MEM and incubated for 5 min at room temperature before being mixed with siRNA-containing medium. The mixture was then added to the cells. On the second day, 200 pmol of siRNA was cotransfected with 1.5 μg of pNL4-3 using the Lipofectamine 2000 reagent (Invitrogen). Briefly, 4 μl of the Lipofectamine 2000 reagent was diluted in 500 μl Opti-MEM, and the solution was incubated at room temperature for 5 min before being added to 500 μl Opti-MEM containing siRNA and plasmid DNA. The lipid-DNA complex solution was incubated at room temperature for 20 min. Cells were washed once with 1 ml of phosphate-buffered saline, and the medium containing DNA-liposome complex was then added. Cells were incubated at 37°C for 4 h, when the medium was replaced with DMEM with 10% fetal calf serum. Supernatants were collected for further analysis at 48 h after cotransfection.
RESULTS
ADAR1 and ADAR2 expression in cell lines and PBMC.
The expression of ADARs in cell lines commonly used to study HIV-1 replication was studied. RNAs from the cell lines 293T, COS7, SupT1, and H9 were harvested, and mRNA of the ADAR1 and ADAR2 genes was amplified. Amplification products of 1.2 kb and 209 bp were detected in all the cell lines for ADAR1 and ADAR2, respectively (Fig. 1 a).
FIG. 1.

(a) RT-PCR amplification of ADAR1 (upper set) or ADAR2 (lower set) transcripts. The RNAs were extracted from cell lines commonly used to study HIV-1 expression: 293T (lane 1), COS7 (lane 2), SupT1 (lane 3), and H9 (lane 4). (b) Expression of the deaminases in CD4+ T lymphocytes. ADAR1 (upper set) and ADAR2 (middle set) transcripts amplified from nonactivated (lane 1) or PHA-activated (lane 2) sorted CD4+ T lymphocytes are analyzed. MLN51 transcript was demonstrated to be stably expressed throughout T-cell differentiation (10) and was used to show the equivalent amount of total RNA input (lower set). Lane M shows a 100-bp DNA marker.
PHA-activated PBMC were generally used to study HIV-1 infection and replication in vitro. Since HIV-1 replicates well in activated T lymphocytes, we further asked whether expression of the enzymes was altered in different stages of the cells. RNA was collected from both nonactivated and PHA-activated T cells, and ADAR RNA was detected in the RNA extract by RT-PCR. Increasing amounts of ADAR1 RNA in PHA-activated CD4+ T lymphocytes were demonstrated, whereas the expression of ADAR2 remained unchanged (Fig. 1b).
ADARs upregulated HIV-1 production.
To study the effect of ADARs on HIV-1 expression, COS-7 and 293T cell lines were cotransfected with an HIV-1 plasmid (pNL4-3) and either ADAR-containing plasmids (pSS151 and pSKW005) or the control plasmid (pSS43). The transfection efficiency was controlled by monitoring the percentage of GFP-positive cells and was always found to be comparable among transfections within the same set of experiments. Overexpression of ADAR1 in pSS151-transfected cells was confirmed by Western blot analysis (Fig. 2b). In the Western blot, two forms of the ADAR1 protein were detected. ADAR1-L and ADAR1-S are produced from alternatively spliced mRNA from the same gene, and they both contain RNA editing activity (17). The levels of the p24 Gag protein from both the supernatant and lysate of cell lines transfected with ADAR-containing plasmids were significantly higher than that from cells transfected with the control plasmid (Fig. 2a and b). Upregulation of HIV-1 p24 by ADARs was shown to be dose related (Fig. 2c). Upregulation of viral p24 by the enzymes was also demonstrated in other cell lines, such as HeLa CD4high CCR5high cells and MT2 (data not show). The upregulation of HIV-1 was believed not to be a fortuitous event of nonspecific editing, since overexpression of ADAR in cells cotransfected with a GFP expression plasmid under the cytomegalovirus promoter did not alter the level of GFP expression (data not shown). We also used a mutant ADAR1 protein that lacks catalytic activity (12) and showed that this mutant could not upregulate HIV-1 p24 expression (Fig. 2d). Accumulation of the mutant ADAR1 protein was confirmed by Western blot analysis, indicating that the mutant enzyme was efficiently expressed and was stable in the transfected cells (Fig. 2d). This suggested that the mechanism of HIV-1 upregulation by ADAR1 involved RNA editing and that the upregulation was specific to the ADARs with editing activity.
FIG. 2.

(a) Overexpression of ADARs by cotransfection of either an ADAR1 (pSS151) or ADAR2 (pSKW005) plasmid with pNL4-3 enhanced HIV-1 p24 production, as shown by p24 ELISA of the culture supernatant of transfected COS-7 cells. The transfection efficiencies, indicated as percentages of GFP-positive cells (%GFP), are shown below the bars. (b) Western blot analysis of the transfected 293T cell lysate and culture supernatant confirmed the upregulation of HIV-1 structural protein expression (upper panel). The overexpression of the ADAR1 protein is also shown by Western blot analysis (lower panel). (c) Dose-dependent enhancement of HIV-1 replication by ADAR1 and ADAR2 is shown in the supernatant of transfected COS-7 cells. (d) In contrast, an editing-deficient ADAR1 mutant does not show enhancing activity for HIV-1 p24 expression in the supernatant of transfected cells. The expression of the mutant ADAR1 protein was confirmed by a Western blot analysis. (e) A similar dose-dependent enhancement of p24 expression by ADAR1 was also observed with a subtype C HIV-1 clone, pMJ4. The graphs show three independent experiments with error bars. One or two asterisks indicate a statistical significance at a P value of <0.05 or <0.01, respectively, as determined by a t test for a comparison to the control. The “‡” symbol in panel d indicates a P value of <0.05 for the comparison between transfections with wild-type and mutant ADAR1.
In order to test whether the effect of ADAR1 on HIV-1 expression was broadly applicable to other HIV-1 strains, we tested another HIV-1 construct, pMJ4, which carried an infectious molecular clone of HIV-1 subtype C (20). This subtype C construct was also upregulated by ADAR1 overexpression in the same manner as pNL4-3 (Fig. 2e).
Silencing ADARs inhibited HIV-1 production.
After demonstrating that overexpression of ADARs could upregulate HIV-1 expression, we further asked whether the enzymes played a role in the viral gene expression at a normal expression level. We therefore measured HIV-1 expression in an ADAR-knocked-down condition. 293T cells were cotransfected with pNL4-3 and either siControl or siADAR1. The siRNA effectively reduced the level of ADAR1 mRNA (see Fig. 5b). In the presence of siADAR1, the levels of both the Gag precursor p55 protein and the capsid p24 protein in the cell lysate and the level of p24 in supernatant were significant lower (Fig. 3a). This indicated that ADAR1 plays a role in HIV-1 expression. In order to show the specificity of the silencing effect, we complemented ADAR1 by cotransfecting the pSS151 plasmid. The ADAR1 expression plasmid was able to rescue the expression of p24 against suppression by siADAR1 (Fig. 3b). This suggested that the suppression of HIV-1 expression by siADAR1 was specific to the reduction in the ADAR1 enzyme level. Comparing pSS151-transfected cells with and without siADAR1, we could also show that siADAR1 effectively reduced the ADAR1 protein level (Fig. 3b).
FIG. 5.

(a) Representative RT-PCR products from cytoplasmic/nuclear RNA of ADAR-suppressed cells. The RT-PCR products of the HIV-1 multiply spliced RNA and of the unspliced RNA are shown in the two upper sets. The RT-PCR products of ADAR1 and ADAR2 show specific RNA reduction by siRNA. GAPDH mRNA and GAPDH pre-mRNA showing proper nuclear/cytoplasmic fractionation are shown as labeled. Lane 1 shows a 100-bp marker; lanes 2 and 3 show RT-PCR products obtained from nuclear RNA of cells transfected with pNL4-3 and either siControl (siC) or siADAR1 (siA1), respectively. In the same arrangement, lanes 4 and 5 show RT-PCR products obtained from cytoplasmic RNA. Lane 6 shows a negative control for RT-PCR. Reactions without reverse transcription were PCR amplified in parallel for all RNA samples and yielded no specific amplification product (not shown). In multiply spliced transcripts, two bands of RT-PCR products are as follows: 315 bp represents tat RNA (upper band), and 150 bp represent rev RNA (lower band). The RT-PCR products of ADAR1, ADAR2, and GAPDH are shown. A schematic diagram of the positions of primers and RT-PCR products is shown. (b) In order to confirm the change in the cytoplasmic unspliced HIV RNA level, cytoplasmic unspliced HIV RNA (upper two graphs) and ADAR1 RNA (lower two graphs) were analyzed by quantitative real-time RT-PCR under ADAR1-silencing (upper graphs) or -overexpressing (lower graphs) conditions. Data are shown as n-fold differences in the ratios of HIV-1 unspliced/GADPH mRNA. One or two asterisks indicate statistical significance at a P value of <0.05 or<0.01, respectively, as determined by a t test for a comparison to the control. For all amplifications, reactions without reverse transcriptase were performed in parallel and found to be negative. Real-time RT-PCR amplification of GAPDH mRNA was performed for each RNA sample, and the levels were found to be similar between the control and siRNA or pSS151-transfected RNA samples (not shown).
FIG. 3.

(a) Silencing ADAR1 with siRNA decreased viral protein expression from transfected 293T cells, as shown by Western blot analysis of cell lysates and culture supernatants. (b) Additionally, cotransfection of the ADAR1 expression plasmid, pSS151, could rescue p24 expression in culture supernatants, as determined by an ELISA. One or two asterisks indicate statistical significance at a P value of <0.05 or <0.01, respectively, as determined by a t test for a comparison to the control. In addition, the reduction of the ADAR1 protein by siADAR1 was demonstrated by Western blot analysis in siADAR1-, pSS151-cotransfected cells compared to results for siControl-, pSS151-cotransfected cells. (c) The siRNA did not induce an interferon response, as shown by the absence of the RT-PCR product of MxA: lane 1 shows a 100-bp marker, lane 2 shows the RT-PCR product of HIV-1-infected PBMC as a positive control, and lane 3 shows the RT-PCR product of cells transfected with siADAR1.
In order to confirm that the downregulation of HIV-1 expression by siADAR was specific and was not due to a nonspecific interferon action, as has been shown to be induced by some siRNAs (2, 23), expression of an interferon-inducible gene, the MxA gene, was analyzed, and the MxA mRNA was not detected in siRNA-transfected cells (Fig. 3c). This indicated that the siRNA did not induce interferon and the decrease in viral protein production was not caused by interferon-mediated viral suppression.
ADARs did not exert an effect on HIV-1 LTR.
It was previously reported, using the model of Xenopus oocytes nuclei, that the HIV-1 TAR stem-loop structure was a substrate for double-stranded-RNA-modifying activity in a Tat-dependent manner (31). So, the mechanism by which ADARs regulated HIV-1 expression was initially speculated to be TAR related. Overexpression of ADARs in COS-7 cells cotransfected with an LTR reporter plasmid (pLTR-LacZ) and a Tat-encoding plasmid was performed. However, the expression of the reporter gene, LacZ, under the control of the Tat-dependent viral promoter on the LTR in either the absence or presence of the Tat protein was not affected by ADAR overexpression (Fig. 4). This suggested that in mammalian cells, ADARs do not enhance HIV-1 replication by acting on the viral promoter.
FIG. 4.
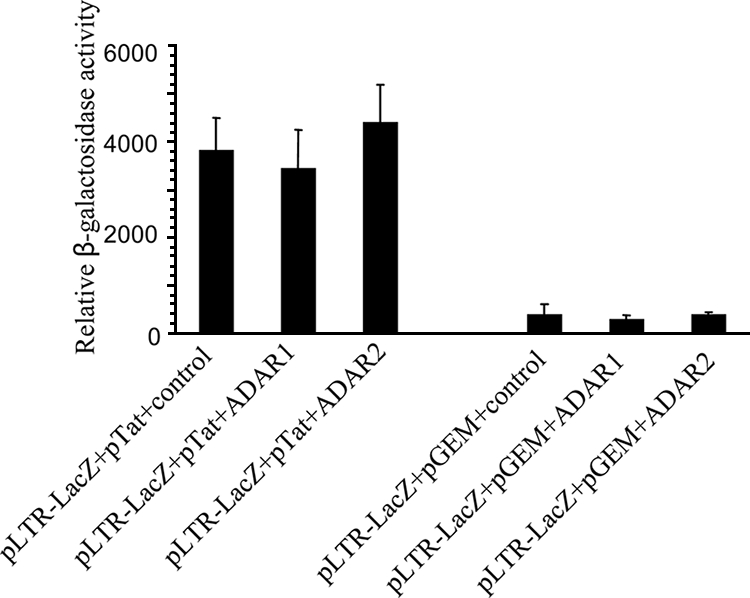
Overexpression of ADARs in COS-7 cells cotransfected with an LTR containing a reporter plasmid. pLTR-LacZ-transfected cells were cotransfected with pTat either alone or in combination with either the ADAR1- or ADAR2-containing plasmid (left set). The right set shows a similar experiment but without pTat.
ADARs altered levels of HIV-1 RNA species.
In order to get further insight into the mechanism of HIV-1 regulation by ADARs, HIV-RNA was analyzed. Viral RNA from cytoplasmic and nuclear fractions of ADAR1-silenced 293T cells was separately analyzed. We found a significant decrease in the level of unspliced viral RNA in the cytoplasmic fraction and an increase in the level of Tat-encoded multiply spliced RNA in both nuclear and cytoplasmic fractions of the siADAR1-transfected cells (Fig. 5a). We confirmed the decrease of cytoplasmic unspliced viral RNA in the presence of siADAR1 by using a quantitative real-time RT-PCR (Fig. 5b). In addition, using the real-time RT-PCR, we found a slight increase in the cytoplasmic level of unspliced viral RNA in pSS151-transfected cells (Fig. 5b).
ADAR edited HIV-1 RNA.
Because ADARs are RNA editing enzymes that target double-stranded RNA, it is plausible to hypothesize that RNA editing may be involved in the upregulation of HIV-1 by ADARs. In order to explore the role of RNA editing, the viral RNA sequences from cells cotransfected with pNL4-3 and the ADAR1-expressing plasmid, pSS151, were studied. We cloned and sequenced viral cDNA at two known double-stranded regions, TAR and RRE, which are likely to be edited and are important viral cis-regulatory elements. At both regions, the viral RNA from cells transfected with pSS151 showed a higher frequency of A-to-G changes than viral sequences from cells transfected with the control plasmid, pSS43 (Table 1). The sequences in and around the TAR did not show any repetitive editing site, whereas 8 out of 30 clones showed A-to-G changes at a three-nucleotide site downstream of RRE (nucleotide positions 8164 to 8166). This high frequency of A-to-G changes at a similar site indicated that this site was specifically recognized and edited by the RNA editing enzyme. The nucleotide position and frequency of each edited site are shown in Table 1.
TABLE 1.
Positions (NL4-3 numbering) and frequencies of A-to-G changes identified in clones of viral RNA amplified from cells cotransfected with pSS151 or pSS43a
| Region | Nucleotide position | No. of sequenced clones | Cotransfected plasmid | Position(s) with A-to-G mutation | No. of occurrences |
|---|---|---|---|---|---|
| TAR | 472-865 | 30 | pSS151 | 487 | 1 |
| 714 | 1 | ||||
| 873 | 1 | ||||
| 472-865 | 30 | pSS43 | 888 | 1 | |
| RRE | 7759-8459 | 30 | pSS151b | 7869 | 3 |
| 7889 | 1 | ||||
| 7914 | 1 | ||||
| 7921 | 1 | ||||
| 7938 | 1 | ||||
| 7944 | 2 | ||||
| 7952 | 1 | ||||
| 7971 | 1 | ||||
| 8014 | 1 | ||||
| 8051 | 1 | ||||
| 8093 | 1 | ||||
| 8099 | 1 | ||||
| 8164 | 3 | ||||
| 8165 | 3 | ||||
| 8166 | 4 | ||||
| 8203 | 2 | ||||
| 8216 | 1 | ||||
| 8226 | 3 | ||||
| 8276 | 1 | ||||
| 7759-8459 | 30 | pSS43c | 7938 | 1 | |
| 8169 | 1 | ||||
| 8258 | 1 | ||||
| 8303 | 1 |
COS-7 cells were cotransfected with pNL4-3 and either the ADAR1-expressing plasmid, pSS151, or the control plasmid, pSS43. Total RNA was extracted 3 days after the transfection, and two fragments of HIV-1 RNA were amplified by RT-PCR. The PCR products were cloned into pGEM-T Easy. Two clones were pooled for a sequencing reaction. A total of 30 clones were sequenced for each transfection.
Pools with multiple changes: (7869, 7914, 7944, 8051, 8164, 8166, 8226), (7869, 7889, 7921, 7938, 7944, 8165), (8165, 8166, 8203, 8216, 8226), (8166, 8203, 8276), (7921, 8014), (8093, 8165), and (8166, 8226). These parenthetical groupings represent pools of sequences with multiple changes within one pool. The numbers within parentheses are positions with an A-to-G change in each pool.
Pools with multiple changes: (8258, 8303). See footnote b.
Edited construct expressed more efficiently.
In order to test whether the editing at nucleotides 8164 to 8166 had a functional consequence, we constructed a pNL4-3 plasmid with A-to-G mutations at positions 8164 and 8166, which were found to be frequently edited. The mutant construct with the edited sequence expressed a higher level of the p24 protein (Fig. 6a) and of viral unspliced RNA (Fig. 6b) than the wild-type construct when transfected into COS7 cells. This suggested that the editing had a functional effect on the HIV-1 expression and may be responsible, at least partly, for the upregulation of HIV-1 expression by ADARs.
FIG. 6.

(a) Comparison of viral protein and RNA expression between the pNL4-3 wild type and the 8164,8166/A-to-G mutant. Viral protein expression was detected by Western blotting in lysates and supernatants of transfected COS7 cells, and the levels of p24 in the supernatants as determined by ELISA are shown below the blot. (b) RNA was isolated, and real-time RT-PCR for unspliced HIV-1 RNA was performed. Data are shown as n-fold differences in the ratios of HIV-1 unspliced/GADPH mRNA.
Effect of ADAR and the edited sequence on viral infectivity.
Although the A-to-G mutation at positions 8164 and 8166 enhanced HIV-1 production from transfected cells, the mutant virus was low in infectivity, indicating that despite the efficient expression, the mutations were not compatible with some other steps in the viral replication. The infectivity defect of the mutant was shown by a reduction in both the GFP reporter signal and the amount of reverse transcript (Fig. 7a and b). The reduction in the reverse transcript suggested that the defect was located in the early phase of the infection. However, viruses produced from cells transfected with either the ADAR1 expression plasmid or siADAR1 were intact in their infectivity compared to the virus from control cells (Fig. 7c). This indicated that the increase in p24 antigen production in ADAR1-transfected cells reflected the increase in production of infectious virus.
FIG. 7.

(a) To test the infectivity of viral preparations from wild-type and edited mutant pNL4-3 genomes, wild-type or mutant virus was inoculated into GHOST-CXCR4 cells. The histograms show a markedly reduced GFP signal in cells inoculated with mutant virus at day 3 postinfection. (b) The reduced infectivity was confirmed by a reduction of reverse transcript in inoculated cells as shown by real-time PCR. (c) In contrast, cells inoculated with NL4-3 viruses produced from cells transfected with wild-type ADAR1, control plasmid, mutant ADAR1, or siADAR1 did not show any difference in infectivity at 24 and 72 h postinfection.
DISCUSSION
A single-stranded DNA editing enzyme that plays a role in HIV-1 replication was previously identified: APOBEG3G edits HIV-1 reverse transcripts (32), causing damage to open reading frames of the virus and degradation of viral DNA (11, 18, 34). However, the role of RNA editing enzymes on HIV-1 production has not been clearly revealed.
There are three members of human ADARs: ADAR1, ADAR2, and ADAR3. The first two are ubiquitously expressed in various tissues and have an A-to-I editing activity, while the latter is found only in the brain and does not have any editing activity. While ADAR2 was constitutively expressed, ADAR1 expression was upregulated when the cells were activated by a mitogen. This is in agreement with the work of Yang et al., who demonstrated upregulation of ADAR1 in tumor necrosis factor alpha and gamma-interferon-activated T lymphocytes (34). These findings raised the importance of ADAR1 for HIV-1 replication. It is known that HIV-1 production is minimal in resting T cells. Although a labile preintegration stage and an absence of transcription initiation are believed to be mainly responsible for the blockage of viral replication, they are not the only obstacles (review in reference 16). Although the viral upregulation by ADAR1 is downstream of these blocks, it is possible that the limitation of ADAR1 expression in resting T cells may further hinder expression and replication of HIV-1 if some escape from the blocks has occurred and hence partially contribute to the blockage of the viral replication.
Previously it was shown that multiply spliced viral RNAs in asymptomatic HIV-1-infected patients were more abundant than unspliced transcripts (30). It has been shown that an interferon-induced form of ADAR1 contains a nuclear export signal, which interacts specifically with CRM1 and RanGTP to form an export complex (8, 26). The CRM1/Exportin pathway is essential for the export of unspliced HIV-1 RNA. By this property, ADAR1 may be involved in the transport of unspliced viral RNA across the nucleus. Our RNA data for siADAR1-transfected cells supported this hypothesis. The increased level of Tat-encoded RNA in the silenced cells may be explained by the decrease in unspliced RNA nuclear export, which resulted in increased nuclear RNA splicing.
Sequencing around the RRE region in the ADAR1-overexpressing cells showed some site-specific editing at nucleotides 8164 to 8166. A-to-G mutations at this site offered an advantage in expression to the virus. This suggested that the adenosine bases at these positions may be a component of an unknown cis-acting element. The nucleotide G at this position was found in up to 51.6% of all the subtype B sequences in the HIV Sequence Database. At the protein level, the region is situated on gp41 and does not overlap with Tat or Rev open reading frames. A-to-G modification at A8164 does not alter the amino acid coding, while that at A8166 causes an amino acid change from Asn to Ser in gp41.
The lower infectivity of viruses produced from the mutant clone with edited sequence may explain why the edited sequence did not accumulate in the viral genome. While editing at this site provided a benefit for viral protein expression, edited RNA might be less efficient as a viral genome. However, since the viruses produced from ADAR1-overexpressing or ADAR1-knocked-down cells exhibited normal infectivity, the enhancement of HIV-1 expression by ADAR1 must have involved either editing at other sites that did not interfere with infectivity or other nonediting mechanism as well.
Our data demonstrate a novel posttranscriptional regulatory mechanism that is essential for the efficient expression of HIV-1. Targeting this regulatory machinery may provide a new therapeutic approach against this virus.
Acknowledgments
We thank David Lazinski for the plasmids pSS43, pSS151, and pSKW005 and rabbit polyclonal anti-ADAR1. The plasmid pNL4-3 was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We thank Boonrat Tassaneetrithep and Thanat Chookajorn for valuable comments.
This project was supported by a research grant from the Ellison Foundation. A.P. was supported by the Medical Scholars (Ph.D.-M.D.) Program of Mahidol University. R.K. was supported by a Siriraj Graduate Thesis scholarship.
Footnotes
Published ahead of print on 27 August 2008.
REFERENCES
- 1.Bourara, K., S. Litvak, and A. Araya. 2000. Generation of G-to-A and C-to-U changes in HIV-1 transcripts by RNA editing. Science 2891564-1566. [DOI] [PubMed] [Google Scholar]
- 2.Bridge, A. J., S. Pebernard, A. Ducraux, A. L. Nicoulaz, and R. Iggo. 2003. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34263-264. [DOI] [PubMed] [Google Scholar]
- 3.Casey, J. L. 2006. RNA editing in hepatitis delta virus. Curr. Top. Microbiol. Immunol. 30767-89. [DOI] [PubMed] [Google Scholar]
- 4.Casey, J. L., and J. L. Gerin. 1995. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J. Virol. 697593-7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cattaneo, R., A. Schmid, D. Eschle, K. Baczko, V. ter Meulen, and M. A. Billeter. 1988. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 55255-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cecilia, D., V. N. KewalRamani, J. O'Leary, B. Volsky, P. Nyambi, S. Burda, S. Xu, D. R. Littman, and S. Zolla-Pazner. 1998. Neutralization profiles of primary human immunodeficiency virus type 1 isolates in the context of coreceptor usage. J. Virol. 726988-6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206935-944. [DOI] [PubMed] [Google Scholar]
- 8.Eckmann, C. R., A. Neunteufl, L. Pfaffstetter, and M. F. Jantsch. 2001. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol. Biol. Cell 121911-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galinski, M. S., R. M. Troy, and A. K. Banerjee. 1992. RNA editing in the phosphoprotein gene of the human parainfluenza virus type 3. Virology 186543-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamalainen, H. K., J. C. Tubman, S. Vikman, T. Kyrola, E. Ylikoski, J. A. Warrington, and R. Lahesmaa. 2001. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Anal. Biochem. 29963-70. [DOI] [PubMed] [Google Scholar]
- 11.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113803-809. [DOI] [PubMed] [Google Scholar]
- 12.Herbert, A., and A. Rich. 2001. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc. Natl. Acad. Sci. USA 9812132-12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holzinger, D., C. Jorns, S. Stertz, S. Boisson-Dupuis, R. Thimme, M. Weidmann, J. L. Casanova, O. Haller, and G. Kochs. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J. Virol. 817776-7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayan, G. C., and J. L. Casey. 2002. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of ADAR1 but not ADAR2 expression. J. Virol. 7612399-12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kingsman, S. M., and A. J. Kingsman. 1996. The regulation of human immunodeficiency virus type-1 gene expression. Eur. J. Biochem. 240491-507. [DOI] [PubMed] [Google Scholar]
- 16.Lassen, K., Y. Han, Y. Zhou, J. Siliciano, and R. F. Siliciano. 2004. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 10525-531. [DOI] [PubMed] [Google Scholar]
- 17.Liu, Y., C. X. George, J. B. Patterson, and C. E. Samuel. 1997. Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase. J. Biol. Chem. 2724419-4428. [DOI] [PubMed] [Google Scholar]
- 18.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 42499-103. [DOI] [PubMed] [Google Scholar]
- 19.Menzo, S., P. Bagnarelli, M. Giacca, A. Manzin, P. E. Varaldo, and M. Clementi. 1992. Absolute quantitation of viremia in human immunodeficiency virus infection by competitive reverse transcription and polymerase chain reaction. J. Clin. Microbiol. 301752-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ndung'u, T., B. Renjifo, and M. Essex. 2001. Construction and analysis of an infectious human immunodeficiency virus type 1 subtype C molecular clone. J. Virol. 754964-4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimori, H., T. Shiratsuchi, T. Urano, Y. Kimura, K. Kiyono, K. Tatsumi, S. Yoshida, M. Ono, M. Kuwano, Y. Nakamura, and T. Tokino. 1997. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 152145-2150. [DOI] [PubMed] [Google Scholar]
- 22.Paterson, R. G., and R. A. Lamb. 1990. RNA editing by G-nucleotide insertion in mumps virus P-gene mRNA transcripts. J. Virol. 644137-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Persengiev, S. P., X. Zhu, and M. R. Green. 2004. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 1012-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phuphuakrat, A., and P. Auewarakul. 2005. Functional variability of Rev response element in HIV-1 primary isolates. Virus Genes 3023-29. [DOI] [PubMed] [Google Scholar]
- 25.Polson, A. G., B. L. Bass, and J. L. Casey. 1996. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature 380454-456. [DOI] [PubMed] [Google Scholar]
- 26.Poulsen, H., J. Nilsson, C. K. Damgaard, J. Egebjerg, and J. Kjems. 2001. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol. Cell. Biol. 217862-7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saltarelli, M. J., E. Hadziyannis, C. E. Hart, J. V. Harrison, B. K. Felber, T. J. Spira, and G. N. Pavlakis. 1996. Analysis of human immunodeficiency virus type 1 mRNA splicing patterns during disease progression in peripheral blood mononuclear cells from infected individuals. AIDS Res. Hum. Retrovir. 121443-1456. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez, A., S. G. Trappier, B. W. Mahy, C. J. Peters, and S. T. Nichol. 1996. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 933602-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato, S., S. K. Wong, and D. W. Lazinski. 2001. Hepatitis delta virus minimal substrates competent for editing by ADAR1 and ADAR2. J. Virol. 758547-8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seshamma, T., O. Bagasra, D. Trono, D. Baltimore, and R. J. Pomerantz. 1992. Blocked early-stage latency in the peripheral blood cells of certain individuals infected with human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 8910663-10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharmeen, L., B. Bass, N. Sonenberg, H. Weintraub, and M. Groudine. 1991. Tat-dependent adenosine-to-inosine modification of wild-type transactivation response RNA. Proc. Natl. Acad. Sci. USA 888096-8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 33.Taylor, D. R., M. Puig, M. E. Darnell, K. Mihalik, and S. M. Feinstone. 2005. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 796291-6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang, J. H., X. Luo, Y. Nie, Y. Su, Q. Zhao, K. Kabir, D. Zhang, and R. Rabinovici. 2003. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 10915-23. [DOI] [PMC free article] [PubMed] [Google Scholar]