

Abstract
The arenavirus envelope glycoprotein (GPC) mediates viral entry through pH-induced membrane fusion in the endosome. This crucial process in the viral life cycle can be specifically inhibited in the New World arenaviruses by the small-molecule compound ST-294. Here, we show that ST-294 interferes with GPC-mediated membrane fusion by targeting the interaction of the G2 fusion subunit with the stable signal peptide (SSP). We demonstrate that amino acid substitutions at lysine-33 of the Junín virus SSP confer resistance to ST-294 and engender de novo sensitivity to ST-161, a chemically distinct inhibitor of the Old World Lassa fever virus. These compounds, as well as a broadly active inhibitor, ST-193, likely share a molecular target at the SSP-G2 interface. We also show that both ST-294 and ST-193 inhibit pH-induced dissociation of the G1 receptor-binding subunit from GPC, a process concomitant with fusion activation. Interestingly, the inhibitory activity of these molecules can in some cases be overcome by further lowering the pH used for activation. Our results suggest that these small molecules act to stabilize the prefusion GPC complex against acidic pH. The pH-sensitive interaction between SSP and G2 in GPC represents a robust molecular target for the development of antiviral compounds for the treatment of arenavirus hemorrhagic fevers.
Arenaviridae comprise a large family of enveloped, negative-strand RNA viruses whose species have coevolved and diversified with their respective rodent hosts (10, 45). Many arenavirus species are nonpathogenic to humans, but several can be transmitted to cause severe acute hemorrhagic fevers. In the so-called Old World group of arenaviruses, Lassa fever virus (LASV) is responsible for up to 300,000 infections annually in western Africa (36). Although arenavirus disease is less prevalent in the Americas (40), four distinct species of New World arenaviruses are recognized to cause fatal hemorrhagic fevers: the Junín (JUNV), Machupo, Guanarito, and Sabiá viruses. New species of disease-associated arenaviruses continue to be identified (8, 13). Without effective treatment or immunization, the hemorrhagic fever arenaviruses remain an urgent public health and biodefense concern.
Arenavirus entry into its host cell is promoted by the virus envelope glycoprotein (GPC) and provides a potential target for therapeutic intervention. The G1 subunit of the GPC complex initiates infection by binding to a cell surface receptor. The pathogenic New World arenaviruses utilize transferrin receptor 1 for entry (41, 42), whereas nonpathogenic New World viruses and Old World viruses bind α-dystroglycan or an unknown receptor (6, 20, 49). Upon receptor binding, the virion is endocytosed (4), and fusion of the viral and cellular membranes is subsequently activated by acidic pH in the maturing endosome (7, 14, 15). The mechanics of membrane fusion are promoted by the transmembrane fusion subunit, G2. As is characteristic of other class I virus fusion proteins (16, 17, 26, 29, 48), activation of the prefusion GPC complex is followed by a structural reorganization of the G2 ectodomain that leads to formation of a highly stable six-helix bundle and to membrane fusion (19, 24, 53).
GPC is unusual among class I envelope glycoproteins in that the mature complex retains its cleaved signal peptide as an essential subunit (5, 18, 58) (Fig. 1). This stable signal peptide (SSP) contains 58 amino acids and spans the membrane twice, with both N and C termini in the cytosol (1). SSP is likely retained in the mature GPC complex by formation of an intersubunit zinc-finger structure with the cytoplasmic domain of G2 (55). Interestingly, amino acid substitutions at a lysine in the short ectodomain loop of SSP (K33) have been shown to modulate the pH at which membrane fusion is activated (57). Because the charge at K33 is itself not altered by acidic pH, we reasoned that the lysine side chain might respond to a titratable pocket in the ectodomain of G2. Thus, SSP and G2 may interact to mediate the pH-induced transition from the metastable prefusion GPC complex to the activated fusion-competent form, whereupon the structural reorganization in G2 is actuated to drive membrane fusion. In this report, we demonstrate that the recently reported small-molecule inhibitor of arenavirus entry, ST-294 (3), prevents membrane fusion by targeting the SSP-G2 interface and stabilizing the prefusion state of GPC against acidic pH.
FIG. 1.

Arenavirus GPC complex. A representation of the JUNV GPC open reading frame is shown at the top. Amino acids are numbered from the initiating methionine, and the SSP, G1, and G2 subunits are indicated. Membrane-spanning regions in SSP (hφ1 and hφ2) (1) and in G2 (TM) are shaded dark gray, and the N- and C-terminal heptad-repeat regions in G2 (53) are in light gray. Long tick marks and the amino acids noted above GPC represent ST-294 resistance mutations described in the text; short tick marks show other mutations examined. Below the schematic, the amino acid sequences of SSP and the membrane-proximal region of G2 in JUNV, TCRV, and LASV are detailed (GenBank accession numbers D10072, M20304 and J04324, respectively). The ST-294 resistance mutations in JUNV are indicated by brackets. At the right of the figure, we illustrate our current model of the subunit organization in the tripartite GPC complex. Thickened sections of lines represent membrane-spanning domains in SSP and G2 and the heptad-repeat regions in G2. The cytosolic N terminus of SSP is myristoylated (thin line) (58), and an intersubunit zinc finger (circle) is thought to link the C terminus of SSP with the cytoplasmic domain of G2 (55). Mutations at K33 in the ectodomain of SSP have previously been shown to modulate the pH of membrane fusion (57). The drawing is not to scale.
MATERIALS AND METHODS
Molecular and chemical reagents and monoclonal antibodies (MAbs).
GPC from the pathogenic JUNV strain MC2 (25) was expressed in Vero cells by transfection using a pcDNA3.1-based plasmid carrying the complete GPC open reading frame or by cotransfection using plasmids encoding CD4sp-GPC (in which SSP is replaced by the conventional signal peptide of CD4) and SSP-term (in which a stop codon is introduced following the C-terminal SSP amino acid T58) (57). These two components associate in trans and reconstitute the native GPC complex (18). The GPC open reading frame of LASV-Josiah (33) and that of the vesicular stomatitis virus (VSV) G glycoprotein (kindly provided by John K. Rose, Yale University) were also expressed in pcDNA3.1 plasmids. Transient expression utilized a recombinant vaccinia virus expressing T7 RNA polymerase (vTF7-3) (23). Mutations in GPC were introduced by QuikChange mutagenesis (Stratagene), and all constructs were verified by DNA sequencing.
The small-molecule fusion inhibitors ST-294, ST-336 (3), ST-193 (33), and ST-161 (unpublished data) were dissolved in dimethyl sulfoxide. Chemical structures are shown in Fig. 2. The murine MAb BF11 (46), directed to G1, was contributed by Tom Ksiasek and Tony Sanchez (Special Pathogens Branch, CDC, Atlanta, GA) and obtained through the NIH Biodefense and Emerging Infections Research Resources Repository.
FIG. 2.
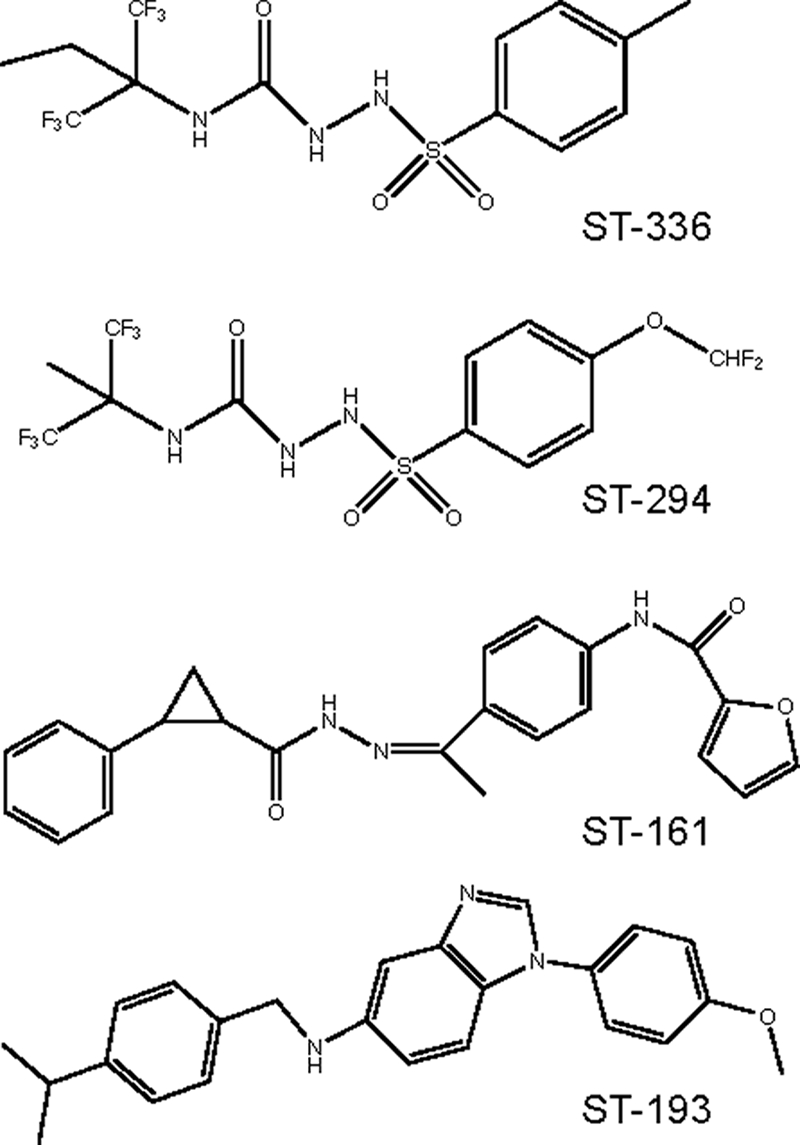
SIGA fusion inhibitors. The New World arenavirus inhibitor ST-294 was developed from the initial hit compound ST-336 (3). ST-161 (unpublished data) and ST-193 (33) were derived from separate hit compounds identified by high-throughput screening using the Old World LASV GPC.
Cell-cell fusion assay.
A vaccinia virus-based β-galactosidase fusion reporter assay (39) was used to characterize the ability of GPC to mediate pH-dependent cell-cell fusion (57, 58). Briefly, Vero cells infected by vTF-7 and expressing GPC were maintained in 10 μM of cytosine arabinoside (araC) to limit vaccinia virus replication. Target cells for syncytium formation were infected with a recombinant vaccinia virus bearing the β-galactosidase gene under the control of a T7 promoter (vCB21R-lacZ) (39) and seeded into 96-well microculture dishes (40,000 cells/well) in the presence of 100 μg rifampin/ml to minimize vaccinia virus assembly and cytopathic effect (28). Prior to use in the cell-cell fusion assay, target cells (40,000 cells/well) were added to the microcultures, allowed to settle for 30 min, and subjected to a brief low-speed centrifugation (∼25 × g) to ensure cell-cell contact. These cocultures were continued for 3 h in medium containing araC and rifampin. Membrane fusion was initiated by incubation in drug-containing medium that had been adjusted to low pH by using HEPES and PIPES [piperazine-N,N′-bis(2-ethyanesulfonic acid)] buffers. After 10 to 30 min at 37°C, the cells were restored to the neutral drug-containing medium and cultured for 5 h to allow for expression of the β-galactosidase fusion reporter gene, which was then quantitated using the chemiluminescent GalactoLite Plus substrate and a Tropix TR717 microplate luminometer. The wide dynamic range of the assay permits robust measurement of ≤1% of wild-type activity. This low level is routinely 10-fold greater than that measured using nonfusogenic GPC molecules (e.g., CD4spGPC in the absence of SSP or a cleavage-defective mutant) (58) or upon treatment of cells expressing wild-type GPC at neutral pH.
For studies of fusion inhibition, cells were incubated with serial dilutions of the compounds developed by SIGA Technologies throughout the initial 3.5-h period of coculture. In these studies, 4 to 6 replicate wells were used for each determination, and control cocultures were treated with the dimethyl sulfoxide solvent (which did not affect the assay at final concentrations of ≤0.25%). In preliminary studies, inhibitor was added prior to, during, or after the pH pulse to show that pretreatment was both necessary and sufficient for inhibition (Fig. 3B).
FIG. 3.

ST-294 inhibits JUNV GPC-mediated membrane fusion. pH-dependent cell-cell fusion was initiated by a pulse of medium adjusted to pH 5.0, detected using a recombinant vaccinia virus-based β-galactosidase-reporter assay, and quantified by chemiluminescence. (A) Vero cells expressing JUNV GPC (filled circles) or VSV G protein (open squares) were incubated with the indicated concentrations of ST-294, washed, and then pulsed for 20 min with medium at pH 5.0. The percentage of fusion relative to that of the control in the absence of ST-294 is indicated. Error bars representing ±1 standard deviation are calculated for all points and in some cases may not be visible on the scale of the graph. (B) Cells expressing JUNV GPC were incubated with ST-294 prior to (pre), during (pulse) or after (post) the pH pulse (the numbers 10 or 50 represent the μM concentration of ST-294). Cell-cell fusion is reported as relative light units (RLUs) in the assay. Pretreatment with ST-294 was necessary and sufficient for inhibition.
G1 shedding.
pH-induced shedding of the G1 subunit from metabolically labeled cells was detected by immunoprecipitation (57, 58) from the low-pH culture supernatant. Vero cells expressing GPC as described above were suspended and pulsed at the indicated pH for 10 min at 37°C. Cells were also treated in medium adjusted to pH 2.5 in an effort to normalize G1 shedding relative to the total amount of G1 eluted under highly stringent conditions. In studies of fusion inhibition, the cell suspension was pretreated for 30 min with the SIGA compounds. The low-pH medium was recovered and neutralized prior to centrifugation to remove cell debris and shed membranes. G1 was recovered from this supernatant by immunoprecipitation using MAb BF11 (46) and protein A Sepharose (Sigma) in buffers containing 1% Triton X-100. The glycoprotein was subsequently deglycosylated by using peptide N-glycosidase F (New England Biolabs) (57) and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions (NuPAGE 4-12% Bis-Tris gel; Invitrogen). G1 shedding was quantitated in phosphostimulated luminescence units by using a Fuji FLA-3000G imager and ImageGauge software.
RESULTS
ST-294 targets the prefusion form of GPC to inhibit cell-cell fusion.
ST-294 was developed by SIGA Technologies through optimization of ST-336 (Fig. 2), a molecule first identified by high-throughput screening for inhibition of cell culture infection with the nonpathogenic New World arenavirus Tacaribe (TCRV) (3). This class of compounds inhibits in vitro infection by the related New World arenaviruses JUNV, Machupo virus, and Guanarito virus at submicromolar concentrations yet is minimally active against the Old World LASV. Studies demonstrated that these inhibitors bind to intact virions and act prior to infection. TCRV isolates selected for in vitro resistance to ST-336 had several mutations in G2 and none in G1 (3). Together, these data suggested that ST-294 acts upon the virion GPC complex prior to the initial stages of cell infection.
To determine whether ST-294 directly inhibits the membrane fusion activity of GPC, we investigated its ability to inhibit syncytium formation among cells expressing a molecularly cloned JUNV GPC (53). In this assay, Vero cells are briefly pulsed with medium adjusted to pH 5.0 and cell-cell fusion is assessed using a β-galactosidase reporter gene (39). As illustrated in the left panel of Fig. 3, we found that the membrane fusion activity of JUNV GPC was sensitive to ST-294, whereas pH-dependent cell-cell fusion by the unrelated G protein of VSV was unaffected. In these studies, cell-cell fusion was reduced to 50% at 4 μM ST-294, whereas previous work had determined a 50% inhibitory concentration (IC50) of 0.3 μM for JUNV plaque reduction (3). The numerical difference in the inhibitory concentrations likely reflects differences inherent in the two assays. Thus, ST-294 prevents arenavirus entry by specifically inhibiting the membrane fusion activity of GPC.
Studies had previously demonstrated that prior exposure of virions to ST-336 was sufficient to block subsequent infection of untreated cells (3). In order to determine the timing requirements for inhibition of cell-cell fusion, GPC-expressing cells were treated with 10 μM ST-294 prior to the pH pulse, during the pH pulse, or upon return to neutrality (Fig. 3, right panel). Inhibition was observed only when ST-294 was included prior to pulsing with pH 5.0. Pretreatment with 10 μM ST-294 reduced cell-cell fusion by 75%, and no additional inhibition was seen if treatment was continued throughout. Further inhibition could be detected upon pretreatment with 50 μM ST-294. These findings support the conclusion that these compounds act prior to infection (3) and demonstrate clearly that ST-294 targets a prefusion form of the GPC complex.
Mutations in G2 and SSP confer resistance to ST-294.
ST-336-resistant TCRV isolates encode mutations in the membrane-proximal ectodomain and transmembrane domain of G2 (3). To determine whether the amino acid changes noted were responsible for viral resistance, we introduced the individual mutations (T416N, I418T, S433I, and F436I) into the homologous positions in JUNV GPC (T418N, L420T, A435I, and F438I). The GPC mutants were expressed, proteolytically matured, and transported to the surface of the cell similarly to the wild type (not shown) (53), and all were able to promote cell-cell fusion, albeit to differing degrees. The relative fusion competence levels of the mutants are indicated on the left panel of Fig. 4A. All four mutants were highly resistant to 10 μM ST-294 (Fig. 4A, right panel). Thus, each of the mutations identified in the membrane-proximal and transmembrane domains of G2 from ST-336-resistant TCRV isolates (3) also rendered JUNV GPC resistant to ST-294.
FIG. 4.
Genetic analysis of ST-294 resistance. JUNV GPC was expressed by cotransfection of plasmids encoding SSP (SSP-term) and CD4sp-GPC (in which SSP is replaced by the conventional signal peptide of human CD4) as previously described (57). Using trans-complementation, we obviate concerns that mutations in SSP may affect signal-peptidase cleavage. The G2 and SSP mutants listed on the left were coexpressed with their respective wild-type partners. pH-dependent membrane fusion was determined in the absence of ST-294 (gray bars) or upon pretreatment with 10 μM ST-294 (black bars). The left panel (% fusion) reports the percentage of fusion by the mutant GPC relative to that by GPC comprising wild-type SSP and CD4sp-GPC. The right panel (% resistance) reflects the extent of membrane fusion in the presence of 10 μM ST-294, relative to that of the untreated GPC. (A) Mutations in G2 originally identified in ST-336-resistant TCRV isolates. The T418N mutation was surprisingly severe in the JUNV background. (B and C) New and existing mutants in G2 and SSP, respectively. All points are representative of results for two or more independent experiments.
Alanine-scanning mutagenesis was used to identify additional amino acid determinants of ST-294 resistance in the membrane-proximal ectodomain of G2. We focused on the large number of charged residues in this region (Fig. 1), some of which may be positioned to interact with the critical K33 side chain in the ectodomain loop of SSP (57; unpublished data). We also included an existing collection of mutations spanning the N- and C-terminal heptad-repeat regions of the G2 ectodomain (53). As in other class I fusion proteins, the N- and C-terminal heptad repeats in the prefusion GPC complex are sequestered in an unknown conformation and refold to form the six-helix bundle structure during membrane fusion.
Of over 30 new and existing G2 mutants examined, 22 were able to mediate cell-cell fusion at levels of ≥10% that of the wild-type GPC (Fig. 4B, left panel). Three additional determinants of ST-294 resistance were identified among these: I347A, D400A, and F427A (Fig. 4B, right panel). I347 lies in the N-terminal heptad repeat of G2, at an interhelical a-position in the postfusion six-helix bundle, and the alanine mutation reduces membrane fusion to ∼40% of wild-type levels (53). D400 is located in the C-terminal heptad repeat (53) at a position expected to reside on the outer surface of the six-helix bundle. D400A reduces cell-cell fusion to ∼75% of the wild type. F427 is located near the ectodomain face of the predicted transmembrane domain, between the L420T and A435I mutations identified in ST-336-resistant TCRV, and F427A is comparable to the wild-type GPC in its membrane-fusion activity. Although the seven resistance mutations span much of the G2 ectodomain, the three-dimensional relationship between these residues in the prefusion GPC complex is unknown.
Six of the ST-294 resistance mutations (D400A, T418N, L420T, F427A, A435I, and F438I), however, lie close in sequence to the nominal ectodomain face of the membrane, where they may be positioned to interact with SSP (57). We therefore extended our analysis of ST-294 resistance to include residues in SSP (1, 56). For the 15 fusion-competent mutants examined, only mutations at N37 and K33 in the ectodomain loop of SSP were able to render GPC resistant to ST-294 (Fig. 4C). The K33R and K33H mutations, which have been shown to reduce the pH threshold for membrane fusion (57), both generated resistance to ST-294 (as did K33Q and K33E [not shown]). Amino acid changes in the second membrane-spanning region of SSP (positions 41 through 54) did not significantly affect membrane fusion activity (1) or sensitivity to ST-294. Mutations in the two cytosolic regions of SSP—G2A (58), E17A and R55A (57), and T58R (54)—did not appreciably alter sensitivity. Thus, the ectodomain loop of SSP that is critical in modulating the pH of fusion activation is also an important determinant of inhibition by ST-294. The K40 residue at the nominal C terminus of the ectodomain loop (57) appears to not participate in determining sensitivity.
The K33H mutation confers sensitivity to Old World-specific inhibitors.
SIGA Technologies has independently identified two distinct chemical classes of Old World arenavirus fusion inhibitors (Fig. 2) through high-throughput screening for molecules that prevent cell entry by pseudotyped retroviruses bearing the LASV GPC (33). We characterized the inhibitory activity of these new compounds, ST-161 and ST-193, by using JUNV and LASV GPCs in cell-cell fusion assays. ST-161 was found to be specific to LASV GPC and did not inhibit cell-cell fusion by JUNV GPC (Fig. 5, left panel), while the broadly active ST-193 molecule (33) was shown to inhibit cell-cell fusion by both LASV and JUNV GPCs (Fig. 5, right panel).
FIG. 5.

JUNV and LASV GPC sensitivities to ST-161 and ST-193. Cells expressing trans-complemented wild-type JUNV GPC (filled circles), L420T JUNV GPC (filled squares), K33H JUNV GPC (filled upright triangles), K33R JUNV GPC (filled inverted triangles), or wild-type LASV GPC (open circles) were treated with the indicated concentrations of ST-161 or ST-193 prior to pulsing with medium at pH 5.0. The percentage of fusion relative to that of the inhibitor-untreated control is indicated. Error bars represent ±1 standard deviation.
In order to probe the molecular basis for the inhibitory activities of ST-161 and ST-193, we first investigated whether these new inhibitors shared determinants of resistance with ST-294. Of the four resistance mutations identified in TCRV, we focused on L420T because of its good fusion activity and its localization in the G2 ectodomain. The L420T mutant of JUNV GPC remained insensitive to the Old World-specific inhibitor ST-161 but was now resistant to the broadly active molecule ST-193 (Fig. 5, left panel). We also examined the effect of ST-294 resistance mutations at K33. Remarkably, we found that the K33H mutant JUNV GPC now exhibited de novo sensitivity to the LASV-specific inhibitor ST-161 (Fig. 5, left panel). Both K33H and K33R mutations also enhanced sensitivity to ST-193 (Fig. 5, right panel).
Together, these results suggest that the three independently identified chemical classes of arenavirus fusion inhibitors share a common molecular target involving both SSP and G2. Sensitivity among the different classes of inhibitors may be determined in part by SSP. However, because K33 is uniformly conserved among naturally occurring arenaviruses (57), species specificity is also determined by the amino acid diversity in G2.
Inhibitors prevent pH-induced G1 shedding from the GPC complex.
We have previously shown that amino acid substitutions at K33 that decrease positive polarity (K33R, K33H, K33Q, and K33E) systematically depress the pH needed for activation of cell-cell fusion (57). Substitution of a nonpolar alanine side chain at this position abolishes all evidence of membrane fusion. In these studies, we also noted that K33A mutant GPC retains much of its G1 subunit during biosynthesis, as opposed to wild-type GPC in which the loss of G1 from the complex is readily discerned (57). This observation suggested that the K33A mutation may limit spontaneous dissociation of the G1 subunit by stabilizing the GPC complex and thereby prevent membrane fusion. Stabilization of the prefusion GPC complex might likewise represent a possible mechanism of action of the SIGA inhibitors.
To investigate this notion, we made use of the observation in class I envelope glycoproteins that the receptor-binding subunit can be shed in response to the activation of membrane fusion (32, 38). Partial dissociation of the receptor-binding subunit is considered an early event in the conformational cascade to membrane fusion in all class I proteins (16, 17, 26, 29, 48). In the absence of a covalent linkage between the subunits, the receptor-binding subunit can be physically shed from the complex concomitant with activation. Although the functional relationship between shedding and membrane fusion is uncertain (22, 37, 51), the degree of dissociation can serve as a useful marker for both the stability of the prefusion complex and its activation toward fusion (21, 32, 47, 52).
To determine whether G1 shedding is related to pH-induced activation of GPC, we quantitated the amount of metabolically labeled G1 shed from the cell surface during a 10-min incubation in low-pH media. Soluble G1 was collected by immunoprecipitation from the neutralized medium, deglycosylated, and resolved by SDS-PAGE. As illustrated in Fig. 6A, shedding by the wild-type GPC complex was first evident at pH 5.0, coincident with optimal membrane fusion. Unlike fusion, however, shedding continued to increase as pH was lowered to pH 4.0. No additional G1 was released upon treatment of the cells at pH 2.5 (Fig. 6A), conditions likely to dissociate all mature G1 from the cell surface. In contrast to wild-type GPC, no G1 was shed from the K33A mutant at pH 5.0 or 4.0, despite ample amounts of G1 present on the cell surface (pH 2.5) (Fig. 6A).
FIG. 6.

Inhibition of pH-dependent G1 shedding by ST-294 and ST-193. (A) G1 shed into the low-pH medium from metabolically labeled cultures expressing wild-type (filled circles) or K33A (open circles) GPC was collected by immunoprecipitation and deglycosylated using peptide N-glycosidase F (New England Biolabs) (57). The G1 polypeptide was resolved by SDS-PAGE (image below graph) and quantitated (graph) using ImageGauge software (Fuji). G1 radioactivity is expressed in phosphostimulated luminescence units. No membrane-associated G1-G2 precursor was detected in the culture supernatant (not shown). The source of a faster-migrating G1 species released at pH 2.5 is unknown. (B) Cells expressing wild-type GPC were incubated in medium containing 50 μM of ST-294 and ST-161 or 25 μM ST-193 for 30 min. G1 shedding at pH 5.0 was determined by immunoprecipitation (image below histogram), and is expressed as the fraction of total amount of G1 eluted from the cell surface at pH 2.5. The data displayed are representative of results obtained from three independent experiments.
Taken together, these results suggest that G1 shedding from the GPC complex is closely correlated with pH-induced activation of membrane fusion. Although the functional significance of G1 shedding within the conformational cascade is unclear, it is likely either a necessary or collateral consequence of the pathway taken by GPC to membrane fusion. At least partial dissociation early in the process would be required to permit exposure of the sequestered G2 fusion subunit, insertion of the G2 fusion peptide into the host-cell membrane, and formation of the six-helix bundle structure (16, 17, 26, 29, 48). The lack of G1 shedding by the K33A mutant at pH 5.0 is consistent with its inability to mediate membrane fusion and supports the notion that this mutation stabilizes the prefusion GPC complex.
Shedding by the wild-type GPC at pH 5.0 was also reduced by pretreatment with either 50 μM ST-294 or 25 μM ST-193 (Fig. 6B). Each compound reduced shedding by ≥20-fold, consistent with the complete inhibition of membrane fusion (Fig. 3 and 5). By contrast, ST-161 is a very poor inhibitor of JUNV membrane fusion activity (20% inhibition at 25 μM [Fig. 5]). Accordingly, pretreatment of JUNV GPC with 50 μM ST-161 reduced G1 shedding minimally, to 50% of the untreated level (Fig. 6B). These findings extend the correlation between G1 shedding and membrane fusion activity and suggest that the SIGA fusion inhibitors act to stabilize the prefusion GPC complex against low pH.
Fusion inhibition can be overcome by reduced pH.
Stabilization of the prefusion GPC complex can be accomplished by mutation at K33 or by inhibitor binding. Substitutions at K33 lower the pH at which membrane fusion is activated (57), and we wanted to determine whether inhibition by these small molecules might be functionally linked to this pH-responsive mechanism in GPC. If the metastable prefusion state of GPC were stabilized by inhibitor binding in opposition to pH-induced destabilization, we reasoned that increased H+ concentration might in some cases shift the balance between these equilibria and overcome inhibition. This effect would manifest as an increase in cell-cell fusion as pH is reduced in the presence of the inhibitor.
To test this hypothesis, we sought a relatively weak inhibitor to establish experimental conditions under which the low binding affinity of the inhibitor might be more readily overcome by an increase in H+ concentration. ST-193 had originally been identified as an inhibitor of LASV GPC and was subsequently found to be active against New World arenaviruses (33). Although this molecule shows nanomolar potency against both Old World and New World GPC in pseudotyped virions (33), the IC50 against JUNV GPC in the cell-cell fusion assay is markedly poorer than that with LASV GPC (0.9 μM versus ≪50 nM, respectively) (Fig. 5, right panel). This may, in part, reflect differences between the two assays. Based on this observation, however, we chose to determine whether inhibition of cell-cell fusion by 25 μM ST-193, which is effective at pH 5.0, can be overcome at lower pH.
Indeed, whereas cell-cell fusion at pH 5.0 was inhibited by 90% using ST-193, membrane fusion was restored at pH 4.5 (Fig. 7A). The maximal extent of fusion in the presence of ST-193 occurred at pH 4.5 and was only slightly reduced relative to that in the absence of inhibitor (at pH 5.0). When the pH was lowered to 4.0, fusion by both treated and untreated GPCs was markedly reduced, possibly due to inactivation of the complex by excess protonation. We observed the same depression in the pH of fusion by the K33H mutant of GPC using ST-161. In the absence of inhibitor, the K33H mutation itself reduces the pH threshold for membrane fusion such that fusion is now maximal at pH 4.5 (Fig. 7B). In the presence of 50 μM ST-161, the optimal pH was further reduced to ≤4.0. The maximum extent of fusion with ST-161 (at pH 4.0) was comparable to that in the absence of inhibitor (at pH 4.5). In contrast to the case for wild-type GPC, the downward shift in pH in the K33H mutant appeared to protect against inactivation at pH 4.0. Together, our findings that inhibition by ST-193 in wild-type GPC and inhibition by ST-161 in the K33H mutant can be overcome experimentally by reduced pH are consistent with a model of the prefusion GPC complex in which the stabilizing effects of inhibitor binding and the destabilizing effects of protonation are opposing and additive.
FIG. 7.

Inhibition of JUNV GPC is sensitive to pH. Cells expressing wild-type JUNV GPC (A and C) or K33H GPC (B) by trans-complementation were treated with 25 μM ST-193 (panel A, open squares), 50 μM ST-161 (panel B, open triangles), 50 μM ST-294 (panel C, open circles), or left untreated (all filled shapes). Cultures were pulsed for 10 min with medium at the indicated pH, and the extent of cell-cell fusion was determined (as RLUs). Error bars represent ±1 standard deviation. In panel D, we plot the pH sensitivities of wild-type GPC (filled circles) and the ST-294-resistant D400A mutant (filled squares). Sensitive mutants shown (open grey symbols) are: E405A (circles), E410A (upright triangles), K417A (inverted triangles), and D424A (diamonds). Fusion activity (in RLUs) is normalized to that at pH 5.0 to remove differences in intrinsic activity among the mutants. Although the results are representative of results for at least three independent partial and complete experiments, the respective GPCs are not statistically distinguishable in these studies. Error bars have been omitted for clarity.
In contrast to ST-193, inhibition of JUNV GPC by ST-294 remained intact throughout the useable pH range (Fig. 7C). It is possible that still-lower pH levels would be needed to counteract the activity of ST-294, but these extreme cell culture conditions were not examined. The basis for the difference in the behaviors of ST-193 and ST-294 is not readily apparent from their respective IC50 values, which are comparable (0.9 and 4 μM). It is possible that overt inhibition involves both the binding affinity of the compound and its inherent ability to interfere in the activation process. Compounds that differ in these components of inhibition may respond differently to pH. These factors may have been differentially selected for during screening and lead optimization. It is worth noting that ST-294 was identified through its inhibition of New World virus replication (3), whereas ST-193 and ST-161 were developed as inhibitors of Old World GPC (33). Furthermore, the pH requirement for viral entry in the endosome is itself undefined, and thus, in vitro measures of inhibitor potency at pH 5.0 may differ from those determined by virus infection.
The observed pH dependence in inhibitor potency raises the possibility that resistance in the D400A mutant GPC (Fig. 4) might also reflect a change in its intrinsic sensitivity to acidic pH. As shown in Fig. 7D, the effect of the D400A mutation on the pH of membrane fusion, if any, was quite small compared to that of K33H. The slight increase in both fusion activity at pH 5.5 and lability at pH 4.5 (relative to those of the wild type) is clearly not correlated with ST-294 resistance in the D400A mutant, as a similar pattern is observed in ST-294-sensitive E410A and D424A mutants. Although E405A and K417A GPCs exhibit the opposite trend in pH sensitivity, we should emphasize that we cannot determine at present whether these charged residues are involved in the activation of membrane fusion.
DISCUSSION
Membrane fusion must be appropriately controlled to enable productive virus entry. The generally accepted model for activation and progression to membrane fusion by class I viral fusion proteins has been developed over several decades of now-classic research (reviewed in references 16, 17, 26, 29, and 48). The class I envelope glycoproteins are assembled as stable but inert precursors that require proteolytic cleavage for fusion activity. The fusion-competent envelope glycoprotein complex is thought to exist in a metastable state, established on proteolytic maturation and maintained by a balance of stabilizing and destabilizing forces within the protein. For viruses such as human immunodeficiency virus type 1 that enter through the plasma membrane, the membrane fusion activity is subsequently triggered by binding to cell surface receptor(s) at neutral pHs. Other viruses, including the arenaviruses and orthomyxoviruses, are endocytosed and exposed to acidic pH in order to activate the fusion process. Upon activation, all class I envelope glycoproteins undergo a structural reorganization that follows a thermodynamically determined path toward formation of the stable six-helix bundle and fusion of the virus and cell membranes. Our results suggest that the SIGA inhibitors prevent membrane fusion by stabilizing the prefusion GPC complex against activation at low pH.
The molecular basis for pH-induced activation of envelope glycoprotein-mediated membrane fusion is largely undefined. Despite detailed structural knowledge of membrane fusion in the best-studied pH-dependent envelope glycoprotein, influenza virus hemagglutinin (HA), key elements of the activation process remain to be completely elucidated (9, 43, 50). Studies of tert-butyl hydroquinone HA fusion inhibitors have demonstrated the metastable nature of the prefusion HA complex and the ability of small molecules to stabilize against acidic pHs (2, 27). In accordance with this mechanism of action, tert-butyl hydroquinone-resistant mutants were found to display an opposing increase in their pH of fusion (27). When mapped onto the atomic structure of the neutral-pH form of HA, these mutations cluster at the HA1-HA2 interface and proximate to the fusion peptide at the N terminus of HA2 (27). The possible role of the fusion peptide and surrounding structures in stabilizing and destabilizing the prefusion HA complex was independently highlighted by directed mutagenesis in this region and in studies of the pH-associated effects of cell-culture adaptation and amantadine resistance (11, 35, 52). It remains uncertain, however, whether the determinant for pH-induced activation of HA is distributed or localized to a specific region.
Our current model of the GPC complex proposes a pH-sensitive interaction between the ectodomains of SSP and G2. This interaction is stable at neutral pHs and thereby maintains the GPC complex in its prefusion state. The interaction is destabilized upon protonation at low pH to initiate the conformational cascade leading to class I type membrane fusion. We now demonstrate that amino acid determinants for inhibition by the SIGA compounds likewise reside in SSP and G2. Together, our observations suggest that these novel fusion inhibitors act by interfering with the ability of the SSP-G2 interface to sense acidic pH or, subsequently, to respond productively. This provides a starting point in efforts to characterize the molecular basis for pH-induced activation in GPC. The abundance of charged residues in the membrane-proximal ectodomain of G2 raises the possibility that the pH-sensing interface may comprise an ensemble of titratable interactions with K33 in SSP, similar perhaps to that recently described in the acid-sensing ion channel 1 (30). Although atomic-resolution structure of the prefusion GPC complex is far in the distance, localization of an element of the pH sensor to SSP may facilitate efforts to characterize the molecular determinants for pH-induced activation and its inhibition by small-molecule compounds.
The unique subunit organization of GPC clearly provides a robust molecular pocket for binding small molecules that are capable of stabilizing the complex against acidic pH. At least three chemically distinct classes of inhibitors, independently identified in different arenavirus species, appear to target a common site at the SSP-G2 interface. As with the chemically diverse nonnucleoside analog inhibitors of human immunodeficiency virus type 1 reverse transcriptase, all of which bind a common hydrophobic pocket (12, 31, 44), it is possible that the bound forms of the SIGA inhibitors share spatial and chemical characteristics. It will surely be of interest to determine whether the chemically distinct GPC-directed fusion inhibitors described by Lee and colleagues (34) bind similarly. If identified, this pharmacophore core may provide scaffolding in the design of potent and broadly active second-generation inhibitors. To date, both ST-294 and ST-193 have shown promise in small-animal efficacy studies (3, 33). Our observation that inhibition by these compounds is profoundly sensitive to pH suggests that coadministration with drugs that reduce acidification in the endosome might further enhance their therapeutic efficacy.
Acknowledgments
We are grateful to Meg Trahey (The University of Montana) and Min Lu (Weill Medical College of Cornell University) for their editorial comments and assistance. J.H.N. is grateful to Tove’ C. Bolken and Dennis E. Hruby (SIGA Technologies) for their enthusiastic support of the work. We also thank Jack Rose (Yale University) for the molecularly cloned VSV G cDNA. The JUNV G1-specific MAb BF11 was obtained from the CDC through the NIH Biodefense and Emerging Infections Research Resources Program.
This work was supported at The University of Montana by NIH research grant AI059355 and by the Rocky Mountain Regional Center of Excellence for Biodefense and Emerging Infectious Diseases at Colorado State University (U54 AI065357).
Footnotes
Published ahead of print on 3 September 2008.
REFERENCES
- 1.Agnihothram, S. S., J. York, M. Trahey, and J. H. Nunberg. 2007. Bitopic membrane topology of the stable signal peptide in the tripartite Junín virus GP-C envelope glycoprotein complex. J. Virol. 814331-4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodian, D. L., R. B. Yamasaki, R. L. Buswell, J. F. Stearns, J. M. White, and I. D. Kuntz. 1993. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 322967-2978. [DOI] [PubMed] [Google Scholar]
- 3.Bolken, T. C., S. Laquerre, Y. Zhang, T. R. Bailey, D. C. Pevear, S. S. Kickner, L. E. Sperzel, K. F. Jones, T. K. Warren, S. Amanda Lund, D. L. Kirkwood-Watts, D. S. King, A. C. Shurtleff, M. C. Guttieri, Y. Deng, M. Bleam, and D. E. Hruby. 2006. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antivir. Res. 6986-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrow, P., and M. B. A. Oldstone. 1994. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 1981-9. [DOI] [PubMed] [Google Scholar]
- 5.Buchmeier, M. J. 2002. Arenaviruses: protein structure and function. Curr. Top. Microbiol. Immunol. 262159-173. [DOI] [PubMed] [Google Scholar]
- 6.Cao, W., M. D. Henry, P. Borrow, H. Yamada, J. H. Elder, E. V. Ravkov, S. T. Nichol, R. W. Compans, K. P. Campbell, and M. B. A. Oldstone. 1998. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 2822079-2081. [DOI] [PubMed] [Google Scholar]
- 7.Castilla, V., and S. E. Mersich. 1996. Low-pH-induced fusion of Vero cells infected with Junin virus. Arch. Virol. 1411307-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 2000. Fatal illnesses associated with a new world arenavirus—California, 1999-2000. MMWR Morb. Mortal. Wkly. Rep. 49:709-711. [PubMed] [Google Scholar]
- 9.Choi, H. S., J. Huh, and W. H. Jo. 2006. Electrostatic energy calculation on the pH-induced conformational change of influenza virus hemagglutinin. Biophys. J. 9155-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clegg, J. C. S. 2002. Molecular phylogeny of the arenaviruses. Curr. Top. Microbiol. Immunol. 2621-24. [DOI] [PubMed] [Google Scholar]
- 11.Daniels, R. S., J. C. Downie, A. J. Hay, M. Knossow, J. J. Skehel, M. L. Wang, and D. C. Wiley. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40431-439. [DOI] [PubMed] [Google Scholar]
- 12.Das, K., S. G. Sarafianos, A. D. Clark, Jr., P. L. Boyer, S. H. Hughes, and E. Arnold. 2007. Crystal structures of clinically relevant Lys103Asn/Tyr181Cys double mutant HIV-1 reverse transcriptase in complexes with ATP and non-nucleoside inhibitor HBY 097. J. Mol. Biol. 36577-89. [DOI] [PubMed] [Google Scholar]
- 13.Delgado, S., B. R. Erickson, R. Agudo, P. J. Blair, E. Vallejo, C. G. Albariño, J. Vargas, J. A. Comer, P. E. Rollin, T. G. Ksiazek, J. G. Olson, and S. T. Nichol. 2008. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog. 4e1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Simone, C., and M. J. Buchmeier. 1995. Kinetics and pH dependence of acid-induced structural changes in the lymphocytic choriomeningitis virus glycoprotein complex. Virology 2093-9. [DOI] [PubMed] [Google Scholar]
- 15.Di Simone, C., M. A. Zandonatti, and M. J. Buchmeier. 1994. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology 198455-465. [DOI] [PubMed] [Google Scholar]
- 16.Earp, L. J., S. E. Delos, H. E. Park, and J. M. White. 2005. The many mechanisms of viral membrane fusion proteins, p. 25-66. In M. Marsh (ed.), Membrane trafficking in viral replication, vol. 285. Springer Verlag, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70777-810. [DOI] [PubMed] [Google Scholar]
- 18.Eichler, R., O. Lenz, T. Strecker, M. Eickmann, H. D. Klenk, and W. Garten. 2003. Identification of Lassa virus glycoprotein signal peptide as a trans-acting maturation factor. EMBO Rep. 41084-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eschli, B., K. Quirin, A. Wepf, J. Weber, R. Zinkernagel, and H. Hengartner. 2006. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J. Virol. 805897-5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flanagan, M. L., J. Oldenburg, T. Reignier, N. Holt, G. A. Hamilton, V. K. Martin, and P. M. Cannon. 2008. New World clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 82938-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Follis, K. E., S. J. Larson, M. Lu, and J. H. Nunberg. 2002. Genetic evidence that interhelical packing interactions in the gp41 core are critical for transition of the human immunodeficiency virus type 1 envelope glycoprotein to the fusion-active state. J. Virol. 767356-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu, Y. K., T. K. Hart, Z. L. Jonak, and P. J. Bugelski. 1993. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J. Virol. 673818-3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuerst, T. R., E. G. Niles, F. W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 838122-8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallaher, W. R., C. DiSimone, and M. J. Buchmeier. 2001. The viral transmembrane superfamily: possible divergence of arenavirus and filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghiringhelli, P. D., R. V. Rivera-Pomar, M. E. Lozano, O. Grau, and V. Romanowski. 1991. Molecular organization of Junin virus S RNA: complete nucleotide sequence, relationship with other members of the Arenaviridae and unusual secondary structures. J. Gen. Virol. 722129-2141. [DOI] [PubMed] [Google Scholar]
- 26.Harrison, S. C. 2005. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 64231-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman, L. R., I. D. Kuntz, and J. M. White. 1997. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J. Virol. 718808-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hruby, D. E., D. L. Lynn, and J. R. Kates. 1980. Identification of a virus-specified protein in the nucleus of vaccinia virus-infected cells. J. Gen. Virol. 47293-299. [DOI] [PubMed] [Google Scholar]
- 29.Hughson, F. M. 1997. Enveloped viruses: a common mode of membrane fusion? Curr. Biol. 7R565-R569. [DOI] [PubMed] [Google Scholar]
- 30.Jasti, J., H. Furukawa, E. B. Gonzales, and E. Gouaux. 2007. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449316-323. [DOI] [PubMed] [Google Scholar]
- 31.Kohlstaedt, L. A., J. Wang, J. M. Friedman, P. A. Rice, and T. A. Steitz. 1992. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 2561783-1790. [DOI] [PubMed] [Google Scholar]
- 32.Krueger, D. K., S. M. Kelly, D. N. Lewicki, R. Ruffolo, and T. M. Gallagher. 2001. Variations in disparate regions of the murine coronavirus spike protein impact the initiation of membrane fusion. J. Virol. 752792-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson, R. A., D. Dai, V. T. Hosack, Y. Tan, T. C. Bolken, D. E. Hruby, and S. M. Amberg. 2008. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol. 8210768-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee, A. M., J. M. Rojek, C. F. Spiropoulou, A. T. Gundersen, W. Jin, A. Shaginian, J. York, J. H. Nunberg, D. L. Boger, M. B. A. Oldstone, and S. Kunz. 2008. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem. 28318734-18742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin, Y. P., S. A. Wharton, J. Martín, J. J. Skehel, D. C. Wiley, and D. A. Steinhauer. 1997. Adaptation of egg-grown and transfectant influenza viruses for growth in mammalian cells: selection of hemagglutinin mutants with elevated pH of membrane fusion. Virology 233402-410. [DOI] [PubMed] [Google Scholar]
- 36.McCormick, J. B., P. A. Webb, J. W. Krebs, K. M. Johnson, and E. S. Smith. 1987. A prospective study of the epidemiology and ecology of Lassa fever. J. Infect. Dis. 155437-444. [DOI] [PubMed] [Google Scholar]
- 37.Moore, J. P., J. A. McKeating, W. A. Norton, and Q. J. Sattentau. 1991. Direct measurement of soluble CD4 binding to human immunodeficiency virus type 1 virions: gp120 dissociation and its implications for virus-cell binding and fusion reactions and their neutralization by soluble CD4. J. Virol. 651133-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore, J. P., J. A. McKeating, R. A. Weiss, and Q. J. Sattentau. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 2501139-1142. [DOI] [PubMed] [Google Scholar]
- 39.Nussbaum, O., C. C. Broder, and E. A. Berger. 1994. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. J. Virol. 685411-5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters, C. J. 2002. Human infection with arenaviruses in the Americas. Curr. Top. Microbiol. Immunol. 26265-74. [DOI] [PubMed] [Google Scholar]
- 41.Radoshitzky, S. R., J. Abraham, C. F. Spiropoulou, J. H. Kuhn, D. Nguyen, W. Li, J. Nagel, P. J. Schmidt, J. H. Nunberg, N. C. Andrews, M. Farzan, and H. Choe. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 44692-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radoshitzky, S. R., J. H. Kuhn, C. F. Spiropoulou, C. G. Albariño, D. P. Nguyen, J. Salazar-Bravo, T. Dorfman, A. S. Lee, E. Wang, S. R. Ross, H. Choe, and M. Farzan. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc. Natl. Acad. Sci. USA 1052664-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Remeta, D. P., M. Krumbiegel, C. A. Minetti, A. Puri, A. Ginsburg, and R. Blumenthal. 2002. Acid-induced changes in thermal stability and fusion activity of influenza hemagglutinin. Biochemistry 412044-2054. [DOI] [PubMed] [Google Scholar]
- 44.Ren, J., R. Esnouf, A. Hopkins, C. Ross, Y. Jones, D. Stammers, and D. Stuart. 1995. The structure of HIV-1 reverse transcriptase complexed with 9-chloro-TIBO: lessons for inhibitor design. Structure 3915-926. [DOI] [PubMed] [Google Scholar]
- 45.Salazar-Bravo, J., L. A. Ruedas, and T. L. Yates. 2002. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 26225-63. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez, A., D. Y. Pifat, R. H. Kenyon, C. J. Peters, J. B. McCormick, and M. P. Kiley. 1989. Junin virus monoclonal antibodies: characterization and cross-reactivity with other arenaviruses. J. Gen. Virol. 701125-1132. [DOI] [PubMed] [Google Scholar]
- 47.Sanders, R. W., M. Vesanen, N. Schuelke, A. Master, L. Schiffner, R. Kalyanaraman, M. Paluch, B. Berkhout, W. C. Olson, M. Lu, and J. P. Moore. 2002. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 768875-8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69531-569. [DOI] [PubMed] [Google Scholar]
- 49.Spiropoulou, C. F., S. Kunz, P. E. Rollin, K. P. Campbell, and M. B. A. Oldstone. 2002. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J. Virol. 765140-5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinhauer, D. A., J. Martin, Y. P. Lin, S. A. Wharton, M. B. Oldstone, J. J. Skehel, and D. C. Wiley. 1996. Studies using double mutants of the conformational transitions in influenza hemagglutinin required for its membrane fusion activity. Proc. Natl. Acad. Sci. USA 9312873-12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thali, M., C. Furman, E. Helseth, H. Repke, and J. Sodroski. 1992. Lack of correlation between soluble CD4-induced shedding of the human immunodeficiency virus type 1 exterior envelope protein and subsequent membrane fusion events. J. Virol. 665516-5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thoennes, S., Z. N. Li, B. J. Lee, W. A. Langley, J. J. Skehel, R. J. Russell, and D. A. Steinhauer. 2008. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370403-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.York, J., S. S. Agnihothram, V. Romanowski, and J. H. Nunberg. 2005. Genetic analysis of heptad-repeat regions in the G2 fusion subunit of the Junin arenavirus envelope glycoprotein. Virology 343267-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.York, J., and J. H. Nunberg. 2007. Distinct requirements for signal peptidase processing and function of the stable signal peptide (SSP) subunit in the Junin virus envelope glycoprotein. Virology 35972-81. [DOI] [PubMed] [Google Scholar]
- 55.York, J., and J. H. Nunberg. 2007. A novel zinc-binding domain is essential for formation of the functional Junín virus envelope glycoprotein complex. J. Virol. 8113385-13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.York, J., and J. H. Nunberg. 2004. Role of hydrophobic residues in the central ectodomain of gp41 in maintaining the association between human immunodeficiency virus type 1 envelope glycoprotein subunits gp120 and gp41. J. Virol. 784921-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.York, J., and J. H. Nunberg. 2006. Role of the stable signal peptide of the Junín arenavirus envelope glycoprotein in pH-dependent membrane fusion. J. Virol. 807775-7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.York, J., V. Romanowski, M. Lu, and J. H. Nunberg. 2004. The signal peptide of the Junín arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1-G2 complex. J. Virol. 7810783-10792. [DOI] [PMC free article] [PubMed] [Google Scholar]